
Abstract
Maize is one of the most important crops globally, and it shows remarkable genetic diversity. Knowledge of this diversity could help in crop improvement; however, gold-standard genomes have been elucidated only for modern temperate varieties. Here, we present a high-quality reference genome (contig N50 of 15.78 megabases) of the maize small-kernel inbred line, which is derived from a tropical landrace. Using haplotype maps derived from B73, Mo17 and SK, we identified 80,614 polymorphic structural variants across 521 diverse lines. Approximately 22% of these variants could not be detected by traditional single-nucleotide-polymorphism-based approaches, and some of them could affect gene expression and trait performance. To illustrate the utility of the diverse SK line, we used it to perform map-based cloning of a major effect quantitative trait locus controlling kernel weight—a key trait selected during maize improvement. The underlying candidate gene ZmBARELY ANY MERISTEM1d provides a target for increasing crop yields.
Similar content being viewed by others

Main
Maize (Zea mays subspecies mays) is one of the most important crops globally, with an annual production greater than 1 billion tons1, and it has been a genetic model system for over a century. Maize was domesticated from teosinte (Z. mays subspecies parviglumis) about 9,000 years ago in a tropical environment in southwestern Mexico2,3, and then migrated north and east to more temperate regions. The remarkable phenotypic and genetic diversity4 between different maize lines is greater than that between humans and chimpanzees5. Structural variants (SVs), including deletions, insertions, inversions and translocations, contribute to genome diversity6,7,8, and play an important role in maize phenotypic variation7,9. However, the contribution of SVs to traits and gene regulation cannot be fully explored in haplotype maps based on a single reference genome. Indeed, characterizing the phenotypic consequences of SVs across the genome and at a population level presents tremendous biological and computational challenges, but reads originating from more complex polymorphisms often align poorly, resulting in biased genotype estimates10. The existing high-quality maize reference genomes are derived from temperate accessions6,11,12,13, and therefore capture only a subset of genetic diversity. Recent studies achieved high-resolution SV mapping in great ape lineages, based on comparative analysis of several high-quality great ape genomes14, and a new algorithmic approach (BayesTyper) enabled more reliable genotyping of SVs using short-read technology10. Here, we present a new and diverse tropical maize reference genome, providing an unprecedented opportunity to explore the structural variations in maize genomes, and to mine novel genetic variation for crop improvement.
A number of common traits, including seed size and weight15, were selected during crop domestication and improvement, and involved changes in a small number of genes16. In maize, tens of seed size genes have been identified by mutagenesis17; however, few quantitative trait loci (QTLs) have been cloned, limiting their application in breeding programs. The small-kernel (SK) line is an inbred line derived from a tropical landrace18 (Supplementary Fig. 1) with small kernels and a low hundred-kernel weight (HKW) value (Fig. 1a). To produce a high-quality genome of this highly divergent line, we combined multiple approaches to produce a de novo assembly that is better than the improved maize B73 version 4 reference6 (denoted B73 hereafter; SK size: 2,161 megabase pairs (Mb) versus 2,106 Mb for B73; contig N50: 15.78 Mb versus 1.18 Mb; gaps: 238 versus 2,522) and thus provide an outstanding resource for the research community. We demonstrate the value of this genome through the fine mapping and cloning of a kernel size and weight QTL, providing a new opportunity for maize breeding.

a, Top, SK plant, ear and kernels. Bottom, 100 kernels of teosinte (Z. mays subspecies parviglumis; ACC.27479 from the International Maize and Wheat Improvement Center), SK and ZHENG58 are shown. Scale bars: plant = 1 m; ear and kernels = 1 cm. b, Overview of the pipeline used for assembly of the SK genome. c, Comparison of SNP markers on the physical map (x axis) with their position on the genetic map (y axis), for chromosome 1 of the SK genome. Each marker is depicted as a dot on the plot. d, Heat map of chromosome 1 chromatin contact matrices generated by aligning a ChIA-PET dataset of RNA polymerase II binding sites to the SK genome. The frequency of interactions was calculated using a window size of 1 Mb. A 1.7-Mb inversion on chromosome 1 between B73 and SK was supported well by the ChIA-PET data. e, Marker alignments between the SK assembly and B73 assembly. The black rectangle represents the 1.7-Mb inversion and the red rectangle indicates the flanking sequence around the inversion. The red arcs indicate putative chromatin interactions in SK and B73. The blue arcs indicate the ‘bow tie’ configuration.
Genome sequencing, assembly and scaffolding
To perform a de novo assembly of the SK genome, we integrated four sequencing and assembly technologies (Fig. 1b). In total, over 84-fold coverage of sequence data was generated using PacBio Sequel technology (196 gigabase pairs (Gb); ~16 million subreads; mean length: 12,026 base pairs (bp)), and ~229-fold coverage of Illumina paired-end and mate-pair reads was generated with libraries constructed from six different insert sizes (532 Gb; Supplementary Table 1). The SK genome size was estimated to be 2.32 Gb based on k-mer analysis. The PacBio reads were first assembled using FALCON19 and later improved by supplementing with Illumina data. We then generated an approximately 290-fold-coverage BioNano optical map to generate a consensus map and a second assembly of 870 scaffolds with an N50 of 25.65 Mb. Gaps in this assembly were filled using PacBio reads with PBjelly20, to generate assembly 3. The final assembly was generated by incorporating ~166-fold coverage of 10x Genomics Chromium sequence for further scaffolding using the assembly roundup by chromium scaffolding (ARCS) pipeline21, and the final assembly yielded a predicted genome length of 2.16 Gb in 708 scaffolds with an N50 of 73.24 Mb and a contig N50 of 15.78 Mb after further gap filling (Table 1). This result, together with a high-density linkage map from a recombinant inbred population between SK and a widely adopted inbred of China, ZHENG58 (ref. 22), allowed the construction of ten pseudo-chromosomes. A total of 47 super scaffolds mapped to these chromosomes (total size: ~2,094 Mb) and an additional 151 scaffolds (total size: 26 Mb) were assigned to chromosomes, but their location and order could not be determined. The 510 remaining scaffolds with a total size of 41 Mb could not be assigned to chromosomes. The SK assembly had 238 gaps, compared with 2,522 gaps in B73 version 4, of which 48.3% (n = 115) had optical map coverage, giving an estimated median gap length of 23.3 kilobase pairs (kb) (Supplementary Table 2).
The quality of the SK genome was evaluated using five methods. First, we assessed the consistency of physical and genetic maps that were constructed with 2,796 representative single nucleotide polymorphism (SNP) loci23. We identified homology in the SK genome for 2,626 SNPs, 2,553 (97.52%) of which were located at their expected positions (Fig. 1c and Supplementary Fig. 2). Second, ten SK BACs were randomly selected from a newly constructed library and sequenced on the PacBio RSII platform. All ten sequences were highly linear with our assembly, with no structural variations and an average sequence identity of 99.64% (Supplementary Fig. 3). Third, 96.4% of the Plantae BUSCO24 genes could be aligned to the assembled SK genome (Supplementary Table 3), similarly to the Mo17 (ref. 12), W22 (ref. 13) and B73 version 4 reference6 genomes. Fourth, we used the LTR Assembly Index (LAI)25—a standard for evaluating the assembly of repeat sequences—to evaluate the assembly continuity. The assembly of SK had the highest LAI score and the best assembly continuity compared with B73 version 4 (ref. 6) and Mo17 (ref. 12) (Supplementary Fig. 4). Fifth, we aligned chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) data for RNA polymerase II26 to the SK genome assembly, and observed that chromatin interactions mainly occurred within close proximity to one another on the same chromosome, with no apparent interchromosomal hotspots, as expected (Fig. 1d and Supplementary Fig. 5). Excellent colinearity was found between the SK and B73 genomes (Supplementary Fig. 6), but we found 22 insertions, deletions or inversions greater than 1 Mb (Supplementary Table 4), and these were supported by ChIA-PET. For example, a characteristic ‘bow tie’27 configuration indicated a 1.7-Mb inversion on chromosome 1 when we mapped the SK ChIA-PET data onto B73 (Fig. 1e). Collectively, these data provide multiple lines of evidence that the SK genome assembly quality is extremely high, facilitating its use as a reference genome for intraspecific comparisons in maize.
Genome annotation
To determine the transposable-element content of the SK assembly, we used a modified approach (Supplementary Note) based on the annotation pipeline used for B73 (ref. 6). We identified ~90% of the genome as transposable-element sequences (the length of annotated transposable elements divided by the length of the SK assembly; Fig. 2), divided into retroelements (long terminal repeat (LTR) retroelements, 76.3%; non-LTR retroelements, 0.8%) and DNA transposons (6.7%) (Supplementary Table 5). We also re-annotated the B73 transposable elements by using the same modified pipeline. In general, the composition and number of transposable-element families was similar, except that there were over twice as many hAT family transposons in SK relative to B73, suggesting that mechanisms to regulate these elements may vary between the two genomes.

Tracks (outer to inner circles) indicate the following: a, contigs and gaps; b, transposable-element content (window size of 1 Mb with a step size of 200 kb); c, gene density (gene numbers per Mb; darker color indicates more genes); d, gene expression level (FPKM; the highest expression of nine sequenced tissues is shown); e, GC content (window size of 1 Mb); and f, SNPs/indels compared with the B73 genome (the outer and inner layers indicate SNPs and indels, respectively). Chr, chromosome. The outer gray circle represents the chromosome length of SK, with units in Mb.
b
A comprehensive strategy combining de novo gene prediction, protein-based homology searches, RNA sequencing (RNA-Seq) and isoform sequencing (Iso-Seq) of nine tissues (Supplementary Table 6) was used to annotate the genes (Supplementary Fig. 7). A total of 42,271 high-confidence protein-coding gene models with 95,938 transcripts were predicted (Fig. 2 and Supplementary Table 7), and 60.2% were supported by full-length transcripts (Supplementary Table 7). Of these, >98% were functionally annotated in public databases (Supplementary Table 8). Comparative analysis with maize B73, rice, Setaria, sorghum and Brachypodium revealed that a core set of 12,196 gene families were shared among all six grass genomes (Supplementary Fig. 8).
Structural variation analyses
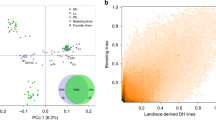
SVs represent a major source of genetic diversity, but they have not been well characterized on a population level in maize. Here, we focused on identifying SVs >10 bp between our tropical SK line and two maize genomes representing the major temperate heterotic groups: B73 (ref. 6; a stiff stalk line) and Mo17 (ref. 12; a non-stiff stalk line) (Supplementary Fig. 1). SVs were identified by mapping contigs of B73 and Mo17 to the SK genome using smartie-sv14. We identified 386,014 SVs ranging from 10–99,330 bp, and there are an additional 108,505 SVs when comparing Mo17 with B73. Next, we genotyped these 386,014 SVs in 521 diverse inbred lines derived from an association mapping panel28 using deep DNA resequencing data, resulting in 80,614 polymorphic SVs (pSVs) (Supplementary Note and Supplementary Fig. 9). By projecting these pSVs onto the SK genome, potential hotspots of structural variation were identified (Supplementary Fig. 10). We checked how frequently the common pSVs (minor allele frequency (MAF) > 5%) were linked to nearby SNPs, to determine whether they represent a previously unassessed source of genetic variation. Surprisingly, 21.9% of the common pSVs showed low linkage disequilibrium with nearby SNPs, suggesting they are a source of genetic diversity not discoverable by SNPs (details in Supplementary Note, Fig. 3a and Supplementary Fig. 11). Variants with high MAF were more often classified as high linkage disequilibrium (Supplementary Fig. 12), suggesting that some were under adaptive selection. To confirm the unique value of newly identified SVs, we used them to re-analyse a genome-wide association study for kernel oil concentration and fatty acid composition29,30. We indeed found a new significant locus for oil concentration and long-chain fatty acid composition (C18_1, C18_2 and C20_1) on chromosome 4 that could not be represented by local SNPs (Fig. 3b, Supplementary Fig. 13 and Supplementary Table 9). A total of 16 expressed genes were identified within the candidate region, including an obvious candidate, Zm00015a017119, which encodes enoyl-acyl carrier protein reductase (ENR), which catalyzes the last enzymatic step in the fatty acid elongation cycle31.

a, Histogram of the number of pSV r2 ranks (0–300) that are above the SNP-based median r2 value for common pSVs. b, Top, Manhattan plot of SNP (with ~11.5 million SNPs obtained from DNA deep resequencing (~20×) of 521 diverse lines) and pSV genome-wide association studies for oil concentration. The red line represents the candidate gene encoding the ENR. Bottom left, the expression of ENR is positively correlated with oil concentration. Bottom right, divergence of oil concentrations between different alleles of the lead pSV. n = 86 for B73 allele; n = 7 for SK allele. c, A 36,320-bp insertion in SK contains three expressed genes not present at the syntenic location in B73. d, A 29-bp SV in the 5′ untranslated region (UTR) was a cis eQTL of Zm00015a006294, and could be the causal variant reducing gene expression. The break points of this SV are shown. TSS, transcription start site. n = 75 for B73 allele; n = 129 for SK allele. In b and d, the lower and upper box edges correspond to the first and third quartiles (the twenty-fifth and seventy-fifth percentiles); the horizontal line indicates the median value; and the lower and upper whiskers correspond to the smallest value at most 1.5× IQR and the largest value no further than 1.5× IQR (where IQR is the inter-quartile range, or distance between the first and third quartiles).
To further ascertain the functional significance of pSVs, we annotated them and found that 1,864 included full-length coding sequences of 2,382 annotated genes, of which 77.6% were present in two or more copies in the genome. A total of 662 genes were deleted from SK relative to B73 and 443 genes were deleted from B73 relative to SK. In addition, 740 genes were deleted from SK relative to Mo17, and 537 genes were deleted from Mo17 relative to SK. One 36,320-bp insertion in SK contained three expressed genes (Fig. 3c) that were not present in B73. Other major large-effect variants, including the creation of 278 stop codons, 171 frame shifts, 1 stop codon loss and 1 start codon loss, were identified in comparisons of the pSVs of B73 versus SK32 (Supplementary Table 10). SVs have also been shown to modulate gene expression27, so we mapped cis expression QTLs (eQTLs) (considering a 1-Mb candidate region upstream and downstream of the coding regions) using 19,707 common pSVs and 11,496,863 SNPs with a MAF > 0.05. We used transcriptome data of 25,008 genes from kernels at 15 d after pollination from 368 inbred lines29 for joint eQTL analysis, and identified 207 eQTLs with a lead SV association and 17,632 with a lead SNP association (P < 10−3). In proportion to the number of variants tested, eQTLs were around sevenfold more likely to be detected by using pSVs compared with SNPs (P = 4.61 × 10−97, one-sided Fisher’s exact test; Supplementary Table 11), similarly to the case in humans8, suggesting that SVs have a disproportionate impact on gene expression. We also found that 3,864 pSVs were in strong linkage disequilibrium, with an additional 1,766 eQTLs with lead associations to SNPs (r2 > 0.5, squared coefficient of correlation). Those 1,973 eQTLs with a larger effect tended to overlap with genic regions (P = 4.4 × 10−4; Supplementary Fig. 14). An example is shown in Fig. 3d, where a 29-bp insertion in the 5′ untranslated region of Zm00015a006294 in SK correlated with decreased expression, and is likely the causal variant of the mapped eQTL (Fig. 3d). In total, 80.8% of the expression-associated pSVs were located in intergenic regions, and may affect chromatin loops. For example, the expression of Zm00015a037064 may be regulated by a 1,794-bp SV and, according to our ChIA-PET data, this could affect interactions with Zm00015a037064 or other flanking sequences (Supplementary Fig. 15). In total, we found 70 expression-associated pSVs that had chromatin interactions with gene-coding regions.
SK genome-assisted genetic dissection of yield traits
Kernel weight is an important yield-related trait that was selected during maize improvement. The HKW of ZHENG58 (an improved maize line with HKW = 28.2 g) is nearly six times higher than that of SK (HKW = 4.9 g), which is only about two times higher than the undomesticated ancestor teosinte (HKW = 2.9 g) (Fig. 1a). Eight QTLs for HKW in a ZHENG58 × SK recombinant inbred line (RIL) population were identified, and could explain 55% of the phenotypic variation22,33 (Fig. 4a), suggesting that a few genes have a major effect on kernel weight. One major QTL, qHKW1 on chromosome 1, explained 18.4% of the phenotypic variation (Fig. 4a). We fine mapped this QTL using approximately 13,800 individuals derived from one heterogeneous inbred family line34 (Supplementary Fig. 16) to an approximately 177-kb region (Fig. 4b). Only one candidate gene, Zm00001d028317, encoding a CLAVATA1 (CLV1)/BARELY ANY MERISTEM (BAM)-related receptor kinase-like protein (Fig. 4c), which localized on the plasma membrane (Supplementary Fig. 17), was identified in this region. Based on the phylogeny, we named it ZmBAM1d (Supplementary Fig. 18). CLV1/BAM genes control shoot meristem size35 and agronomic traits, such as kernel row number in maize or fruit size in tomato36,37, but have not been associated with seed size.

a, Eight QTLs (arrows) were identified for HKW in a ZHENG58 × SK RIL population. The y axis is the logarithm of the odds (LOD) value. b, qHKW1 was mapped to a ~177-kb region between markers M49 and M40 using ~13,800 individuals. c, The candidate region of qHKW1 contains a single gene, ZmBAM1d. The numbers inside circles indicate the seven large indels >100 bp. d, Left, plant architecture of overexpression (OE), CRISPR–Cas9 (Cas9) and wild-type plants (WT). Bar: 20 cm. Right, ZmBAM1d had no effect on kernel length, but there was a significant difference in kernel width between NILSK and NILZHENG58. Bar: 1 cm. e, Expression pattern of ZmBAM1d at different stages during seed development (n = 6 for seeds 5 and 10 d after pollination (DAP); n = 3 for the other stages; **P < 0.01). Data are shown as means ± s.d. f, Overexpression of ZmBAM1d resulted in a significant increase of HKW. n = 8 for OE (–); n = 11 for OE (+); **P < 0.01. g, Sequence alignment of the region covering ZmBAM1d. Numbers with circles indicated four large indels (>5 kb) found in the upstream of ZmBAM1d. Indels with the same number in c and g are the same. h, The 8.9-kb indel was positively associated with HKW (n = 261 for 0 kb; n = 170 for 8.9 kb; *P < 0.05). i, The DNA methylation level of the promoter region of ZmBAM1d (red box in c) was significantly higher in SK than B73 (n = 3; **P < 0.01). Data are shown as means ± sd. CHG and CHH indicate cytosine methylation in other sequence contexts (where H is A, T or C). In f and h, the lower and upper box edges correspond to the first and third quartiles (the twenty-fifth and seventy-fifth percentiles); the horizontal line indicates the median value; and the lower and upper whiskers correspond to the smallest value at most 1.5× IQR and the largest value no further than 1.5× IQR.
Next, we used NIL lines to test whether variation in ZmBAM1d was responsible for HKW variation. As expected, we found a significant difference in kernel size between NILSK and NILZHENG58 (P = 1.27 × 10−3) (Fig. 4d and Supplementary Table 12). The expression of ZmBAM1d was significantly higher in the big kernel line NILZHENG58 than in NILSK (measured at 20 d after pollination; 3.8-fold difference; P = 1.34 × 10−3; Fig. 4e). To confirm that higher expression of this gene increased the kernel weight, we overexpressed a ZmBAM1d-YFP fusion (Fig. 4f) using the ubiquitin promoter, and observed an approximately 1.9 g increase in HKW (P = 1.76 × 10−4; Fig. 4f), which is greater than its additive effect (~1.2 g) in NILs. This observation suggested that ZmBAM1d was the causal gene for qHKW1. ZmBAM1d overexpression or clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) knockout had no measurable effect on other agronomic traits, such as plant height, leaf number, ear height or tassel branch number, similar to the NIL lines (Fig. 4d, Supplementary Table 13 and Supplementary Table 14), suggesting it has the potential for future crop improvement.
The ZHENG58 genome is not available, but it shares an identical-by-state segment in the qHKW1 region with B73, based on high-density marker analysis29. We therefore compared the ZmBAM1d regions between the B73 and SK genomes, and seven indels >100 bp were identified in the ~40-kb upstream region (Fig. 4c), suggesting that structural variation underlies the phenotypic differences. We found chromatin interactions between the ZmBAM1d coding region and two of the five insertions in B73, which were missing in SK (Fig. 4g, red lines). Indel 4 (8.9-kb insertion; Fig. 4c) was significantly associated with HKW (P < 0.05; Fig. 4h) by candidate gene-association analysis, while another two small indels (indels 6 and 7) were not. We also found that DNA methylation was much higher in the promoter region of ZmBAM1d (indicated by the red box in Fig. 4c) in SK than in B73 (Fig. 4i and Supplementary Note). These results suggest that the large indels affect chromatin interactions and methylation levels, enhancing ZmBAM1d expression and HKW.
To ascertain which pathways might be controlled by ZmBAM1d, we performed RNA-Seq analysis on overexpression lines using embryos at 20 d after pollination. In total, 551 differentially expressed genes (DEGs) were detected (fold change > 2), and were significantly enriched in 20 Gene Ontology terms (P < 6.9 × 10−4), many of which were related to carbohydrate metabolism (Supplementary Fig. 19). Similar Gene Ontology enrichment was found in DEGs comparing ZmBAM1d-CRISPR-edited and control plants (P < 4.8 × 10−4) (Supplementary Fig. 19). Comparison of DEGs in overexpression and CRISPR lines also revealed knotted1-like homeobox and MADS-domain (named after the proteins MINICHROMOSOME MAINTENANCE 1, AGAMOUS, DEFICIENS and SERUM RESPONSE FACTOR) transcription factors. Collectively, these results suggest that ZmBAM1d regulates seed development through pathways affecting determinacy and carbohydrate metabolism.
Discussion
Given the vast diversity of maize, the available reference genomes of temperate varieties are insufficient for pan-genome characterization. Our sequencing and assembly of a tropical maize reference genome with only 238 gaps provides an excellent resource that we used to identify and genotype >80,000 pSVs across 521 diverse inbred lines, revealing an abundance of previously uncharacterized genetic variation in maize. We demonstrate that pSVs have the potential to regulate gene expression by affecting regulatory elements and chromatin loops, indicating their agronomically important role in genetic diversity not previously detected by SNP-based assessments. Combining our SK genome with the other eight public maize genomes, we found that the present variations (Supplementary Fig. 20) still did not reach saturation (Supplementary Note). With the decreasing cost of third-generation sequencing, the construction of a pan-genome based on more reference-quality genomes, not only of maize but also of its ancestor teosinte, becomes possible. We suggest that more than 20 reference genomes of maize and teosinte, including different subspecies, will provide better coverage of genetic variations of the Zea genus. This information will provide more understanding about SVs—especially their important unknown functions in domestication, adaptation and improvement.
We also demonstrate the utility of this new genome by using it to clone the first maize kernel weight QTL, ZmBAM1d, which was targeted for selection during maize improvement16. BAM genes have not previously been associated with seed size, although some of their candidate ligands, encoded by CLAVATA3/ESR (CLE) genes, were described as seed-expressed genes more than 15 years ago38. The SK genome has potential to identify novel traits and pathways that may have been lost during maize improvement, and thus may serve as a novel source of variation in future breeding programs.
Methods
Genome assembly and annotation
SK sequencing and assembly
We sequenced the inbred line SK, derived from a tropical landrace (BioSample accession code: SAMC036455). High-molecular-weight DNA extraction and purification was performed using a DNeasy Plant Maxi Kit (Qiagen). DNA concentration was measured using NanoDrop (Thermo Fisher Scientific) and Qubit 2.0 (Invitrogen) instruments. A total of 43 single-molecule real-time cells were run on the PacBio Sequel instrument by BGI using Kit 2.0 chemistry, generating 19.7 million reads with a total length of 199 Gb. The PacBio data were de novo assembled using FALCON assembler19 and polished with the Arrow program (https://www.pacb.com/support/software-downloads/). DNA was also sequenced using an Illumina HiSeq 3000 machine. Paired-end libraries with insert sizes of 410 and 670 bp, as well as mate-pair libraries with insert sizes of 2, 5, 10 and 20 kb, were constructed, following a standard protocol provided by Illumina. We also used Illumina data to improve the assembly result by Pilon39—an integrated tool for comprehensive variant detection and genome assembly improvement.
Construction of optical genome maps
Based on standard BioNano protocols40, nicking, labeling, repair and staining processes were implemented. Specifically, DNA was digested by the single-stranded nicking endonuclease Nt.BspQI. Optical maps were assembled with BioNano IrysView41 analysis software; only single molecules with a minimum length of 100 kb and six labels per molecule were used.
PacBio sequence gap filling and gap filling result correction
The gaps in the BioNano assembly result were closed by PBjelly version 15.2.20 (ref. 20) with the PacBio sequence using default parameters. Then, the filled regions were polished with Plion39.
Scaffold construction using 10x Genomics data
The Chromium Genome Reagent Kit42 (10x Genomics) was used for indexing prepared samples and partitioning barcoded libraries. Sequencing was conducted with Illumina HiSeq X Ten to generate linked reads. Scaffolding was performed using 10x Genomics linked reads based the ARCS pipeline. Linked reads with barcodes that did not match the company’s barcode whitelist were filtered out. ARCS was run with sensitive parameters, as specified in a previous study21. To examine the linked scaffold, we used a consensus approach that contained evidence from three different sources: (1) Irys optical maps; (2) PacBio long-read alignments to the scaffolds; and (3) Illumina HiSeq read alignments to the scaffolds. We found that Irys supported the linking 110 paired scaffolds with each other, and there were 62 paired scaffolds that did not align with the Irys optical map. All of the conflicts were disconnected.
Anchoring of the assembled scaffolds
To anchor the scaffolds, a high-density genetic linkage map was developed using the RIL population with 263 recombination inbred lines derived from an SK × Zheng58 cross and genotyped with a 56,000 SNP array43. The genetic map spanned 1,858.9 cM and contained 2,796 bins derived from 13,883 high-quality SNPs. The sequences of probes from the Illumina MaizeSNP50 array43 were mapped to the 10x Genomics assembly result using BLAT44. Around 2.095 Gb (47 scaffolds) could be anchored to ten chromosomes by genetic linkage mapping, which made up 96.90% of the 10x Genomics assembly result. Genotype-by-sequencing probes of high-resolution genetic mapping of the maize pan-genome45 were also mapped to the 10x Genomics assembly result using BLAT software; 151 scaffolds could be assigned to a chromosome, but they could not be located and ordered within the chromosome. The size of the 151 scaffolds was 26 Mb.
Further gap filling
We allocated the corrected PacBio long reads to ten chromosomes by mapping them onto the ten pseudo-chromosomes and then reassembling them respectively. We aligned the contigs resulting from reassembly onto the ten pseudo-chromosomes and filled the gaps manually.
BioNano map-assisted gap filling
The BioNano de novo assembly and BioNano molecules were used to estimate the gap length. Then, we filled the gaps using corrected PacBio long reads with PBjelly20. Finally, the filled regions were polished with Plion39. Irys optical maps and Illumina HiSeq reads were used to examine these areas again.
Genome annotation
Transposable elements found in the SK genome were the result of the integration of independent de novo predictions (LTRharvest46, LTRdigest47, SINE-Finder48 and HelitronScanner49), and of homolog searching from RepeatMasker using P-MITE50 and Repbase databases51 as repeat libraries.
The pipeline for gene prediction included de novo and evidence-based predictions using MAKER-P52 and PASA53 on the repeat-masked genome (Supplementary Fig. 7). For homolog evidence, we collected the protein sequences of Arabidopsis thaliana, Brachypodium distachyon, Oryza sativa, Setaria italica, Sorghum bicolor and Z. mays. Transcript evidence included high-quality, full-length transcripts from Iso-Seq and Trinity-assembled transcripts from the RNA-Seq of nine tissues (male spikelet, female spikelet, internode, seedling root, seedling leaf, mature pollen, unpollinated silks, kernels 15 d after pollination, and vegetative meristem). For de novo gene prediction, we used Augustus54 and FGENESH (http://www.softberry.com/berry.phtml) trained on 2,000 homolog genes, which were supported by Iso-Seq full-length transcripts and monocots. All of the evidence was submitted to MAKER-P52, and the output of MAKER-P52 was refined again by PASA53.
SV calling
To call SVs, we used the smartie-sv pipeline14, which aligns, compares and calls insertions, deletions and inversions (https://github.com/zeeev/smartie-sv). At the core of the code is a modified version of BLASR, which was designed to align large divergent contigs against a reference genome. We called SVs (>10 bp; deletions and insertions) using smartie-sv. We applied two filters to the raw SV calls. First, we omitted SVs that were smaller than 10 bp or within the centromere. Second, regions (1 Mb windows) with more than 50 alignments were also excluded from the analysis. Third, contigs of <200 kb were also excluded. Furthermore, we confirmed >96% of 29 events (from 10 bp to 2 kb in size) by Sanger sequencing (Supplementary Table 15). For larger SVs, we randomly selected 12 SVs (from 5–70 kb) for visual inspection and good collinearty were shown between two genomes of the flanking sequence of SVs (Supplementary Fig. 21). As an initial dataset for identification of pSVs (Supplementary Note), the accuracy of 386,014 SVs should be acceptable, although there might be some false positives in them.
RNA-Seq data analysis and eQTL mapping
RNA-Seq data were obtained from our previous published dataset (SRP026161). A total of 11,496,863 high-quality SNPs were obtained from DNA deep resequencing (~20×) of 521 diverse inbred lines. We referred to a previously published method to conduct the quantification of gene expression and eQTL mapping55. Raw reads were trimmed, to remove adapters and low-quality reads, with Trimmomatics (version 0.36)56. Trimmed reads were mapped to the SK reference genome using STAR57. Read counts of each gene were calculated using HTSeq58 and normalized by library sequencing depth using the R package DESeq2 (ref. 59). After filtering the gene without expression in more than 100 samples, expression counts were normalized using Box–Cox transformation. Before eQTL mapping, 69 hidden factors were calculated using PEER60 and were used as covariates together with five multidimensional scaling coordinates calculated form the SNP dataset. Using these covariates, SNP eQTL and SV eQTL were mapped using Matrix eQTL61.
QTL mapping and transgenic validation of qHKW1
We planted heterozygous individuals derived from one heterogeneous inbred family line to screen new recombinant events34. The plants were planted in the field in Hainan (Sanya; 18.3° N, 109.5° E) and grown in 2.5 m rows, spaced 0.5 m apart, with 11 individuals in each row. The markers used for fine mapping of qHKW1 are listed in Supplementary Table 16. Progeny tests were performed by comparing the HKW of NILSK and NILZHENG58 homozygous individuals from F3 families for each new recombinant. We used one-way analysis of variance in Excel to test whether there was a significant difference in HKW between two NILs. We fused Zm00001d028317 with yellow fluorescent protein and overexpressed it into maize inbred line ZC01 with the ubiquitin promoter. One-way analysis of variance analysis was used to test whether there were significant differences in expression levels or HKWs between overexpression transgenic-positive and -negative lines. We also performed CRISPR–Cas9-based gene editing of Zm00001d028317, with two guide RNAs targeting the first exon of Zm00001d028317 inserted into pCPB-ZmUbi-hspCas9 (ref. 62). Both of the overexpression and gene-editing transgenic vectors were transformed into C01 with Agrobacterium tumefaciens EHA105 (China National Seed Group). The transgenic lines were planted in a greenhouse in Yunnan province, China (21.9° N, 100.7° E). To avoid the effect of environment, we planted these transgenic materials and controls in the same greenhouse, with 30 cm plant-to-plant and 50 cm row-to-row distances. The primers used for transgenic experiments are listed in Supplementary Table 16.
Expression quantification of Zm00001d028317 and RNA-Seq
We extracted total RNA from the seeds, endosperm and embryos of two NILs, and the leaves of overexpression transgenic lines using a Quick RNA Isolation Kit (Huayueyang Biotech, Beijing, China). First-strand complementary DNA was synthesized using an EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech). Real-time fluorescence quantitative PCR with SYBR Green Master Mix (Vazyme Biotech) on a CFX96 Real-Time System was used to quantify the expression level of Zm00001d028317. Each set of experiments was repeated three times, and the relative quantification method (2−ΔΔCT) used to evaluate quantitative variation. The primers used for quantitative PCR with reverse transcription are listed in Supplementary Table 16. The RNA, extracted from embryos at 20 d after pollination, of the overexpression-positive and -negative lines and CRISPR-edited and control lines was used to perform RNA-Seq. For each genotype, we performed RNA-Seq of three replicates at Annoroad Gene Technology (Beijing, China). One sample of the overexpression-positive line was excluded from further analysis due to its low global Pearson correlation (r < 0.95) with the other two samples.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All datasets reported in this study have been deposited in GenBank (NCBI) with the following accession codes: genome assembly, PRJNA531547; the 521 inbred lines, PRJNA531553; ChIA-PET, PRJNA531751; and RNA-Seq of ZmBAM1, PRJNA532237. All datasets have also been deposited in the Genome Warehouse of the BIG Data Center at the Beijing Institute of Genomics, Chinese Academy of Sciences, under the following accession numbers: SK PacBio long reads, CRA001371; SK BioNano data, CRA001370; SK Illumina short reads, CRA001366; SK 10x Genomics data, CRA001365; SK ChIA-PET data, CRA001369; SK Iso-Seq data for nine tissues, CRA001337; SK RNA-Seq data for nine tissues, CRA001367; resequencing data of the 521 inbred lines, CRA001363; and RNA-Seq data on overexpression and CRISPR of ZmBAM1d, CRA001368. These data are also available in the CNGB Nucleotide Sequence Archive (https://db.cngb.org/cnsa/) with the following accession codes: genome assembly, CNP0000417; the 521 inbred lines, CNP0000418; SK ChIA-PET data, CNP0000419; and RNA-Seq of ZmBAM1d, CNP0000420. The SK genome and annotation are publicly accessible under accession number GWHAACS00000000. The SK genome and annotation can also be accessed at http://mmgdb.hzau.edu.cn/maize/index.php. The SV map and results of each step in Supplementary Fig. 9 are available at http://www.maizego.org/Resources.html. The seeds of SK are publicly available on request.
References
FAOSTAT, Production (Food and Agriculture Organization of the United Nations, 2014, accessed 5 April, 2016); http://faostat3.fao.org/browse/Q/QC/E
Matsuoka, Y. et al. A single domestication for maize shown by multilocus microsatellite genotyping. Proc. Natl Acad. Sci. USA 99, 6080–6084 (2002).
Van Heerwaarden, J. et al. Genetic signals of origin, spread, and introgression in a large sample of maize landraces. Proc. Natl Acad. Sci. USA 108, 1088–1092 (2011).
Yan, J. B., Warburton, M. & Crouch, J. Association mapping for enhancing maize genetic improvement. Crop Sci. 51, 433–449 (2011).
Buckler, E. S. & Stevens, N. M. in Darwin’s Harvest (eds Motley, T. J., Zerega, N. & Cross, H.) 67–90 (Columbia Univ. Press, 2005).
Jiao, Y. et al. Improved maize reference genome with single-molecule technologies. Nature 546, 524–527 (2017).
Yang, N. et al. Contributions of Zea mays subspecies mexicana haplotypes to modern maize. Nat. Commun. 8, 1874 (2017).
Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Saxena, R. K., Edwards, D. & Varshney, R. K. Structural variations in plant genomes. Brief. Funct. Genom. 13, 296–307 (2014).
Sibbesen, J. A., Maretty, L. The Danish Pan-Genome Consortium. & Krogh, A. Accurate genotyping across variant classes and lengths using variant graphs. Nat. Genet. 50, 1054–1059 (2018).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
Sun, S. et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 50, 1289–1295 (2018).
Springer, N. M. et al. The maize W22 genome provides a foundation for functional genomics and transposon biology. Nat. Genet. 50, 1282–1288 (2018).
Kronenberg, Z. N. et al. High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018).
Doebley, J. F., Gaut, B. S. & Smith, B. D. The molecular genetics of crop domestication. Cell 127, 1309–1321 (2006).
Hufford, M. B. et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 44, 808–811 (2012).
Doll, N. M., Depège-Fargeix, N., Rogowsky, P. M. & Widiez, T. Signaling in early maize kernel development. Mol. Plant 10, 375–388 (2017).
Xiao, Y. et al. Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 210, 1095–1106 (2016).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Yeo, S., Coombe, L., Warren, R. L., Chu, J. & Birol, I. ARCS: scaffolding genome drafts with linked reads. Bioinformatics 34, 725–731 (2018).
Raihan, M. S. et al. Multi-environment QTL analysis of grain morphology traits and fine mapping of a kernel-width QTL in Zheng58 × SK maize population. Theor. Appl Genet. 129, 1465–1477 (2016).
Pan, Q. et al. Genome-wide recombination dynamics are associated with phenotypic variation in maize. New Phytol. 210, 1083–1094 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46, e126 (2018).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnology 31, 1119–1125 (2013).
Spielmann, M., Lupiáñez, D. G. & Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 19, 453–467 (2018).
Yang, X. H. et al. Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol. Breed. 28, 511–526 (2011).
Li, H. et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50 (2013).
Yang, N. et al. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 10, e1004573 (2014).
Massengo-Tiassé, R. P. & Cronan, J. E. Diversity in enoyl-acyl carrier protein reductases. Cell. Mol. Life Sci. 66, 1507–1517 (2009).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Liu, J. et al. The conserved and unique genetic architecture of kernel size and weight in maize and rice. Plant Physiol. 175, 774–785 (2017).
Liu, N. et al. Intraspecific variation of residual heterozygosity and its utility for quantitative genetic studies in maize. BMC Plant Biol. 18, 66 (2018).
Nimchuk, Z. L., Zhou, Y., Tarr, P. T., Peterson, B. A. & Meyerowitz, E. M. Plant stem cell maintenance by transcriptional cross-regulation of related receptor kinases. Development 142, 1043–1049 (2015).
Somssich, M., Je, B. I., Simon, R. & Jackson, D. CLAVATA-WUSCHEL signaling in the shoot meristem. Development 143, 3238–3248 (2016).
Janocha, D. & Lohmann, J. U. From signals to stem cells and back again. Curr. Opin. Plant Biol. 45, 136–142 (2018).
Cock, J. M. & McCormick, S. A large family of genes that share homology with CLAVATA3. Plant Physiol. 126, 939–942 (2001).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
VanBuren, R. et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527, 508–511 (2015).
Pendleton, M. et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015).
Weisenfeld, N. I. et al. Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017).
Ganal, M. W. et al. A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 6, e28334 (2011).
Kent, W. J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 12, 656–664 (2002).
Lu, F. et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nat. Commun. 6, 6914 (2015).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Steinbiss, S., Willhoeft, U., Gremme, G. & Kurt, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013 (2009).
Wenke, T. et al. Targeted identification of short interspersed nuclear element families shows their widespread existence and extreme heterogeneity in plant genomes. Plant Cell 23, 3117–3128 (2011).
Xiong, W., He, L., Lai, J., Dooner, H. K. & Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl Acad. Sci. USA 111, 10263–10268 (2014).
Chen, J. et al. P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res. 42, D1176–D1181 (2013).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Campbell, M. S. et al. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524 (2014).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Kremling, K. A. G. et al. Dysregulation of expression correlates with rare-allele burden and fitness loss in maize. Nature 555, 520–523 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-Seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15, 550 (2014).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507 (2012).
Shabalin, A. A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012).
Li, C. et al. RNA-guided Cas9 as an in vivo desired-target mutator in maize. Plant Biotechnol. J. 15, 1566–1576 (2017).
Acknowledgements
We thank J. Li from the China Agricultural University for providing the seeds of SK, X. Li for helping to conduct ChIA-PET sequencing and K. Kremling for critical comments on the manuscript. This research was supported by the National Natural Science Foundation of China (91735301, 31525017 and 31961133002), National Key Research and Development Program of China (2016YFD0101003) and Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
J.Y. designed and supervised the study. J.H. and W.L. managed the field work and prepared the samples. N.Y., Q.G., L.Y., L.C., T.D., Y.W., L.H., J.Luo., P.X., Y.P., Z.S., Z.M., S.G. and X.Y. performed the data analysis. J.Liu., L.Lan. and L.Liu. performed the fine mapping of the HKW QTL. J.Liu., L.Liu. and D.J. performed the transgenic experiment and RNA-Seq data analysis. Q.Z., M.B. and S.L. performed the sequencing work. N.Y., J.Liu. and J.Y. prepared the manuscript. D.J. edited the manuscript. All of the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have filed a patent application (China patent number CN108484741A) on ZmBAM1d related to the potential utilization in breeding described in the article.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note, Supplementary Figs. 1–21 and Supplementary Tables 1, 3–5, 7–10, 12–14 and 16
Supplementary Table 2
Gap estimation in the SK genome
Supplementary Table 6
Summary of Iso-Seq/RNA-Seq data
Supplementary Table 11
The enrichment analysis of lead SV eQTL
Supplementary Table 15
SV validation Sanger sequencing
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, N., Liu, J., Gao, Q. et al. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet 51, 1052–1059 (2019). https://doi.org/10.1038/s41588-019-0427-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-019-0427-6
This article is cited by
-
Bacillus species are core microbiota of resistant maize cultivars that induce host metabolic defense against corn stalk rot
Microbiome (2024)
-
Mapping and functional characterization of structural variation in 1060 pig genomes
Genome Biology (2024)
-
Cytoplasmic genome contributions to domestication and improvement of modern maize
BMC Biology (2024)
-
Plant pangenomes for crop improvement, biodiversity and evolution
Nature Reviews Genetics (2024)
-
High-throughput single-molecule long-read RNA sequencing analysis of tissue-specific genes and isoforms in lettuce (Lactuca sativa L.)
Communications Biology (2024)
