
Abstract
Millions of human genomes and exomes have been sequenced, but their clinical applications remain limited due to the difficulty of distinguishing disease-causing mutations from benign genetic variation. Here we demonstrate that common missense variants in other primate species are largely clinically benign in human, enabling pathogenic mutations to be systematically identified by the process of elimination. Using hundreds of thousands of common variants from population sequencing of six non-human primate species, we train a deep neural network that identifies pathogenic mutations in rare disease patients with 88% accuracy and enables the discovery of 14 new candidate genes in intellectual disability at genome-wide significance. Cataloging common variation from additional primate species would improve interpretation for millions of variants of uncertain significance, further advancing the clinical utility of human genome sequencing.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Change history
17 December 2018
In the version of this article originally published, the name of author Serafim Batzoglou was misspelled. The error has been corrected in the HTML and PDF versions of the article.
References
MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014).
Rehm, H. L. et al. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 372, 2235–2242 (2015).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755 (2011).
Rehm, H. L. Evolving health care through personal genomics. Nat. Rev. Genet. 18, 259–267 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Mallick, S. et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016).
Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Human. Mutat. 32, 894–899 (2011).
Chimpanzee Sequencing Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005).
Takahata, N. Allelic genealogy and human evolution. Mol. Biol. Evol. 10, 2–22 (1993).
Asthana, S., Schmidt, S., & Sunyaev, S. A limited role for balancing selection. Trends Genet. 21, 30–32 (2005).
Leffler, E. M. et al. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339, 1578–1582 (2013).
Samocha, K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014).
Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 246, 96–98 (1973).
Reich, D. E. & Lander, E. S. On the allelic spectrum of human disease. Trends Genet. 17, 502–510 (2001).
Whiffin, N. et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 19, 1151–1158 (2017).
Prado-Martinez, J. et al. Great ape genome diversity and population history. Nature 499, 471–475 (2013).
Klein, J., Satta, Y., O’HUigin, C., & Takahata, N. The molecular descent of the major histocompatibility complex. Annu. Rev. Immunol. 11, 269–295 (1993).
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge Univ. Press, Canbridge, UK, 1983).
de Manuel, M. et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477–481 (2016).
Locke, D. P. et al. Comparative and demographic analysis of orang-utan genomes. Nature 469, 529–533 (2011).
Rhesus Macaque Genome Sequencing Analysis Consortium. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316, 222–234 (2007).
Worley, K. C. et al. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 46, 850–857 (2014).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Schrago, C. G., & Russo, C. A. Timing the origin of New World monkeys. Mol. Biol. Evol. 20, 1620–1625 (2003).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–868 (2016).
Brandon, E. P., Idzerda, R. L. & McKnight, G. S. Targeting the mouse genome: a compendium of knockouts (Part II). Curr. Biol. 5, 758–765 (1995).
Lieschke, J. G. & Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353–367 (2007).
Sittig, L. J. et al. Genetic background limits generalizability of genotype-phenotype relationships. Neuron 91, 1253–1259 (2016).
Bazykin, G. A. et al. Extensive parallelism in protein evolution. Biol. Direct 2, 20 (2007).
Ng, P. C., & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Chun, S. & Fay, J. C. Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561 (2009).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Reva, B., Antipin, Y., & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118 (2011).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N., & Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 14 Suppl 3, S3 (2013).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688 (2012).
Gulko, B., Hubisz, M. J., Gronau, I., & Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 47, 276–283 (2015).
Shihab, H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543 (2015).
Quang, D., Chen, Y., & Xie, X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763 (2015).
Bell, C. J. et al. Comprehensive carrier testing for severe childhood recessive diseases by next generation sequencing. Sci. Transl. Med. 3, 65ra64 (2011).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Smedley, D. et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in mendelian disease. Am. J. Hum. Genet. 99, 595–606 (2016).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Jagadeesh, K. A. et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48, 1581–1586 (2016).
Grimm, D. G. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Human. Mutat. 36, 513–523 (2015).
He, K., Zhang, X., Ren, S., & Sun, J. Deep residual learning for image recognition. in Proc. IEEE Conf. Comput. Vis. Pattern Recognit. 770–778 (2016).
Heffernan, R. et al. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 5, 11476 (2015).
Wang, S., Peng, J., Ma, J. & Xu, J. Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 6, 18962–18962 (2016).
Harpak, A., Bhaskar, A., & Pritchard, J. K. Mutation rate variation is a primary determinant of the distribution of allele frequencies in humans. PLoS Genet. 12 e1006489 (2016).
Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011).
Shen, H. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326 (2017).
Nakamura, K. et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 81, 992–998 (2013).
Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89, 10915–10919 (1992).
Li, W. H., Wu, C. I. & Luo, C. C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Molec. Evol. 21, 58–71 (1984).
Grantham, R. Amino acid difference formula to help explain protein evolution. Science 185, 862–864 (1974).
LeCun, Y., Bottou, L., Bengio, Y., & Haffner, P. Gradient based learning applied to document recognition. Proc. IEEE 86, 2278–2324 (1998).
Vissers, L. E., Gilissen, C., & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18 (2016).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015).
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014).
Zhu, X., Need, A. C., Petrovski, S. & Goldstein, D. B. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat. Neurosci. 17, 773–781, https://doi.org/10.1038/nn.3713 (2014).
Leffler, E. M. et al. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol. 10, e1001388 (2012).
Estrada, A. et al. Impending extinction crisis of the world’s primates: why primates matter. Sci. Adv. 3, e1600946 (2017).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Tyner, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017).
Kabsch, W., & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Joosten, R. P. et al. A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–419 (2011).
He, K., Zhang, X., Ren, S., & Sun, J. Identity mappings in deep residual networks. in 14th European Conference on Computer Vision – ECCV 2016. ECCV 2016. Lecture Notes in Computer Science, vol 9908; 630–645 (Springer, Cham, Switzerland; 2016).
Ionita-Laza, I., McCallum, K., Xu, B., & Buxbaum, J. D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 48, 214–220 (2016).
Li, B. et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750 (2009).
Lu, Q. et al. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 5, 10576 (2015).
Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Human. Mutat. 34, 57–65 (2013).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025 (2010).
Liu, X., Wu, C., Li, C., & Boerwinkle, E. dbNSFPv3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Human. Mutat. 37, 235–241 (2016).
Jain, S., White, M., Radivojac, P. Recovering true classifier performance in positive-unlabeled learning. in Proceedings Thirty-First AAAI Conference on Artificial Intelligence. 2066–2072 (AAAI Press, San Francisco; 2017).
de Ligt, J. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 (2012).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
Rauch, A. et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682 (2012).
Epi, K. C. et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013).
EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370 (2014).
Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014).
Lelieveld, S. H. et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196 (2016).
Famiglietti, M. L. et al. Genetic variations and diseases in UniProtKB/Swiss-Prot: the ins and outs of expert manual curation. Human. Mutat. 35, 927–935 (2014).
Horaitis, O., Talbot, C. C.Jr., Phommarinh, M., Phillips, K. M., & Cotton, R. G. A database of locus-specific databases. Nat. Genet. 39, 425 (2007).
Stenson, P. D. et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9 (2014).
Acknowledgements
The authors would like to thank J. K. Pritchard, M. E. Hurles, J. W. Belmont, and R. E. Green for insightful discussions. The authors would like to thank the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute (grant number WT098051). The views expressed in this publication are those of the authors and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. L.S., S.R.P., Y.L. and X.L. were partially supported by R01GM110240 from the National Institute of General Medical Sciences and National Science Foundation (grant number CNS-1747783, CNS-1624782, and OAC-1229576).
Author information
Authors and Affiliations
Contributions
K.K.F., L.S., H.G., S.R.P., and J.F.M. designed the study and wrote the manuscript. L.S., S.R.P., Y.L., N.F., J.H., A.D., J.S., J.X., S.B., X.L., and K.K.F. performed the deep learning analysis. H.G., J.F.M., L.S., S.R.P., J.A.K., and K.K.F. performed the genetics analysis. L.S. and H.G. are co-first authors.
Corresponding author
Ethics declarations
Competing interests
Authors with Illumina affiliations were employees of Illumina, Inc., at the time of this work.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Expected missense:synonymous ratios across the human allele frequency spectrum in the absence of purifying selection.
Shaded gray bars represent the number of synonymous variants, and dark green bars represent the number of missense variants. The dotted line shows the baseline formed by synonymous variants. Missense:synonymous ratios are indicated for each allele frequency category. The expected missense and synonymous counts in each allele frequency category were calculated by taking intronic variants from the ExAC/gnomAD dataset consisting of 123,136 exomes and using them to estimate the fraction of variants expected to fall into each of the four allele frequency categories, based on the trinucleotide context of the variant, which controls for mutational rate and GC bias in gene conversion.
Supplementary Figure 2 Missense:synonymous ratios for CpG and non-CpG variants.
a,b, Missense:synonymous ratios for CpG (a) and non-CpG (b) variants across the human allele frequency spectrum, using all variants from the ExAC/gnomAD exomes. c,d, Missense:synonymous ratios for CpG (c) and non-CpG (d) variants across the human allele frequency spectrum, restricted to only human variants that are identical by state with chimpanzee common polymorphisms.
Supplementary Figure 3 Missense:synonymous ratios of human variants identical by state with six primates.
Patterns of missense:synonymous ratios across the human allele frequency spectrum for ExAC/gnomAD variants that are identical by state with variation present in chimpanzee, bonobo, gorilla, orangutan, rhesus, and marmoset.
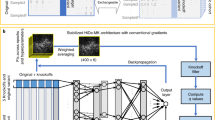
Supplementary Figure 4 Schematic illustration of PrimateAI, the deep learning network architecture for pathogenicity classification.
The inputs to the model include 51 amino acids (AA) of flanking sequence for both the reference sequence and the sequence with the variant substituted in, conservation represented by three 51-AA-length position-weighted matrices from primate, mammal, and vertebrate alignments, and the outputs of networks for training secondary structure and solvent accessibility (also 51 AA in length).
Supplementary Figure 5 Detailed illustration of the deep learning network architecture used for predicting secondary structure and solvent accessibility of proteins.
The input to the model is a position-weighted matrix using conservation generated by the RaptorX software (for training on Protein Data Bank sequences) or the 99-vertebrate alignments (for training and inference on human protein sequences). The output of the second to last layer, which is 51 AAs in length, becomes the input for the deep learning network for pathogenicity classification.
Supplementary Figure 6 Correcting for the effect of sequencing coverage on the ascertainment of common primate variants.
The probability of observing a given variant in a nonhuman primate species is inversely correlated with the sequencing depth at that position in the ExAC/gnomAD exome dataset. In contrast, the lower gnomAD read depth did not affect the probability of observing a common human variant at that position (>0.1% allele frequency) because the large number of human exomes sequenced makes ascertainment of common variation almost guaranteed. When picking matched variants for each of the primate variants for training the network, the probability of picking a variant was adjusted for the effects of sequencing depth, in addition to matching for trinucleotide context to control for mutational rate and gene conversion.
Supplementary Figure 7 Recognition of protein motifs by the neural network.
a, To illustrate the neural network’s recognition of protein domains, we show the average PrimateAI scores for variants at each amino acid position in three different protein domains. Top, the collagen strand of COL1A2, with glycine in a repeating GXX motif highlighted. Clinically identified mutations in collagen genes are largely due to missense mutations in glycine in GXX repeats, as these interfere with the normal assembly of collagen and exert strong dominant-negative effects. Middle, the active site of the IDS sulfatase enzyme (highlighted), which contains a cysteine at the active site that is post-translationally modified to formylglycine. Bottom, the bHLHzip domain of the MYC transcription factor. The basic domain contacts DNA via positively charged arginine and lysine residues (highlighted) that interact with the negatively charged sugar-phosphate backbone. The leucine-zipper domain consists of leucine residues spaced seven amino acids apart (highlighted), which are crucial for dimerization. b, Line plot showing the effect of perturbing each position in and around the variant on the predicted DL score for the variant. We systematically zeroed out the inputs at nearby amino acids (positions –25 to + 25) around the variant and measured the change in the neural network’s predicted pathogenicity of the variant. The plot shows the average change in predicted pathogenicity score for perturbations at each nearby amino acid position for 5,000 randomly selected variants.
Supplementary Figure 8 Correlation patterns of weights mimic BLOSUM62 and Grantham score matrices.
Correlation patterns of weights from the first three layers of the secondary-structure DL network show correlations between amino acids that are similar to BLOSUM62 and Grantham score matrices. The left heat map shows the correlation of parameter weights from the first convolutional layer following two initial upsampling layers of the secondary-structure DL network between amino acids encoded using a one-hot representation. The middle heat map shows BLOSUM62 scores between pairs of amino acids. The right heat map shows Grantham distance between amino acids. The Pearson correlation between DL weights and BLOSUM62 scores is 0.63 (P = 3.55 × 10–9). The correlation between DL weights and Grantham scores is –0.59 (P = 4.36 × 10–8). The correlation between BLOSUM62 and Grantham scores is –0.72 (P = 8.09 × 10–13).
Supplementary Figure 9 Performance evaluation of the deep learning network and other classifiers.
a, Accuracy of the deep learning network PrimateAI at predicting a benign consequence for a test set of 10,000 primate variants that were withheld from training and comparison with other classifiers, including SIFT, PolyPhen-2, CADD, REVEL, M-CAP, LRT, MutationTaster, MutationAssessor, FATHMM, PROVEAN, VEST3, MetaSVM, MetaLR, MutPred, DANN, FATHMM-MKL_coding, Eigen, GenoCanyon, integrated_fitCons, and GERP. The y axis represents the percentage of primate variants classified as benign, based on normalizing the threshold for each classifier to its 50th-percentile score using a set of 10,000 randomly selected variants that were matched to the primate variants for trinucleotide context to control for mutational rate and gene conversion. b, Comparison of the performance of the PrimateAI network in separating de novo missense variants in DDD cases versus controls, along with the 20 existing methods listed above. The y axis shows the P values of the Wilcoxon rank-sum test for each classifier. c, Comparison of the performance of the PrimateAI network in separating de novo missense variants in DDD cases versus unaffected controls within 605 disease-associated genes, with the 20 methods listed above. The y axis shows the P values of the Wilcoxon rank-sum test for each classifier.
Supplementary Figure 10 Distribution of the prediction scores of four classifiers.
Histograms of the prediction scores of four classifiers, including SIFT, PolyPhen-2, CADD, and REVEL, for de novo missense variants occurring in DDD cases versus unaffected controls, with corresponding Wilcoxon rank-sum P values.
Supplementary Figure 11 Comparing the accuracy of the PrimateAI network and other classifiers at separating pathogenic and benign variants in 605 disease-associated genes.
a, Scatterplot showing the performance of each of the classifiers on DDD cases versus controls (y axis) and benign prediction accuracy on the withheld primate dataset (x axis). b, Comparison of different classifiers at separating de novo missense variants in cases versus controls within the 605 genes, shown on a receiver operator characteristic (ROC) curve, with area under the curve (AUC) indicated for each classifier. c, Classification accuracy and AUC for the PrimateAI network and the 20 classifiers listed in Supplementary Fig. 9. The classification accuracy shown is the average of the true positive and true negative rates, using the threshold where the classifier would predict the same number of pathogenic and benign variants as expected based on the enrichment in Fig. 4a. The maximum achievable AUC for a perfect classifier is indicated with a dotted line, assuming that de novo missense variants in DDD cases are 67% pathogenic variants and 33% benign, and de novo missense variants in controls are 100% benign.
Supplementary Figure 12 Correlation between classifier performance on human expert-curated ClinVar variants and performance on empirical datasets.
a, Scatterplot showing the classification accuracy (y axis) on ClinVar variants and on the 10,000 withheld primate variants (x axis) for each of the 20 other classifiers and the PrimateAI network trained with human-only or human + primates data. Shown are the Spearman correlation coefficient rho and associated P value. To limit the evaluation to data that were not used for training the classifiers, we only used ClinVar variants that were added between January 2017 and November 2017, and excluded common human variants from ExAC/gnomAD (>0.1% allele frequency). The ClinVar classification accuracy shown is the average of the true positive and true negative rates, using the threshold where the classifier would predict the same number of pathogenic and benign variants as observed in the ClinVar dataset. b, Scatterplot showing the classification accuracy (y axis) on ClinVar variants and the DDD cases versus controls full dataset (x axis) for each of the 20 other classifiers and the PrimateAI network trained with human-only or human + primates data.
Supplementary Figure 13 Simulation showing saturation of new common missense variants discovered by increasing the size of the human cohorts surveyed.
In simulations, the genotypes of each sample were sampled according to gnomAD allele frequencies. The fraction of gnomAD common variants discovered is averaged across 100 simulations for each sample size from 10 to 100,000.
Supplementary Figure 14 Accuracy of PrimateAI across different conservation profiles in the genome.
The x axis represents the percentage alignability of the 51 AA around a sequence with the 99-vertebrate alignments. The y axis represents the classification performance of PrimateAI for variants in each of the conservation bins, benchmarked on the test dataset of 10,000 withheld primate variants.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–14, Supplementary Tables 5, 8–12, 14–17 and 19, Supplementary Data 1–4 and Supplementary Note
Supplementary Tables
Supplementary Tables 1–4, 6, 7, 13, 18, 20 and 21
Rights and permissions
About this article

Cite this article
Sundaram, L., Gao, H., Padigepati, S.R. et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 50, 1161–1170 (2018). https://doi.org/10.1038/s41588-018-0167-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0167-z
This article is cited by
-
MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach
Genome Medicine (2024)
-
DVA: predicting the functional impact of single nucleotide missense variants
BMC Bioinformatics (2024)
-
Enhancing missense variant pathogenicity prediction with protein language models using VariPred
Scientific Reports (2024)
-
Prioritizing genomic variants pathogenicity via DNA, RNA, and protein-level features based on extreme gradient boosting
Human Genetics (2024)
-
PdmIRD: missense variants pathogenicity prediction for inherited retinal diseases in a disease-specific manner
Human Genetics (2024)
