
Abstract
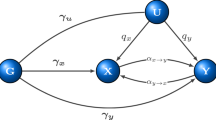
Mendelian randomization, a method to infer causal relationships, is confounded by genetic correlations reflecting shared etiology. We developed a model in which a latent causal variable mediates the genetic correlation; trait 1 is partially genetically causal for trait 2 if it is strongly genetically correlated with the latent causal variable, quantified using the genetic causality proportion. We fit this model using mixed fourth moments \({\it{E}}({\it{\alpha }}_1^2{\it{\alpha }}_1{\it{\alpha }}_2)\) and \({\it{E}}\left( {{\it{\alpha }}_2^2{\it{\alpha }}_1{\it{\alpha }}_2} \right)\) of marginal effect sizes for each trait; if trait 1 is causal for trait 2, then SNPs affecting trait 1 (large \({\it{\alpha }}_1^2\)) will have correlated effects on trait 2 (large α1α2), but not vice versa. In simulations, our method avoided false positives due to genetic correlations, unlike Mendelian randomization. Across 52 traits (average n = 331,000), we identified 30 causal relationships with high genetic causality proportion estimates. Novel findings included a causal effect of low-density lipoprotein on bone mineral density, consistent with clinical trials of statins in osteoporosis.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
UK Biobank summary statistics are publicly available at http://data.broadinstitute.org/alkesgroup/UKBB/.
Change history
06 November 2018
In the version of this article originally published, there were errors in equations. In the HTML and PDF, the initial term of equation 10 was estimated GCP but should have been estimated standard error, while a ‘hat’ was missing from the first alpha in the second term of the expression at the end of the paragraph following equation (6) in the Methods. In addition, in the abstract in the PDF, a subscript 1 was used instead of a subscript 2 for the final term of the first fourth-moment expression. These errors have been corrected in the HTML, PDF and print versions of the paper.
References
Davey Smith, G. & Ebrahim, S. Mendelian randomization: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 32, 1–22 (2003).
Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–R98 (2014).
Voight, B. F. et al. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet 380, 572–580 (2012).
Do, R. et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 45, 1345–1352 (2013).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Kang, H. et al. Instrumental variables estimation with some invalid instruments and its application to Mendelian randomization. J. Am. Stat. Assoc. 111, 132–144 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J. et al. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the phenome. eLife 7, e34408 (2018).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998 (2017).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Cohen, J. C., Boerwinkle, E., Mosley, T. H. Jr & Hobbs, H. H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 (2006).
Paaby, A. B. & Rockman, M. V. The many faces of pleiotropy. Trends Genet. 29, 63–73 (2013).
VanderWeele, T. J. et al. Methodological challenges in Mendelian randomization. Epidemiology 25, 427–435 (2014).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Welsh, P. et al. Unraveling the directional link between adiposity and inflammation: a bidirectional Mendelian randomization approach. J. Clin. Endocrinol. Metab. 95, 93–99 (2010).
Vimaleswaran, K. S. et al. Causal relationship between obesity and vitamin D status: bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 10, e1001383 (2013).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Yang, J. et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120 (2015).
Kolesar, M. et al. Identification and inference with many invalid instruments. J. Bus. Econ. Stat. 33, 474–484 (2015).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389 (2017).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. Genome-wide genetic data on ~500,000 UK Biobank participants. Preprint at bioRxiv https://doi.org/10.1101/166298 (2017).
Loh, P. R. et al. Mixed-model association for biobank-scale datasets. Nat. Genet. 50, 906–908 (2018).
Holmes, M. V., Ala-Korpela, M. & Davey Smith, G. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat. Rev. Cardiol. 14, 577–590 (2017).
Davey Smith, G. et al. The association between BMI and mortality using offspring BMI as an indicator of own BMI: large intergenerational mortality study. BMJ 339, b5043 (2009).
Nordestgaard, B. G. et al. The effect of elevated body mass index on ischemic heart disease risk: causal estimates from a Mendelian randomisation approach. PLoS Med. 9, e1001212 (2012).
Hägg, S. et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int. J. Epidemiol. 44, 578–586 (2015).
Holmes, M. V. et al. Causal effects of body mass index on cardiometabolic traits and events: a Mendelian randomization analysis. Am. J. Hum. Genet. 94, 198–208 (2014).
Klein, I. & Ojamaa, K. Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 344, 501–509 (2001).
Grais, I. M. & Sowers, J. R. Thyroid and the heart. Am. J. Med. 127, 691–698 (2014).
Zhao, J. V. & Schooling, C. M. Thyroid function and ischemic heart disease: a Mendelian randomization study. Sci. Rep. 7, 8515 (2017).
Monzani, F. et al. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 86, 1110–1115 (2001).
Meier, C. et al. TSH-controlled l-thyroxine therapy reduces cholesterol levels and clinical symptoms in subclinical hypothyroidism: a double blind, placebo-controlled trial (Basel Thyroid Study). J. Clin. Endocrinol. Metab. 86, 4430–4863 (2001).
Monzani, F. et al. Effect of levothyroxine replacement on lipid profile and intima-media thickness in subclinical hypothyroidism: a double-blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 89, 2099–2106 (2004).
Razvi, S. et al. The beneficial effect of l-thyroxine on cardiovascular risk factors, endothelial function, and quality of life in subclinical hypothyroidism: randomized, crossover trial. J. Clin. Endocrinol. Metab. 92, 1715–1723 (2007).
Nagasaki, T. et al. Decrease of brachial-ankle pulse wave velocity in female subclinical hypothyroid patients during normalization of thyroid function: a double-blind, placebo-controlled study. Eur. J. Endocrinol. 160, 409–415 (2009).
Chaker, L. et al. Thyroid function and risk of type 2 diabetes: a population-based prospective cohort study. BMC Med. 14, 150 (2016).
Brenta, G. et al. Acute thyroid hormone withdrawal in athyreotic patients results in a state of insulin resistance. Thyroid 19, 665–669 (2009).
Wang, Z. et al. Effects of statins on bone mineral density and fracture risk: a PRISMA-compliant systematic review and meta-analysis. Medicine 95, e3042 (2016).
Yerges, L. M. et al. Decreased bone mineral density in subjects carrying familial defective apolipoprotein B-100. J. Clin. Endocrinol. Metab. 98, E1999–E2005 (2013).
Sanjak, J. S. et al. Evidence of directional and stabilizing selection in contemporary humans. Proc. Natl Acad. Sci. USA 115, 151–156 (2018).
Price, G. R. Selection and covariance. Nature 227, 520–521 (1970).
Clarke, T. K. et al. Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol. Psychiatry 21, 419–425 (2016).
Davies, G. et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N = 112,151). Mol. Psychiatry 21, 758–767 (2016).
Keller, M. C. & Miller, G. Resolving the paradox of common, harmful, heritable mental disorders: which evolutionary genetic models work best? Behav. Brain Sci. 29, 385–404 (2006).
Mullins, N. et al. Reproductive fitness and genetic risk of psychiatric disorders in the general population. Nat. Commun. 8, 15833 (2017).
Ware, J. J. et al. Genome-wide meta-analysis of cotinine levels in cigarette smokers identifies locus at 4q13.2. Sci. Rep. 6, 20092 (2016).
Burgess, S. et al. Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int. J. Epidemiol. 44, 484–495 (2014).
Schoech, A. et al. Quantification of frequency-dependent genetic architectures and action of negative selection in 25 UK Biobank traits. Preprint at bioRxiv https://doi.org/10.1101/188086 (2017).
Mokry, L. E. et al. Vitamin D and risk of multiple sclerosis: a Mendelian randomization study. PLoS Med. 12, e1001866 (2015).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Lyall, D. M. et al. Association of body mass index with cardiometabolic disease in the UK Biobak: a Mendelian randomization study. JAMA Cardiol. 2, 882–889 (2017).
Child, D. The Essentials of Factor Analysis. (A&C Black, London, 1990).
Comon, P. Independent component analysis, a new concept? Signal Process. 36, 287–314 (1994).
UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Acknowledgements
We are grateful to B. Neale, S. Raychaudhuri, C. Patel, S. Kathiresan, B. Pasaniuc, and H. Finucane for helpful discussions and to P.-R. Loh and S. Gazal for producing BOLT-LMM summary statistics for UK Biobank traits. This research was conducted using the UK Biobank Resource under Application #16549 and was funded by National Institutes of Health grants R01 MH107649, U01 CA194393, and R01 MH101244.
Author information
Authors and Affiliations
Contributions
L.J.O. and A.L.P. conceived the methods, designed the analyses, and wrote the manuscript. L.J.O. performed the analyses.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Null and causal simulations with no LD and LCV model violations.
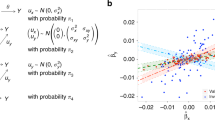
a–g, We report the positive rate (α = 0.05 for null simulations, α = 0.001 for causal simulations) for two-sample MR, MR-Egger, Bidirectional MR and LCV. a–c correspond to Gaussian mixture model extensions of the models in Fig. 2b-d. f and g correspond to causal analogs of the models in a and d, respectively. We also display scatterplots illustrating the bivariate distribution of true SNP effect sizes on the two traits. a, Null simulation with nonzero SNP effects drawn from a mixture of Gaussian distributions; one mixture component has correlated effects on each trait. b, Null simulation with SNP effects drawn from a mixture of Gaussian distributions, and differential polygenicity between the two traits. c, Null simulation with SNP effects drawn from a mixture of Gaussian distributions and unequal power between the two traits. d, Null simulation with two intermediaries having different effects on each trait. e, Null simulation with two intermediaries having different effects on each trait and unequal polygenicity for the two intermediaries. f, Causal simulation with SNP effects drawn from a mixture of Gaussian distributions; all SNPs affecting trait 1 also affect trait 2, but the relative effect sizes were noisy. g, Causal simulation with an additional genetic confounder (i.e., a second intermediary) mediating part of the genetic correlation. Results for each panel are based on 1,000 simulations. Numerical results are reported in Supplementary Tables 4 and 5, which also include comparisons to MR-WME and MR-MBE.
Supplementary Figure 2 Mean GCP estimates in simulations with LCV model violations.
Error bars show s.d. based on 1,000 simulations. a, Null simulation with two intermediaries having possibly unequal polygenicity. The two intermediaries had either a slightly, moderately, or highly heterogenous effect on the two traits; that is, when heterogeneity was high, intermediary 1 had a much larger effect on trait 1 while intermediary 2 had a much larger effect on trait 2. Then, we specified a certain difference in polygenicity between the two traits (measured by the proportion of causal SNPs). b, Causal simulation with an additional latent confounder. The latent confounder explained a low, medium, or high proportion of the genetic correlation. We varied the polygenicity of the confounder and of the causal trait, such that a 16× difference in polygenicity indicates that 16× more SNPs were causal for the causal trait than for the genetic confounder.
Supplementary Figure 3 Unbiasedness of posterior mean GCP estimates in simulations with LD and random true GCP values.
Estimated values of GCP were binned and averaged, and mean true values of GCP are plotted for each bin, with standard errors. Points above the line indicate that GCP estimates were downward biased (toward –1). a, Ascertained simulations (43%) with significant genetic correlation (P < 0.05) and evidence for partial causality (P < 0.001). Only bins with a count of at least 10 are plotted. b, All 10,000 simulations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–3, Supplementary Tables 2–11 and 13–17, and Supplementary Note
Supplementary Table 1
Simulation parameters
Supplementary Table 12
LCV and MR results for all trait pairs
Rights and permissions
About this article

Cite this article
O’Connor, L.J., Price, A.L. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 50, 1728–1734 (2018). https://doi.org/10.1038/s41588-018-0255-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0255-0
This article is cited by
-
Genetic evidence of the causal relationship between chronic liver diseases and musculoskeletal disorders
Journal of Translational Medicine (2024)
-
Evaluation of the causal effects of blood metabolites on irritable bowel syndrome: Mendelian randomization
BMC Gastroenterology (2024)
-
Phenome-wide causal associations between osteoarthritis and other complex traits through the latent causal variable analysis
BMC Musculoskeletal Disorders (2024)
-
The causal relationship between human blood metabolites and the risk of visceral obesity: a mendelian randomization analysis
Lipids in Health and Disease (2024)
-
Adolescent substance use initiation and long-term neurobiological outcomes: insights, challenges and opportunities
Molecular Psychiatry (2024)
