
Abstract
Roses have high cultural and economic importance as ornamental plants and in the perfume industry. We report the rose whole-genome sequencing and assembly and resequencing of major genotypes that contributed to rose domestication. We generated a homozygous genotype from a heterozygous diploid modern rose progenitor, Rosa chinensis ‘Old Blush’. Using single-molecule real-time sequencing and a meta-assembly approach, we obtained one of the most comprehensive plant genomes to date. Diversity analyses highlighted the mosaic origin of ‘La France’, one of the first hybrids combining the growth vigor of European species and the recurrent blooming of Chinese species. Genomic segments of Chinese ancestry identified new candidate genes for recurrent blooming. Reconstructing regulatory and secondary metabolism pathways allowed us to propose a model of interconnected regulation of scent and flower color. This genome provides a foundation for understanding the mechanisms governing rose traits and should accelerate improvement in roses, Rosaceae and ornamentals.
Similar content being viewed by others


Main
Roses are among the most commonly cultivated ornamental plants worldwide. They have been cultivated by humans since antiquity, for example, in China. Ornamental features as well as therapeutic and cosmetic value have certainly motivated rose domestication. The genus Rosa contains approximately 200 species, more than half of which are polyploid1. Roses have undergone extensive reticulate evolution with interspecific hybridization, introgression and polyploidization. Only 8 to 20 rose species are thought to have contributed to the present complex hybrid rose cultivars, namely Rosa × hybrida2. The Chinese rose R. chinensis (diploid) was introduced to Europe in the eighteenth century. This species is considered one of the main species that participated in the subsequent extensive process of hybridization with roses from the European, Mediterranean and Middle Eastern (mostly tetraploid) sections (Supplementary Note 1). These crosses gave rise to hybrid tea rose cultivars, which are the parents of the modern roses with extraordinarily diverse traits3. Among the breeding traits originating from Chinese roses, the capacity of recurrent flowering as well as color and scent signatures are key4. Despite recent progress5, the lack of a rose genome sequence has hampered the discovery of the molecular and genetic determinants of these traits and of their breeding history.
Owing to natural autoincompatibility and recent interspecific hybridization, all roses have highly heterozygous genomes6 that are challenging to assemble7 despite their relatively small size (560 Mb)8. To date, attempts to assemble rose genomes with short reads have led to highly fragmented assemblies composed of thousands of scaffolds (83,139 (ref. 9) and 15,938 (this study)). To overcome these bottlenecks to producing a reference genome, we obtained a homozygous genome that we sequenced with long-read sequencing technology. We developed an original in vitro culture protocol combining fine-tuned starvation, cold stress and hormonal treatments to induce R. chinensis ‘Old Blush’ microspores to switch from gametophyte to sporophyte development. This approach allowed microspores to initiate divisions, form homozygous cell clusters and develop embryogenic callus from which homozygous plantlets could be regenerated (Supplementary Note 2 and Supplementary Fig. 1).
The homozygous rose line was sequenced on the PacBio RS II platform. A sequencing coverage of 80× was obtained with 40 single-molecule real-time cells. Preliminary assembly of the rose data with a single assembler generated several hundred contigs, thus illustrating the challenge of assembling plant genomes, even with long-read data10,11. A key step in improving the contiguity of the assembly is the detection and the filtering of spurious edges in the graph of overlaps. The assembler CANU implements filter parametrization at the read level, thus leading to more accurate and contiguous assemblies12. We developed software called til-r, which implements similar and alternate heuristics to clean the graph of overlaps of the FALCON assembler13 (Supplementary Fig. 2; URLs). We then used CANU to perform meta-assembly of six complementary raw assemblies generated by CANU and FALCON/TIL-R (Supplementary Note 3; URLs). The final assembly was composed of 82 contigs for an N50 of 24 Mb (where N50 is the contig length, such that 50% of the entire assembly is contained in contigs equal or larger than this value), thus increasing the contiguity metrics of a simple assembly by threefold and demonstrating the power of meta-assembly approaches (Supplementary Fig. 2).
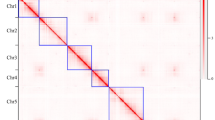
The seven pseudochromosomes were built by integrating 86.4% of the 25,695 markers of the K5 rose high-density genetic map14. A large fraction of the assembly (97.7%, 503 Mb) was oriented with Pearson’s correlation coefficients ranging from 0.986 to 0.996, thus illustrating the high congruence between sequence and genetic data. The genome structure and quality were confirmed by mapping of Hi-C chromosomal-contact-map data (Fig. 1 and Supplementary Fig. 3). With its very few remaining gaps and high consistency between genetics and sequence data, the rose genome assembly is one of the most contiguous obtained to date for a plant genome.

a, R. chinensis ‘Old Blush’ mature flowers. b, Representations of chromosome connections between the physical positions on the reconstructed chromosome and genetic-map positions (left panels). Scatter plots with dots representing the physical position on the chromosome (Chr) (x axis) versus the map position (y axis) are shown. Rho (ρ) is the Pearson correlation coefficient (middle panels). A Hi-C intrachromosomal contact map is shown for each chromosome (right panels). The intensity of pixels represents the count of Hi-C links between 400-kb windows on chromosomes on a logarithmic scale. Darker red color indicates higher contact probability. LG, linkage group.
The rose genome comprises 36,377 inferred protein-coding genes and 3,971 long noncoding RNAs. Annotation assessment with the Plantae BUSCO v2 dataset15 identified 96.5% complete gene models. BUSCO analyses using the assembled heterozygous genome of R. chinensis ‘Old Blush’ (Supplementary Note 4) identified 93.5% complete genes (Supplementary Data 1). On the basis of transcriptomic data from pooled tissues, 207 miRNA precursors were predicted. Transposable elements (TEs) spanned 67.9% of the assembly, and 50.6% were long-terminal-repeat retrotransposons (Supplementary Note 5, Supplementary Fig. 4 and Supplementary Table 1). The web portal RchiOBHm-V2 (see URLs) provides access to the reference genome integrating annotations, polymorphisms, transcriptomic data and the first rose epigenome on rose petals (Supplementary Note 6).
Comparative genomic investigation allowed us to assess rose paleohistory within the Rosaceae family (Supplementary Note 7). Conserved gene adjacencies identified an ancestral Rosaceae karyotype consisting of nine protochromosomes with 8,861 protogenes (Supplementary Fig. 5a). Our evolutionary scenario established that the ancestral Rosoideae karyotype of the strawberry and Rosa genomes, structured into eight protochromosomes with 13,070 protogenes, was derived from the ancestral Rosaceae karyotype through one ancestral chromosome fission and two fusions. Interestingly, the strawberry genome experienced an extra ancestral chromosome fusion from the ancestral Rosoideae karyotype to reach its modern genome structure, whereas the Rosa sp. went through one fission and two fusions, independently of strawberry, to reach its modern genome structure. A phylogeny based on 748 gene sequences showed that Rosa, Fragaria and Rubus diverged within a short timeframe, thus suggesting an evolutionary radiation inside the Rosoideae subfamily (Supplementary Fig. 5b).
To gain insight into the makeup of modern roses, we resequenced representatives of three sections (Synstylae, Chinenses and Cinnamomeae; Supplementary Table 2) that were involved in the domestication and breeding that led to rose hybrid cultivar creation (Supplementary Notes 1 and 8). We observed discrete levels of variant density along the genomes of hybrid cultivars (Fig. 2b) that may reflect different introgression histories. We used the changes in variant density to segment the genome into 35 intervals (2–56 Mb) and studied their genetic structure through principal component analysis (Fig. 2c and Supplementary Fig. 6). We focused on the modern Rosa × hybrida ‘La France’, which is considered to be among the first created hybrids combining the growth-vigor traits of European species and the recurrent blooming of Chinese species.

a, Genealogy of resequenced genotypes. Sections: CIN, Cinnamomeae; SYN, Synstylae; CHI, Chinenses. Genotypes: PEN, Rosa pendulina; RUG, Rosa rugosa; MAJ, Rosa majalis; ARV, Rosa arvensis; MOS, Rosa moschata; WIC, R. wichurana; SPO, R. chinensis ‘Spontanea’; GIG, Rosa gigantea; MUT, R. chinensis ‘Mutabilis’; SAN, R. chinensis ‘Sanguinea’; GAL, Rosa gallica; DAM, Rosa damascena; OB, R. chinensis ‘Old Blush’; HUM, R. chinensis ‘Hume’s Blush’; FRA, Rosa × hybrida ‘La France’ (flower photo). b, Genetic structure and variant density. 1, circular representation of pseudomolecules. 2, schematic representation of the contribution of Cinnamomeae, Synstylae and Chinenses sections to ‘La France’ in 35 chromosomal segments: light red, CHI; light green, SYN; light blue, CIN; multiple bands, mixed origin in the fragment. 3–8, density in heterozygote and homozygote variants (light and dark shades respectively) in 1-Mb sliding windows in ‘La France’, R. gigantea, ‘Hume’s Blush’, ‘Mutabilis’, ‘Sanguinea’ and ‘Old Blush’ heterozygote genotypes, respectively. c, Principal component analyses of genetic variation in three illustrative genomic segments. Orange dot, ‘La France’; blue, CIN; green, SYN; red, CHI; black, other cultivars. y axis, first component; x axis, second component. The number indicated in each plot refers to the genomic fragments analyzed (e.g., 4.3 is the third segment of chromosome 4; Supplementary Fig. 6).
Patterns of diversity along the seven chromosomes showed that the genome of ‘La France’ is a complex mosaic formed by DNA fragments transmitted by the three ancestral pools of diversity represented in the targeted rose sections (Fig. 2, Supplementary Note 8, Supplementary Fig. 6 and Supplementary Data 2). For example, chromosome 4 haplotypes are structured by a combination of Cinnamomeae, Synstylae and Chinenses genomes, whereas chromosome 7 haplotypes have been transmitted by Synstylae and Chinenses ancestors, without an apparent contribution of Cinnamomeae.
We took advantage of the transmission of genomic bits of Chinenses hybrids to ‘La France’ to identify new candidate genes potentially involved in recurrent blooming. The insertion of a TE in TFL1 (RoKSN), a repressor of floral transition responsive to activation by gibberellic acid, is considered a major determinant of recurrent blooming16. We found that this TE was transmitted to ‘La France’ by R. chinensis cultivars and thus may participate in its recurrent blooming. A recent segregation analyses of R. chinensis ‘Old Blush’ × Rosa wichurana backcross progeny has shown that recurrent blooming probably involves at least a second independent locus17. This second locus may have been transmitted to ‘La France’ by only R. chinensis and thus may be located in chromosomal segments such as those originating from the Chinenses section, e.g., segments 2.4 and 5.1 (Fig. 2). On these segments, we identified the putative homologs of the transcription factor SPT (segment 2.4, Fig. 3a), which is known to control flowering in Arabidopsis18,19, and of DOG1 (segment 5.1, Fig. 3a), which is known to modify flowering by acting on miR156 (ref. 20). These genes are thus additional promising candidates that may determine recurrent blooming in roses.

a, Schematic representation of the rose chromosomes together with the positions of candidate genes potentially affecting anthocyanin pigments, volatile-molecule biosynthesis and flowering. Chromosome segments 2.4, 3.2–3.6 and 5.1 originating only from R. chinensis are indicated in light red. Red, anthocyanin-synthesis genes; blue, terpene-biosynthesis genes; black, flowering-time genes; green, development genes. b, Schematic representation of interconnections between color (pink background) and scent (blue background) pathways. Gene expression data show the anticorrelation between expression of miR156 and SPL9 genes during petal development. RT–qPCR was performed on petals harvested at three successive stages: noncolored petals early during development (St. 1); petals at the onset of anthocyanin synthesis (St. 2); and fully colored petals (St. 3). Black arrows, biosynthetic steps reported in rose; red arrows, biosynthetic steps reported in other species but not in rose; green arrows, putative steps with unknown enzymes; dashed black arrow, several enzymatic steps; maroon arrows, gene regulation reported in Arabidopsis thaliana but not in rose; dashed maroon arrow, putative gene regulation. IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; DFR, dihydroflavonol-4-reductase; ANS, anthocyanidin synthase; 3GT, anthocyanidin 3-O-glucosyltransferase; GT1, anthocyanidin 3,5-diglucosyltransferase; GPPS, geranyl diphosphate synthase; FPPS, farnesyl diphosphate synthase; GGPPS, geranylgeranyl diphosphate synthase; GDS, germacrene D synthase; TPS, terpene synthase; NES, linalool/nerolidol synthase; CCD1/CCD4, carotenoid cleavage dioxygenases 1/4; NUDX1, nudix hydrolase 1.
Roses exhibit a high diversity of flower fragrance and color, of which biochemical and regulatory determinants have been only partially elucidated (Supplementary Note 9 and Supplementary Fig. 7). Data mining of the rose genome combined with in-depth biochemical and molecular analyses of volatile organic compounds permitted identification of at least 22 biosynthetic steps in the terpene pathway that have not been characterized in the rose, two of which have not previously been characterized in other species (Supplementary Note 9 and Supplementary Fig. 7).
To study the relationships between color and scent pathways, we performed biochemical and molecular analyses on cyanidin, whose glucosylated derivatives represent more than 99% of the total anthocyanin pigments21, and on germacrene D, a volatile organic compound produced in petal cells of R. chinensis ‘Old Blush’ (Supplementary Data 3). Our analyses suggest that coordinated biosynthesis of these two compounds is achieved through the miR156–SPL9 regulatory module. In Arabidopsis, SPL9 is a repressor of anthocyanin synthesis in the cells of aging plants22. miR156 negatively regulates SPL9 in the cells of young plants, thereby enabling the formation of a MYB–bHLH–WD40 protein complex that activates anthocyanin production22. Analysis of this module in the petals of ‘Old Blush’ indicated that the expression of SPL9 peaked before maximum ANTHOCYANIDIN SYNTHASE (ANS) expression (Supplementary Fig. 8). In fully colored petals, we observed induced expression of miR156, which correlated with downregulation of SPL9 expression and upregulation of ANS expression (Fig. 3b, Supplementary Fig. 8 and Supplementary Fig. 9). The maximum expression of GDS, which encodes the enzyme catalyzing germacrene D synthesis, also correlated with miR156 and ANS activation and with SPL9 downregulation (Fig. 3 and Supplementary Fig. 8). This observation, together with a previous demonstration that ANS and GDS can be activated in rose petals by expression of the Arabidopsis AtPAP1 MYB transcription factor23, suggests that the biosynthesis of anthocyanin and germacrene D may be modulated by the miR156–SPL9 regulatory module, possibly through action on a MYB–bHLH–WD40 complex. Although PAP1 is not expressed in ‘Old Blush’ petals, we found that the expression pattern of RhMYB10, which has been described as a regulator of the anthocyanin-biosynthetic pathway in Rosaceae24, is compatible with a role in coactivation of the synthesis of cyanidin and germacrene D in petal epidermal cells (Supplementary Fig. 8).
The biosynthesis of terpenes, major scent compounds in roses, has been shown to involve TERPENE SYNTHASE (TPS) proteins, such as NEROLIDOL SYNTHASE (NES)25. A search for TPS in the rose genome revealed a cluster of NES genes on chromosome 5 that has a counterpart in Fragaria26. These genes were not substantially expressed in rose petals (Supplementary Data 4). In Arabidopsis, the expression of some TPS is activated by SPL9 (ref. 27). In rose petals, the downregulation of SPL9 through activation of miR156 (Fig. 3b and Supplementary Fig. 8) might explain the absence of expression of NES genes and probably explains why they do not participate in the production of some terpenes in rose flowers. Our data provide hints as to why alternative routes to produce terpenes, such as the one involving NUDX1 (ref. 28), have been used in rose flowers.
Here, we propose that the miR156–SPL9 regulatory hub orchestrates the coordination of production of both colored anthocyanins and certain terpenes, by permitting the complexation of preexisting MYB–bHLH–WD40 proteins, which in turn modulate different components of both pathways (Fig. 3). Therefore, anthocyanin synthesis in rose flowers may be linked to the production of some volatile compounds, thus providing a regulatory explanation for the evolution of nonstandard terpene-biosynthesis pathways. Moreover, this co-regulation may hinder the combination of pigmentation and specific scents in rose hybrids.
The very high-quality rose genome sequence reported in this study, combined with an expert annotation of the main pathways of interest for the rose (Supplementary Notes 9–13, Supplementary Figs. 7–23, Supplementary Table 3 and Supplementary Data 5–10), provides new insights into the genome dynamics of this woody ornamental and offer a basis to disentangle the seemingly mandatory trait associations or exclusions. Furthermore, access to candidate genes, such as those involved in abscisic acid synthesis and signaling, paves the way for improving rose quality with better water-use efficiency and increased vase life. Breeding for other characteristics such as increased resistance to pathogens should also benefit from these data and may lead to decreased use of pesticides.
URLs.
Genome browser and genomic resources, https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/; MetExplore, https://metexplore.toulouse.inra.fr/metexplore2/?idBioSource=5104/; EuGene plant pipeline, http://eugene.toulouse.inra.fr/Downloads/egnep-Linux-x86_64.1.4.tar.gz; tbl2asn2, https://www.ncbi.nlm.nih.gov/genbank/tbl2asn2/; REPET, https://urgi.versailles.inra.fr/Tools/REPET/; miRanda, http://www.microrna.org/; til-r, http://lipm-bioinfo.toulouse.inra.fr/download/til-r/.
Methods
Production of homozygous rose line derived from heterozygous R. chinensis ‘Old Blush’
Flower buds were harvested from R. chinensis ‘Old Blush’ plants when most microspores were at the mid–late uninucleate/early bicellular development stages (Supplementary Fig. 1). Microspores were aseptically isolated from anthers, suspended in starvation medium and pretreated at 4 °C in darkness for 21 d. Approximately 160,000 microspores were suspended in AT12 medium corresponding to AT3 medium29 supplemented with 4.5 µM 2,4-D and 0.44 µM BAP, pH 5.8, and then incubated at 25 °C in the dark. Developing microcalli (~0.5 mm diameter) were observed after approximately 11 weeks and were then subcultured individually under the same conditions (Supplementary Note 2). Developed calli were then plated onto solid MS salt medium complemented with B5 vitamins, 30 g/L sucrose, 2.5 mM MES, 4.5 µM 2,4-D, 0.44 µM BAP and 6.5 g/L VitroAgar (Kalys Biotechnologie), pH 5.8. A callus that displayed somatic embryos (designated RcHzRDP12; Supplementary Fig. 1g) was selected. The homozygosity status and ploidy level of this callus were confirmed by DNA genotyping and fluorescence-activated cell-sorting analysis, respectively, as previously described30.
Sample preparation and sequencing
High-quality nuclear DNA was prepared from RcHzRDP12 homozygous callus propagated on callus-maintenance medium (Supplementary Note 2) as previously described31 with the following modifications. Ten percent fresh weight of PVP40 was added to callus cells that had been ground in liquid nitrogen. Purified nuclei pellets were processed with a Qiagen DNeasy Plant kit (Qiagen). DNA integrity was verified via gel electrophoresis (0.7% agarose), and total DNA was quantified through fluorometry with Picogreen (Applied Biosystems/Life Technologies).
To sequence the R. chinensis ‘Old Blush’ genome, we used in vitro–cultured plants obtained through adventitious shoot organogenesis from type 1 somatic embryo (RcOBType1), as previously described32. Axenic in vitro R. chinensis ‘Old Blush’ plantlets were ground in liquid nitrogen, and nuclei were purified as previously described31. Nuclei pellets were then processed with a Qiagen DNeasy Plant kit (Qiagen), according to the protocol provided by the supplier.
High-quality DNA was extracted from leaf samples of Rosa species and cultivars grown at ENS-Lyon, at the Lyon botanical garden, in the rose garden ‘La Bonne Maison, O. Masquier, Lyon, France’ or in the rose garden ‘Jardin Expérimental de Colmar, France’ (Supplementary Note 8).
DNA integrity was verified by gel electrophoresis (0.7% agarose), and DNA was then quantified by fluorometry with Picogreen (Applied Biosystems/Life Technologies).
Paired-end-sequencing DNA libraries were constructed with Illumina’s TruSeq DNA LT kit according to the manufacturer’s recommendations (Supplementary Tables 4 and 5). The distributions of DNA-fragment lengths in the libraries were verified with Agilent BioAnalyzer High Sensitivity DNA chip assays. Whole-genome sequencing of R. chinensis ‘Old Blush’ was performed on an Illumina HiSeq 2000 instrument. Sequences from paired-end and mate-pair reads of the multiple libraries were assembled in ALLPathsLG software33 (Supplementary Table 6).
Three-dimensional proximity information obtained by chromosome conformation capture sequencing (Hi-C)
Leaf tissues were fixed in 1% (vol/vol) formaldehyde and were then used for preparation of two independent in situ Hi-C libraries. Nuclei extraction, nuclei permeabilization, chromatin digestion and proximity-ligation treatments were performed essentially as previously described34. DpnII was used as a restriction enzyme. The recovery of Hi-C DNA and subsequent DNA manipulations were performed as previously described35. Libraries were sequenced on an Illumina NextSeq instrument with 2 × 75-bp reads. Hi-C libraries were independently analyzed in HiC-Pro pipeline (default parameters and LIGATION_SITE = GATCGATC36). Valid ligation products from each library were merged for interaction-matrix construction. The genome was divided into bins of equal size, and the number of contacts was determined between each pair of reported bins. Finally, contact maps were plotted in HiCPlotter software37.
Genome assembly
The program til-r was developed to implement heuristics aiming at filtering the graph of overlap generated by FALCON (Supplementary Note 3). A meta-assembly combining two CANU and four FALCON assemblies was generated in CANU 1.4 (Supplementary Fig. 2 and Supplementary Note 3).
Pseudomolecule building
Pseudomolecules were built by anchoring the 82 contigs to the K5 SNP genetic linkage map14 in ALLMAPS software38. Four chimeric breakpoints were identified and corrected by identifying the primary contigs in which the problematic regions were not merged. Three chimeric breakpoints were absent in CANU assemblies, and the fourth was absent in all primary assemblies. Finally, ALLMAPS was applied on the corrected meta-assembly, thus enabling building of seven pseudomolecules corresponding to the rose haploid chromosome number by anchoring and orienting 97.7% of the contigs (503 Mb) based on 86.4% of the genetic markers. The final assembly consists of seven pseudochromosomes and the mitochondrial and chloroplast genomes plus 46 unanchored contigs spanning 11.2 Mb (Supplementary Fig. 2a).
The genome was first polished in quiver39 with stringent alignment cutoffs (--minLength 3000 --maxHits 1). Then, a run of pilon40 (version 1.21, --mindepth 30 --fix bases) with homozygous ‘Old Blush’ Illumina paired-end reads edited 7,444 SNPs, 107,249 small insertions and 33 small deletions. The final genome assembly is composed of 515,588,973 nt including the 3,300 ‘N’ for the 33 gaps, seven of which represent centromeres. Biological centromeres were located by identifying tandem repeats in TRF software41, selecting patterns of an over-represented length in the genome, assembling them in contigs and visually inspecting their distribution along the pseudomolecules (Supplementary Note 3).
Localization of putative crossovers and segmental conservation between genotypes
Identification of putative loci of crossovers was performed by mapping Illumina reads from the heterozygous genome (five distinct libraries) on the constructed pseudochromosomes in BWA software42 and by counting pairs in which only one read had a match, in 10-kb-long windows. We observed 50 windows with over-represented one-end-mapped pairs in at least two libraries and kept them as candidate crossover loci (Supplementary Fig. 12, yellow frame). To confirm them, when possible, we used the sequence conservation with genotypes related to the inferred parents of ‘Old Blush’ (Supplementary Fig. 12, red plots; Supplementary Note 4.2).
Annotation of protein-coding genes and lncRNAs
Gene models were predicted with a fully automated and parallelized pipeline, egn-ep (see URLs), that carries out probabilistic sequence model training, genome masking, transcript and protein alignment computation and integrative gene modeling in EuGene software43 (release 4.2a). The configuration of the egn-ep pipeline is detailed in Supplementary Note 5. The inferred mRNAs were assessed in BUSCO v2 (ref. 15), which found 1,389 complete, 23 fragmented and 28 missing gene models (96.5%, 1.6% and 1.9% respectively). 36,377 genes were retained after the removal of annotated repeated elements (described below). The correspondence between gene models in homozygous and heterozygous annotations was established on the basis of best reciprocal hits (Supplementary Table 7 and Supplementary Data 1).
Functional annotation of protein-coding genes
The protocol described by Schläpfer et al.44 was used to annotate enzymes and build the metabolic network. Two cutoffs were modified to increase stringency: the BLAST e-value cutoff was lowered to 10−5, and the pathway-prediction score was set to 0.3 in pathway-tools. Nineteen pathways considered to be false positives were removed. A MetExplore instance45 is available to visualize the network (see URLs).
Protein-coding genes were annotated through integration of five sources, depending on their expected accuracy. Priorities were successively given to (i) a search of reciprocal best hits with the 218 Rosaceae proteins tagged as ‘reviewed’ in the UniProt database (90% span, 80% identity)46, (ii) the description of the 8,512 previously annotated enzymes, (iii) transcription factors and kinases identified (2,414 and 1,885 respectively) by ITAK47, (iv) the 3,954 transcription factors identified by PlantTFCat48 and (v) the InterPro analysis matching 31,853 proteins49. Finally, the annotations were tested and edited when needed to follow consistency rules defined by GenBank (see URLs).
De novo transposable-element and repeat annotation
The pseudochromosomes were deconstructed into ‘virtual’ contigs by removal of stretches of >11 undefined bases (Ns) to exclude gaps. We generated 2,742 virtual contigs with an N50 of 22 Mb for a total length of 515 Mb. The TEdenovo pipeline50,51 from the REPET package v2.5 (see URLs) was used to detect TEs in these contigs and to build a consensus sequence for each TE family with a minimum of five sequences per group. A library was generated containing 28,545 consensus sequences, classified according to structural and functional features (similarities with characterized TEs from the RepBase database v21.01 (ref. 52) and domains from Pfam27.0). After removal of redundancy and filtering consensus sequences classified as satellites (labeled SSR) and unclassified consensus sequences constructed with fewer than ten copies in the genome, a library of 8,226 consensus sequences was used to annotate TE copies in the whole homozygote genome with the TEannot pipeline with default parameters53. To refine TE annotation, consensus sequences showing no full-length fragments (i.e., fragments covering more than 95% of the consensus sequence) in the genome were filtered out, and a subset of 3,933 consensus sequences was used to run a second TEannot iteration. After a manual curation step to reclassify some consensus sequences, the final annotation files were renamed with this new classification, and this library was used to annotate the heterozygote genome (15,938 scaffolds for a total length without Ns of 746 Mb) with the TEannot pipeline. Consensus sequences classified as potential host genes bearing Pfam domains were manually curated and removed from the TE set (453 consensus sequences).
Annotation of miRNA precursors and mature miRNAs
To identify R. chinensis miRNA genes, an RNA library was constructed with mixed RNAs from pooled organs. After adaptor cleaning and removal of rRNA/tRNA-related sequences, we identified 38 million putative small RNAs displaying a size distribution ranging between 20 and 25 nt, with two peaks at 21 nt (17 million) and 24 nt (11.8 million). Genome-wide annotation of miRNA precursors was performed with an updated version of the pipeline described by Formey et al.54, which was modified to integrate stringent criteria proposed by miRBase (for example, expression of both mature 5p and 3p miRNAs)55. A total of 207 miRNA precursor loci were predicted to correspond to 636 expressed mature precursors (328 5p and 308 3p). miRNA targets were predicted with miRanda v3.0 (see URLs). Known mature miRNAs not found by the automatic and stringent process were annotated with blastn.
Genetic structure and genome segmentation
Illumina data mapping and SNP calling were performed as described in Supplementary Note 8. The number of homozygote and heterozygote variants in sliding windows of 1 Mb was computed on genic SNPs for each genotype, with functions of the bedtools suite (bedtools makewindows, bedtools intersect and bedtools groupby)56. To compute the density of variants per window, the number of variants was divided by the number of informative sites (mapping coverage between 5 and 60 for the 14 resequenced species and between 50 and 300 for the heterozygote Old Blush genotype). We use the term variants in tetraploid species to refer both to allelic differences and to differences between homeologs (i.e., between genes of different subgenomes). Owing to vegetative multiplication of rose cultivars, limited recombination has occurred after hybridization, and the size of introgressed fragments should be large. If the genomes or subgenomes involved in hybridization events have different distances with respect to the reference genome, genomic regions with different introgression histories should display different levels of variant density in resequenced hybrid cultivars. We used the changes in variant density in the genotypes FRA, GIG, HUM, MUT and SAN to segment the genome into 35 intervals (ranging from 2 to 56 Mb). The genomic boundaries were defined as the start of the windows corresponding to the inflexion points in density files. For each of the 35 genome segments, the genetic structure was inferred on biallelic SNPs with no missing data and not overlapping with repeat elements. Principal component analyses57 were performed with the glPCA function of the adegenet package (version 2.0.1)58. Axes 1 and 2 of the PCA explained a significant proportion of the variance (29.29% to 40.53% and 12.07% to 19.89%, respectively). Therefore, we present only the analyses of these two axes.
Rose and Rosaceae paleogenomics
Two parameters were defined as previously described59 to increase the stringency and significance of BLAST sequence alignment by parsing BLAST results and rebuilding high-scoring pairs or pairwise sequence alignments to identify accurate paralogous and orthologous relationships between Rosa (7 chromosomes, 49,767 genes), apricot (8 chromosomes, 31,390 genes), peach (8 chromosomes, 27,864 genes), apple (17 chromosomes, 63,514 genes), pear (17 chromosomes, 42,812 genes) and strawberry (7 chromosomes, 32,831 genes). From the previous orthologous and paralogous relationships, ancestral karyotypes were reconstructed as defined by Salse59, such that the ancestral genome is a ‘median’ or ‘intermediate’ genome consisting of a clean reference-gene order common to the extant species investigated.
Biochemical analyses of scent composition in roses
Volatile compounds were extracted with hexane from petals and stamens of roses of the different genotypes, mainly as previously described28 (Supplementary Note 9). Camphor was used as an internal standard to estimate compound quantities. Hexane sample fractions were analyzed with a gas chromatograph coupled to an electron ionization mass spectrometer detector (Agilent 6850) operated under an ion-source temperature of 230 °C, a trap emission current of 35 µA and a 70-eV ionization energy. All experiments were performed at least twice. Chromatographs were analyzed in Agilent Data Analysis software, and the volatile substances were identified by screening the WILEY 275, NIST 08 and CNRS libraries to compare MS spectra. The Kovats retention index of each substance was calculated with data of the injection of a homologous set of n-alkane (C8–C20) according to the Kovats formula60. Mass-spectra similarities together with Kovats-retention-index values were then used for compound identification. Concentrations were calculated through comparison of the camphor area as the internal standard.
ChIP–seq assays
Petals were collected from R. chinensis ‘Old Blush’ and fixed in 1% (vol/vol) formaldehyde. ChIP assays were performed with anti-H3K9ac (Millipore, 07-352) or anti-H3K27me3 (Millipore, 07-449) according to a procedure adapted from Veluchamy et al.61. Library quality was assessed with an Agilent 2100 Bioanalyzer (Agilent), and the libraries were subjected to high-throughput sequencing on an Illumina NextSeq 500 instrument. After trimming, reads were aligned to the R. chinensis genome in bowtie2 (ref. 62) with a maximum mismatch of 1 bp and unique mapping reported. To determine the target regions of H3K9ac ChIP–seq, model-based analysis of ChIP–seq (MACS2)63 was used. Detection of H3K27me3-modified regions was performed with SICER64. HOMER65 was used to annotate H3K9ac peaks with nearby genes if peaks were located in windows −2 kb to +1 kb around the gene TSS. For H3K27me3 peaks, bedtools intersect56 was used, and only genes that overlapped with this specific modification were kept. Clustering of H3K9ac and H3K27me3 peaks was performed with SeqMINER66. Rstudio, Circos67 and NGSplot68 were used for graphic representation of histone modifications.
RNA preparation and qPCR analyses
Total RNA and small RNAs were prepared from petals at three developmental stages: noncolored petals early during development (closed bud; stage 1); petals at the onset of anthocyanin synthesis (closed bud; stage 2); and fully colored petals with maximum anthocyanin content (bud opening; stage 3). Total RNA was prepared as previously described69. One microgram of RNA was used in reverse-transcription assays, and qPCR was performed as previously described70 with gene-specific primers (Supplementary Note 10 and Supplementary Tables 8 and 9). Small RNAs were extracted with a Macherey-Nagel NucleoSpin miRNA kit. Contaminating DNA was removed with an Ambion DNA-free kit. RNA concentrations were measured with a NanoDrop ND-1000 Micro-Volume spectrophotometer (NanoDrop Technologies) before and after DNase treatment. Small-RNA quantification was performed with stem-loop RT–PCR as previously described71. Reverse transcription was performed with a RevertAid kit (Thermo Fisher Scientific). Primers specific to 5.8S rRNA or stem-loop RT-primer for miR156 (Supplementary Note 10 and Supplementary Table 8) were used. 5.8S rRNA and miR156 expression were quantified with a QuantStudio 6 Flex Real-Time PCR 384 instrument (Applied Biosystems) with a Fast SYBR Green Master Mix kit (Roche Diagnostic) and specific primers (Supplementary Note 10). Data were collected for three independent biological replicates.
Code availability
Source code (in C) and linux binaries of the til-r software are available at http://lipm-bioinfo.toulouse.inra.fr/download/til-r/ under the GPL license.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary.
Data availability
The R. chinensis ‘Old Blush’ homozygous genome has been deposited in DDBJ/ENA/GenBank under accession number PDCK00000000. PacBio raw data have been deposited in the Sequence Read Archive (SRA) under study accession number SRP119907. The R. chinensis ‘Old Blush’ heterozygous genome has been deposited under BioProject accession number PRJEB24406.
Resequencing sequence reads have been deposited in the SRA under study accession number SRP119986.
Hi-C data have been deposited under SRA accession numbers SRR6189546 and SRR6189547, and ChIP–seq data have been deposited under SRA accession numbers SRR6167310, SRR6167311, SRR6167312 and SRR6167313 and under Gene Expression Omnibus accession number GSE109433.
References
Fougère-Danezan, M., Joly, S., Bruneau, A., Gao, X. F. & Zhang, L. B. Phylogeny and biogeography of wild roses with specific attention to polyploids. Ann. Bot. 115, 275–291 (2015).
De Vries, D. P. & Dubois, L. Rose breeding: past, present, prospects. Acta Hortic. 424, 241–248 (1996).
Martin, M., Piola, F., Chessel, D., Jay, M. & Heizmann, P. The domestication process of the modern rose: genetic structure and allelic composition of the rose complex. Theor. Appl. Genet. 102, 398–404 (2001).
Hurst, C. C. Notes on the origin and evolution of our garden roses. J. R. Hort. Soc. 66, 73–82 (1941).
Bendahmane, M., Dubois, A., Raymond, O. & Bris, M. L. Genetics and genomics of flower initiation and development in roses. J. Exp. Bot. 64, 847–857 (2013).
Esselink, G. D., Smulders, M. J. & Vosman, B. Identification of cut rose (Rosa hybrida) and rootstock varieties using robust sequence tagged microsatellite site markers. Theor. Appl. Genet. 106, 277–286 (2003).
Zharkikh, A. et al. Sequencing and assembly of highly heterozygous genome of Vitis vinifera L. cv Pinot Noir: problems and solutions. J. Biotechnol. 136, 38–43 (2008).
Yokoya, K., Roberts, A. V., Mottley, J., Lewis, R. & Brandham, P. E. Nuclear DNA amounts in roses. Ann. Bot. 85, 557–561 (2000).
Nakamura, N. et al. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. https://doi.org/10.1093/dnares/dsx042 (2017).
Badouin, H. et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546, 148–152 (2017).
VanBuren, R. et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527, 508–511 (2015).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Bourke, P. M. et al. Partial preferential chromosome pairing is genotype dependent in tetraploid rose. Plant J. 90, 330–343 (2017).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Iwata, H. et al. The TFL1 homologue KSN is a regulator of continuous flowering in rose and strawberry. Plant J. 69, 116–125 (2012).
Li, S. et al. Inheritance of perpetual blooming in Rosa chinensis ‘Old Blush’. Hortic. Plant J. 1, 108–112 (2015).
Mouradov, A., Cremer, F. & Coupland, G. Control of flowering time: interacting pathways as a basis for diversity. Plant Cell 14 Suppl, S111–S130 (2002).
Vaistij, F. E. et al. Differential control of seed primary dormancy in Arabidopsis ecotypes by the transcription factor SPATULA. Proc. Natl Acad. Sci. USA 110, 10866–10871 (2013).
Huo, H., Wei, S. & Bradford, K. J. DELAY OF GERMINATION1 (DOG1) regulates both seed dormancy and flowering time through microRNA pathways. Proc. Natl. Acad. Sci. USA 113, E2199–E2206 (2016).
Han, Y. et al. Comparative RNA-seq analysis of transcriptome dynamics during petal development in Rosa chinensis. Sci. Rep. 7, 43382 (2017).
Gou, J. Y., Felippes, F. F., Liu, C. J., Weigel, D. & Wang, J. W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23, 1512–1522 (2011).
Zvi, M. M. et al. PAP1 transcription factor enhances production of phenylpropanoid and terpenoid scent compounds in rose flowers. New Phytol. 195, 335–345 (2012).
Lin-Wang, K. et al. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 10, 50 (2010).
Aharoni, A. et al. Gain and loss of fruit flavor compounds produced by wild and cultivated strawberry species. Plant Cell 16, 3110–3131 (2004).
Shulaev, V. et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116 (2011).
Yu, Z. X. et al. Progressive regulation of sesquiterpene biosynthesis in Arabidopsis and Patchouli (Pogostemon cablin) by the miR156-targeted SPL transcription factors. Mol. Plant 8, 98–110 (2015).
Magnard, J. L. et al. Biosynthesis of monoterpene scent compounds in roses. Science 349, 81–83 (2015).
Touraev, A. & Heberle-Bors, E. Microspore embryogenesis and in vitro pollen maturation in tobacco. Methods Mol. Biol. 111, 281–291 (1999).
Brioudes, F., Thierry, A. M., Chambrier, P., Mollereau, B. & Bendahmane, M. Translationally controlled tumor protein is a conserved mitotic growth integrator in animals and plants. Proc. Natl. Acad. Sci. USA 107, 16384–16389 (2010).
Carrier, G. et al. An efficient and rapid protocol for plant nuclear DNA preparation suitable for next generation sequencing methods. Am. J. Bot. 98, e13–e15 (2011).
Vergne, P. et al. Somatic embryogenesis and transformation of the diploid rose Rosa chinensis cv ‘Old Blush’. Plant Cell Tissue Organ Cult. 100, 73–81 (2010).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 108, 1513–1518 (2011).
Zhu, W. et al. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biol. 18, 157 (2017).
Wang, C. et al. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res. 25, 246–256 (2015).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 16, 198 (2015).
Tang, H. et al. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 16, 3 (2015).
Chin, C. S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Foissac, S. et al. Genome annotation in plants and fungi: EuGene as a model platform. Curr. Bioinform. 3, 87–97 (2008).
Schläpfer, P. et al. Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 173, 2041–2059 (2017).
Cottret, L. et al. MetExplore: a web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res. 38, W132–W137 (2010).
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45 D1, D158–D169 (2017).
Zheng, Y. et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670 (2016).
Dai, X., Sinharoy, S., Udvardi, M. & Zhao, P. X. PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics 14, 321 (2013).
Finn, R. D. et al. InterPro in 2017: beyond protein family and domain annotations. Nucleic Acids Res. 45 D1, D190–D199 (2017).
Flutre, T., Duprat, E., Feuillet, C. & Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS One 6, e16526 (2011).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PLoS One 9, e91929 (2014).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Quesneville, H. et al. Combined evidence annotation of transposable elements in genome sequences. PLOS Comput. Biol. 1, 166–175 (2005).
Formey, D. et al. The small RNA diversity from Medicago truncatula roots under biotic interactions evidences the environmental plasticity of the miRNAome. Genome Biol. 15, 457 (2014).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Pearson, K. LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 2, 559–572 (1901).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Salse, J. Ancestors of modern plant crops. Curr. Opin. Plant Biol. 30, 134–142 (2016).
Adams, R. P. Identification of Essential Oil Components By Gas Chromatography/Mass Spectrometry.. 4th edn. (Allured Publishing Corporation, Carol Stream, IL, USA, 2007).
Veluchamy, A. et al. LHP1 regulates H3K27me3 spreading and shapes the three-dimensional conformation of the arabidopsis genome. PLoS One 11, e0158936 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Zang, C. et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958 (2009).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Ye, T. et al. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 39, e35 (2011).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Shen, L., Shao, N., Liu, X. & Nestler, E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284 (2014).
Dubois, A. et al. Tinkering with the C-function: a molecular frame for the selection of double flowers in cultivated roses. PLoS One 5, e9288 (2010).
Dubois, A. et al. Genomic approach to study floral development genes in Rosa sp. PLoS One 6, e28455 (2011).
Marcial-Quino, J. et al. Stem-loop RT-qPCR as an efficient tool for the detection and quantification of small RNAs in Giardia lamblia. Genes (Basel) 7, E131 (2016).
Acknowledgements
We thank J. Thomas, T. Goujon and C. Bendahmane for critical reading of the manuscript. We thank A. Meilland for helpful discussions. We thank A. Lacroix, P. Bolland and J. Berger (ENS de Lyon, France) for plant handling. We thank the Lyon Botanical Garden–France and the rose garden La Bonne Maison O. Masquelier, Lyon, France, for providing plant material. We thank the Genotoul bioinformatics platform Toulouse Midi-Pyrenées for providing help and computing resources and L. Taulelle and E. Quemener (ENS de Lyon) for assistance with computing. We gratefully acknowledge support from the Pôle Scientifique de Modélisation Numérique of the ENS de Lyon for computing resources. We thank the epigenomic platform of the IPS2-University Paris-Sud-Orsay France. We thank the platforms ‘AniRA-Cytometry’ and ‘Analyse Génétiques et Cellulaire’ of the IFR BioScience Lyon (UMS3444/US8) for HRM and flow cytometry experiments. The Get-Plage platform was supported by the GET-PACBIO program (programme operationnel FEDER-FSE MIDI-PYRENEES ET GARONNE 2014-2020). This work was supported by funds from the French National Institute of Agronomic Research (INRA); the program Fonds Recherche of Ecole Normale Supérieure-Lyon-France to M. Bendahmane and O.R.; the Genoscope to P.W.; the French National Research Agency programs DODO (ANR-16CE20-0024-03 to M. Bendahmane and M. Vandenbussche) and AUXIFLO (ANR-12-BSV6-0005 to T.V); and the European Research Council (ERC-SEXYPARTH) and the Labex Saclay Plant Sciences–SPS (ANR-10-LABX-0040-SPS) to A. Bendahmane.
Author information
Authors and Affiliations
Contributions
O.R. and C.B. performed DNA extraction. P. Vergne and P. Villand produced the rose homozygous line. C.L.-R. and M. Bendahmane performed PacBio sequencing data production. J. Szécsi performed flow cytometry experiments. P.H. provided Rosa material. M. Verdenaud, D.L., M. Benhamed and M.P. performed epigenome analysis. O.R., X.F., S.-H.Y., A.D., M.L.B. and M. Bendahmane performed DNA/RNA sample collection and data production. J.J., M. Verdenaud, M.L., L.F. and O.R. performed RNA-seq and analyses of gene expression. M. Benhamed, M. Verdenaud, C.L., A. Boualem and A. Bendahmane performed chromosome conformation capture Hi-C. M. Benhamed and M. Verdenaud integrated the assembly and genetic map to build pseudochromosomes. J.G. developed bioinformatics tools and assembled the PacBio homozygous genome. M. Benhamed and M. Verdenaud validated the assembly with Hi-C and genetic data. P.W. and A. Lemainque performed Illumina sequencing. A. Boualem and A. Bendahmane provided resequencing sequencing data. A.C., J.-M.A., A.M., K.L. and P.W. assembled the heterozygous rose Illumina sequencing data. N.C., H.Q. and J.J. conducted repetitive DNA analysis. J.G., N.C. and J.J. annotated protein-coding genes, TEs and miRNAs. J.G., H.B., J.J. and L.C. performed bioinformatics analyses. S.C. built the Rosa web portal. J. Salse and C.P. conducted paleoevolution analyses. J.J. and O.R. conducted miRNA analyses. A. Larrieu, T.V. and J.J. performed integrated analyses on auxin genes. S.M., J.-C.C. and S.B. performed gas chromatography–mass spectrometry analyses of scent compounds. S.M., J.-C.C., S.B., O.R. and J.J. performed integrated analyses on scent genes. O.R., L.P., F.P., L.F. and M. Verdenaud performed integrated analyses on flowering genes. M. Vandenbussche performed integrated analyses on MADS transcription-factor genes. O.R., L.F., J.J., L.P., J. Szécsi. and V.B. performed integrated analyses on color genes. M.L.-B., B.G. and Y.L. performed integrated analyses on meiosis genes. H.B., O.R. J.G., J.J. and F.P. performed diversity analysis. M. Bendahmane and J.G. coordinated the rose genome consortium. M. Bendahmane, O.R., H.B. and J.G. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–23, Supplementary Notes 1–13 and Supplementary Tables 1–9
Supplementary Data
Supplementary Data 1–10
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raymond, O., Gouzy, J., Just, J. et al. The Rosa genome provides new insights into the domestication of modern roses. Nat Genet 50, 772–777 (2018). https://doi.org/10.1038/s41588-018-0110-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0110-3
This article is cited by
-
Haplotype-resolved genome assembly of the diploid Rosa chinensis provides insight into the mechanisms underlying key ornamental traits
Molecular Horticulture (2024)
-
Chromosome-scale genome assembly and annotation of Cotoneaster glaucophyllus
Scientific Data (2024)
-
Rose FT homologous gene overexpression affects flowering and vegetative development behavior in two different rose genotypes
Plant Cell, Tissue and Organ Culture (PCTOC) (2024)
-
Molecular profiling of Greek native germplasm collection of Rosa canina L. for enhanced fruit extract production: a comprehensive approach utilizing neutral, gene, and exon-based markers
Genetic Resources and Crop Evolution (2024)
-
Coordination among flower pigments, scents and pollinators in ornamental plants
Horticulture Advances (2024)
