
Abstract
Hypopigmentation along Blaschko’s lines is a hallmark of a poorly defined group of mosaic syndromes whose genetic causes are unknown. Here we show that postzygotic inactivating mutations of RHOA cause a neuroectodermal syndrome combining linear hypopigmentation, alopecia, apparently asymptomatic leukoencephalopathy, and facial, ocular, dental and acral anomalies. Our findings pave the way toward elucidating the etiology of pigmentary mosaicism and highlight the role of RHOA in human development and disease.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Change history
07 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
14 October 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Sybert, V. P. J. Invest. Dermatol. 103, 141S–143S (1994).
Pavone, P., Praticò, A. D., Ruggieri, M. & Falsaperla, R. Neurol. Sci. 36, 1173–1180 (2015).
Canman, J. C. et al. Science 322, 1543–1546 (2008).
Hall, A. Science 279, 509–514 (1998).
Vetter, I. R. & Wittinghofer, A. Science 294, 1299–1304 (2001).
Pan, Z. K. et al. J. Immunol. 160, 3038–3045 (1998).
Zhao, X. et al. Cancer Res. 69, 483–491 (2009).
Maekawa, M. et al. Science 285, 895–898 (1999).
Mirzaa, G. M. et al. JAMA Neurol. 73, 836–845 (2016).
van Steensel, M. A. M. J. Pediatr. Genet. 4, 144–153 (2015).
Lek, M. et al. Nature 536, 285–291 (2016).
Blomen, V. A. et al. Science 350, 1092–1096 (2015).
Wang, T. et al. Science 350, 1096–1101 (2015).
Happle, R. Hum. Genet. 72, 280 (1986).
Fernández, L. C., Torres, M. & Real, F. X. Nat. Rev. Cancer 16, 43–55 (2016).
Kuentz, P. et al. Genet. Med. 19, 989–997 (2017).
Zhang, S., Zhou, X., Lang, R. A. & Guo, F. PLoS ONE 7, e33773 (2012).
Tong, L. & Tergaonkar, V. Biosci. Rep. 34, e00115 (2014).
Ordóñez-Morán, P. et al. J. Cell Biol. 183, 697–710 (2008).
Rodrigues, P. et al. Nat. Commun. 5, 5458 (2014).
Thevenon, J. et al. Clin. Genet. 89, 700–707 (2016).
Rivière, J.-B. et al. Nat. Genet. 44, 934–940 (2012).
Cooper, G. M. et al. Nat. Methods 7, 250–251 (2010).
Kircher, M. et al. Nat. Genet. 46, 310–315 (2014).
Acknowledgements
We thank the participants and families involved in the study. We also thank the University of Burgundy Centre de Calcul (https://haydn2005.u-bourgogne.fr/dsi-ccub/) for technical support and management of the informatics platform. This work was funded by the Agence Nationale de la Recherche (grant no. ANR-13-PDOC-0029 to J.-B.R.), the Programme Hospitalier de Recherche Clinique National 2010 (grant no. NCT01950975 to P.V.), the NIH (grant no. HD067244 to M.E.R.) and the Société Française de Dermatologie. G.B. has received a Research Scholar Junior 1 (2012–2016) salary award from the Fonds de Recherche du Québec en Santé and the New Investigator salary award (2017–2022) from the Canadian Institute for Health Research (no. MOP-G-287547, file no. 24805). V.K. was supported by the Wellcome Trust and by the NIHR.
Author information
Authors and Affiliations
Contributions
P.V. and J.-B.R. designed the study. A.S., J.S.-O., P.K., J.-B.C., V.C., S.P. and V.A.K. performed the genetics experiments. J.-B.R., Y.D. and P.G. performed the bioinformatics experiments. S.S.K. performed the functional experiments. P.V., A.S., B.Demeer, D.B., O.B., A.B., G.C., E.C., C.T., S.P., F.F., V.A.K., B.Devauchelle, D.G., C.G.-J., A.L., M.M.-D., J.T. and L.F. recruited and evaluated the study participants. L.G., G.B. and W.B.D. analyzed the brain MRI. P.V., L.F., M.E.R. and J.-B.R. supervised the study. P.V., A.S., S.S.K, M.E.R. and J.-B.R. wrote the manuscript. All authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
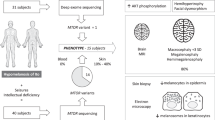
Supplementary Fig. 1 Main clinical features of affected individuals.
a, Craniofacial appearance of affected individuals showing hemifacial microsomia in all subjects. b, Patchy alopecia of hair and beard. c, Teeth anomalies consisting of oligodontia, microdontia, and conical teeth. d, Variable degrees of acral anomalies including symmetric or asymmetric brachydactyly, syndactyly, and polydactyly. Photograph of subject S3 was taken after surgery. e, Linear hypopigmentation following Blaschko’s lines on trunk, limbs, and face. See Supplementary Table 1 for additional details. We obtained written consent to publish photographs of these individuals.
Supplementary Fig. 2 Brain magnetic resonance imaging (MRI) for subjects S1, S2 and S4.
Brain magnetic resonance imaging (MRI) for subjects S1 (at 15 years and 3 months (first row) and six months later (second row)), S2 (third row), and S4 (fourth row). MRI revealed bilateral and symmetrical marked and diffuse hyperintensities on FLAIR images, with sparing of the subcortical white matter/U-fibers. White matter signal abnormalities extend to the anterior limb of the internal capsule, with sparing of the posterior limb of the capsule, associated with either cystic formation (S1) or relative dilatation of the Virchow-Robin spaces (S1, S2, and S4). Note that the corpus callosum is spared. No significant anomalies of thalami and infratentorial cerebellar matter were noted in patients S2 and S4. A posterior fossa cyst is observed in 2 out of 3 patients (S2 and S4), with minimal mass effect on the cerebellum. One focal lesion of the cerebellar white matter was noticed in S1, with a hyperintensity on T2 weighted images. Lesions were stable on the 6-months follow-up MRI for subject S1. It is of interest to note that a leukoencephalopathy with enlarged Virchow-Robin spaces has been described in patients with mutations of PTEN (Vanderver, A. et al. Am. J. Med. Genet. A. 164A, 627–633 (2014)), a gene that encodes an inhibitor of the PI3K-AKT-mTOR signaling pathway and is known to be regulated by RHOA (Li, Z. et al. Nat. Cell Biol. 7, 399–404 (2005)).
Supplementary Fig. 3
Integrative Gemomics Viewer (IGV) screenshots of the de novo postzygotic RHOA c.139G>A substitution (encoding p.(Glu47Lys)) in subject S1 and her parents.
Supplementary Fig. 4
IGV screenshots of the de novo postzygotic RHOA c.139G>A substitution (encoding p.(Glu47Lys)) in subject S2 and his parents.
Supplementary Fig. 5
IGV screenshots of the de novo postzygotic RHOA c.211C>T substitution (encoding p.(Pro71Ser)) in subject S3 and her parents.
Supplementary Fig. 6
IGV screenshots of the NC_012920.1:m.11778G>A, MT-ND4 c.1019G>A, p.(R340H) substitution in S2 (left) and his mother (right).
Supplementary Fig. 7 Expression levels of overexpressed wild-type or mutant myc-tagged RhoA, and endogenous RhoA, total MYPT1, phospho-MYPT1 (pThr696), total MLC2, actin, and phospho-MLC2 (pThr19).
a, Representative Western blots show similar protein loading (endogenous total MYPT1, actin, endogenous RhoA, and total MLC2) and similar overexpression of myc-tagged wild-type and mutant RhoA (three independent experiments). There is a visible reduction in phosphorylated MYPT1(pThr696) and MLC2(pThr19) when RhoA(Thr19Asn) or RhoA(Glu47Lys) are overexpressed. b,c, Levels of phosphorylated MYPT1(pThr696) and MLC2(pThr19) in endogenous and transfected NIH/3T3 cells. Cumulative data of average density values (overlaid with dots from the three independent experiments) for phosphorylated MLC2(pThr19) (b) and MYPT1(pThr696) (c) normalized to total MLC2 and MYPT1, respectively, indicate reduction in MLC2(pThr19) and MYPT1(pThr696) upon RhoA(Thr19Asn) or RhoA(Glu47Lys) overexpression.
Supplementary Fig. 8
Full scans of blot used in Fig. 2.
Supplementary information
Supplementary Information
Supplementary Figs. 1–8 and Tables 1–10
Rights and permissions
About this article

Cite this article
Vabres, P., Sorlin, A., Kholmanskikh, S.S. et al. Postzygotic inactivating mutations of RHOA cause a mosaic neuroectodermal syndrome. Nat Genet 51, 1438–1441 (2019). https://doi.org/10.1038/s41588-019-0498-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0498-4
This article is cited by
-
Monogenic causes of pigmentary mosaicism
Human Genetics (2022)
-
RHO GTPases: from new partners to complex immune syndromes
Nature Reviews Immunology (2021)
-
Postzygotic inactivating mutation of KIF13A located at chromosome 6p22.3 in a patient with a novel mosaic neuroectodermal syndrome
Journal of Human Genetics (2021)
-
Clinical spectrum of MTOR-related hypomelanosis of Ito with neurodevelopmental abnormalities
Genetics in Medicine (2021)
-
Rare genetic causes of complex kidney and urological diseases
Nature Reviews Nephrology (2020)
