
Abstract
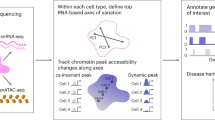
Regulatory variants are often context specific, modulating gene expression in a subset of possible cellular states. Although these genetic effects can play important roles in disease, the molecular mechanisms underlying context specificity are poorly understood. Here, we identified shared quantitative trait loci (QTLs) for chromatin accessibility and gene expression in human macrophages exposed to IFNγ, Salmonella and IFNγ plus Salmonella. We observed that ~60% of stimulus-specific expression QTLs with a detectable effect on chromatin altered the chromatin accessibility in naive cells, thus suggesting that they perturb enhancer priming. Such variants probably influence binding of cell-type-specific transcription factors, such as PU.1, which can then indirectly alter the binding of stimulus-specific transcription factors, such as NF-κB or STAT2. Thus, although chromatin accessibility assays are powerful for fine-mapping causal regulatory variants, detecting their downstream effects on gene expression will be challenging, requiring profiling of large numbers of stimulated cellular states and time points.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Li, Y. et al. Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat. Med. 22, 952–960 (2016).
Barreiro, L. B. et al. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosisinfection. Proc. Natl Acad. Sci. USA 109, 1204–1209 (2012).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014).
Kim, S. et al. Characterizing the genetic basis of innate immune response in TLR4-activated human monocytes. Nat. Commun. 5, 5236 (2014).
Lee, M. N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014).
Çalışkan, M., Baker, S. W., Gilad, Y. & Ober, C. Host genetic variation influences gene expression response to rhinovirus infection. PLoS Genet. 11, e1005111 (2015).
Nédélec, Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–66 .e21 (2016).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Kim-Hellmuth, S. et al. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun. 8, 266 (2017).
Degner, J. F. et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482, 390–394 (2012).
Jin, F., Li, Y. & Ren, B. & Natarajan, R. PU.1 and C/EBP(alpha) synergistically program distinct response to NF-kappaB activation through establishing monocyte specific enhancers. Proc. Natl. Acad. Sci. USA 108, 5290–5295 (2011).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Heinz, S. et al. Effect of natural genetic variation on enhancer selection and function. Nature 503, 487–492 (2013).
Wang, A. et al. Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell 16, 386–399 (2015).
Chow, N. A., Jasenosky, L. D. & Goldfeld, A. E. A distal locus element mediates IFN-γ priming of lipopolysaccharide-stimulated TNF gene expression. Cell Rep. 9, 1718–1728 (2014).
Shin, H. Y. et al. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 48, 904–911 (2016).
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Banovich, N. E. et al. Impact of regulatory variation across human iPSCs and differentiated cells. Genome Res. 28, 122–131 (2017).
Pique-Regi, R. et al. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 21, 447–455 (2011).
Sherwood, R. I. et al. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 32, 171–178 (2014).
Alasoo, K. et al. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci. Rep. 5, 12524 (2015).
Kilpinen, H. et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546, 370–375 (2017).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Hu, X. & Ivashkiv, L. B. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31, 539–550 (2009).
Qiao, Y. et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity 39, 454–469 (2013).
Kumasaka, N., Knights, A. J. & Gaffney, D. J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 48, 206–213 (2016).
Grubert, F. et al. Genetic control of chromatin states in humans involves local and distal chromosomal interactions. Cell 162, 1051–1065 (2015).
Waszak, S. M. et al. Population variation and genetic control of modular chromatin architecture in humans. Cell 162, 1039–1050 (2015).
Cheng, C.S. et al. Genetic determinants of chromatin accessibility and gene regulation in T cell activation across human individuals. Preprint at https://www.biorxiv.org/content/early/2017/06/08/090241/ (2017).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Morris, A. P. et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990 (2012).
Jansen, R. et al. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum. Mol. Genet. 26, 1444–1451 (2017).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464 (2015).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Farh, K. K.-H. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Guo, H. et al. Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum. Mol. Genet. 24, 3305–3313 (2015).
Chun, S. et al. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat. Genet. 49, 600–605 (2017).
Battle, A. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Javierre, B. M. et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369–1384.e19 (2016).
Jin, F. et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503, 290–294 (2013).
Ghavi-Helm, Y. et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512, 96–100 (2014).
Pai, A. A. et al. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 8, e1003000 (2012).
Mullen, A. C. et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576 (2011).
Trompouki, E. et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147, 577–589 (2011).
Ostuni, R. et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013).
Magnani, L., Eeckhoute, J. & Lupien, M. Pioneer factors: directing transcriptional regulators within the chromatin environment. Trends Genet. 27, 465–474 (2011).
Ramirez-Carrozzi, V. R. et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128 (2009).
Bojcsuk, D., Nagy, G. & Balint, B. L. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 45, 3693–3706 (2017).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Schroder, K., Hertzog, P. J., Ravasi, T. & Hume, D. A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 (2004).
Ongen, H., Buil, A., Brown, A. A., Dermitzakis, E. T. & Delaneau, O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 32, 1479–1485 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Jun, G. et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 91, 839–848 (2012).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285 (2012).
Hansen, K. D., Irizarry, R. A. & Wu, Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216 (2012).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 7, 500–507 (2012).
Jiang, H., Lei, R., Ding, S.-W. & Zhu, S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997/ (2013).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Castel, S. E., Levy-Moonshine, A., Mohammadi, P., Banks, E. & Lappalainen, T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 16, 195 (2015).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Davis, J. R. et al. An efficient multiple-testing adjustment for eqtl studies that accounts for linkage disequilibrium between variants. Am. J. Hum. Genet. 98, 216–224 (2016).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Tan, G. & Lenhard, B. TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics 32, 1555–1556 (2016).
Weirauch, M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, New York, 2009).
Alasoo, K. wiggleplotr: Make Read Coverage Plots From BigWig Files. R package version 1.2.0 (Bioconductor, 2017).
Acknowledgements
We thank L. Parts, J. Schwartzentruber, C. Wallace, L. Milani, K. Lepik and H. Peterson for helpful comments on the manuscript. We thank R. Nelson for assistance and early access to HipSci iPSC lines. We thank R. Kreuzhuber for providing access to the imputed genotype data from the Fairfax study. We thank C. D. Brown for helpful comments on the manuscript. We also thank WTSI DNA Pipelines and Cytometry Core Facility for their sequencing and flow cytometry services. This work was supported by Wellcome Trust grant WT098051 (G.D. and D.J.G.). K.A. was supported by a PhD fellowship from the Wellcome Trust (WT099754/Z/12/Z) and a postdoctoral fellowship from the Estonian Research Council (MOBJD67). The iPSC lines were generated at the Wellcome Trust Sanger Institute, under the Human Induced Pluripotent Stem Cell Initiative funded by a strategic award (WT098503) from the Wellcome Trust and Medical Research Council. We also acknowledge Life Science Technologies Corporation as the provider of cytotune.
Author information
Authors and Affiliations
Consortia
Contributions
K.A. and D.J.G. wrote the paper with input from all authors. K.A. and J.R. performed the macrophage differentiation experiments. J.R. and A.J.K. performed the chromatin accessibility assays. A.L.M. and K.K. assisted with disease colocalization and enrichment analysis. K.A., S.M. and C.H. optimized the stimulation experiments. K.A. analyzed the data. K.A., S.M., G.D. and D.J.G. designed the experiments. G.D. and D.J.G. supervised research. The HIPSCI Consortium generated and provided early accesss to the iPSC lines used in this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–19, Supplementary Tables 2, 3 and 9, and Supplementary Note
Supplementary Tables
Supplementary Tables 1 and 4–8
Rights and permissions
About this article

Cite this article
Alasoo, K., Rodrigues, J., Mukhopadhyay, S. et al. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50, 424–431 (2018). https://doi.org/10.1038/s41588-018-0046-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-018-0046-7
This article is cited by
-
PICALO: principal interaction component analysis for the identification of discrete technical, cell-type, and environmental factors that mediate eQTLs
Genome Biology (2024)
-
Epigenetic variation impacts individual differences in the transcriptional response to influenza infection
Nature Genetics (2024)
-
Fundamentals for predicting transcriptional regulations from DNA sequence patterns
Journal of Human Genetics (2024)
-
Investigating the role of common cis-regulatory variants in modifying penetrance of putatively damaging, inherited variants in severe neurodevelopmental disorders
Scientific Reports (2024)
-
A landscape of gene expression regulation for synovium in arthritis
Nature Communications (2024)
