
Abstract
The sea lamprey (Petromyzon marinus) serves as a comparative model for reconstructing vertebrate evolution. To enable more informed analyses, we developed a new assembly of the lamprey germline genome that integrates several complementary data sets. Analysis of this highly contiguous (chromosome-scale) assembly shows that both chromosomal and whole-genome duplications have played significant roles in the evolution of ancestral vertebrate and lamprey genomes, including chromosomes that carry the six lamprey HOX clusters. The assembly also contains several hundred genes that are reproducibly eliminated from somatic cells during early development in lamprey. Comparative analyses show that gnathostome (mouse) homologs of these genes are frequently marked by polycomb repressive complexes (PRCs) in embryonic stem cells, suggesting overlaps in the regulatory logic of somatic DNA elimination and bivalent states that are regulated by early embryonic PRCs. This new assembly will enhance diverse studies that are informed by lampreys’ unique biology and evolutionary/comparative perspective.
Similar content being viewed by others

Main
The sea lamprey is a member of an ancient lineage that diverged from the vertebrate stem approximately 550 million years ago (MYA). By virtue of this deep evolutionary perspective, lamprey has served as a critical model for understanding the evolution of several conserved and derived features that are relevant to broad fields of biology and biomedicine. Studies have used lampreys to provide perspective on the evolution of developmental pathways that define vertebrate embryogenesis1,2, vertebrate nervous and neuroendocrine systems2,3, genome structure4, immunity5, clotting6 and other features7. These studies show aspects of vertebrate biology that have been conserved over deep evolutionary time and identify evolutionary modifications that gave rise to novel features that emerged within the jawed vertebrate lineage (gnathostomes). Lampreys also possess several features that are not observed in gnathostomes, which could represent either aspects of ancestral vertebrate biology that have not been conserved in the gnathostomes or features that arose since the divergence of the ancestral lineages that gave rise to lampreys and gnathostomes. These include the ability to achieve full functional recovery after complete spinal cord transection, the deployment of evolutionarily independent yet functionally equivalent adaptive immune receptors, and the physical restructuring of the genome during development known as programmed genome rearrangement (PGR).
PGR results in the physical elimination of ~0.5 Gb of DNA from the organism’s ~2.3-Gb genome8,9,10. The elimination events that mediate PGR are initiated at the 7th embryonic cell division and are essentially complete by 3 days post fertilization11,12. As a result, lampreys are effectively chimeric, with germ cells possessing a full complement of genes and all other cell types possessing a smaller, reproducible fraction of the germline genome. Previous analyses support the idea that the somatic genome lacks several genes that contribute to the development and maintenance of germ cells but are potentially deleterious if misexpressed in somatic lineages. However, our understanding of the mechanisms and consequences of PGR remains incomplete, as only a smaller, reproducible fraction of the genome (lacking 0.5 Gb of sequence that is invariably specific to the germline) has been sequenced to date.
In contrast to the germline genome, the somatically retained portions of the genome are relatively well characterized. Because it was not known until 2009 that lampreys were subject to PGR8, sequencing efforts focused on somatic tissues from which DNA or intact nuclei could be readily obtained (e.g., blood and liver)13. Sequencing of the sea lamprey somatic genome followed an approach that had proven successful for other vertebrate genomes before the advent of next-generation sequencing technologies (Sanger sequencing of clone ends, fosmid ends and BAC ends). Because of the abundance of highly identical interspersed repetitive elements and moderately high levels of polymorphism (approaching 1%), assembly of the somatic genome resulted in a consensus sequence that was substantially more fragmentary than other Sanger-based vertebrate assemblies14. Nonetheless, this initial assembly yielded significant improvements in our understanding of the evolution of vertebrate genomes and fundamental aspects of vertebrate neurobiology, immunity and development1,2,3,4,5,6,7.
Here we present the first assembly of the sea lamprey germline genome. Through extensive optimization of assembly pipelines, we identified a computational solution that allowed us to generate an assembly from next-generation sequence data (Illumina and Pacific Biosciences reads) that surpasses the existing Sanger-based somatic assembly. Analysis of the resulting assembly identifies several hundred genes that are eliminated from somatic tissues by PGR and sheds new light on the evolution of genes and functional elements in the wake of ancient large-scale duplication events.
Results
Assembly and annotation of the sea lamprey genome
Several shotgun-sequencing and scaffolding data sets were generated in order to permit assembly of the sea lamprey germline genome (>100× sequence coverage in Illumina paired-end reads, >300× physical coverage in 4-kb Illumina mate pairs and >600× physical coverage in 40-kb Illumina mate pairs). Previous analyses demonstrated that the lamprey genome is highly repetitive, and initial analysis of Illumina shotgun sequence data confirmed that the repeat content of lamprey (~60% high-identity repeats) is substantially higher than that of human (Fig. 1). To enable the development of a highly contiguous assembly, we also generated ~17× genome coverage in single-molecule long-read data (Pacific Biosciences XL/C2 chemistry, N50 read length = 5,424) and performed hybrid assembly using DBG2OLC15. This approach yielded an assembly with contiguity statistics (23,286 contigs, N50 = 164,585 bp) that rivaled those of a previously published Sanger-based assembly of the lamprey somatic genome13. To further improve the large-scale structure of this assembly, we integrated scaffolding data (~56× coverage in BioNano optical mapping: >150 kb molecules, and 325 million Chicago (Dovetail) linked read pairs: 2 × 152 bp), as well as published meiotic mapping data4. Linkages identified through these three independent data sets were cross-validated and integrated using AllMaps (Fig. 2)16. This integrated scaffolding approach allowed us to further increase the contiguity of the assembly (12,077 contigs, N50 = 12 Mb, N50 contig number = 34). In total, 74.8% of the current germline genome assembly is anchored to one of 94 previously defined linkage groups4, and >80% of the assembly is present in super-scaffolds that are 1 Mb or longer. Given that the sea lamprey has 99 pairs of chromosomes in its germline, this integrated assembly appears to approach chromosome-scale contiguity.

a, The spectrum of error corrected 25-mers shows a modal count of 68 and a second hump at half of this value, corresponding to allelic k-mers. k-mer multiplicity is defined as the number of times a k-mer was observed in the sequence data set. b, Less than 40% of the lamprey genome can be represented by single-copy 25-mers, whereas >75% of the human genome can be represented by single-copy k-mers of this same length. The x axis is plotted on a log scale to aid the visualization of patterns at lower estimated copy numbers.

Data from three independent strategies were used to place contigs on larger chromosomal structures. Data from meiotic maps (Smith and Keinath, 20154; blue), Dovetail maps (red) and optical maps (green) complement and extend one another. a, Information used to generate super-scaffold 5. b, Ordering of anchors along super-scaffold 5. c, Information used to generate super-scaffold 21. d, Ordering of anchors along super-scaffold 21. ρ = Pearson correlation coefficient based on the following numbers of markers: a (top to bottom), n = 18, 28, 14, 10, 34, 156, 78 and 162 independent scaffolding anchors; b (top to bottom), n = 10, 22, 36, 196 and 79 independent scaffolding anchors.
Our long-range scaffolding approach used three independent methods that extend and cross-validate one another (Fig. 2), and we consider strong agreement among these three methods as evidence that the large-scale structure of the assembly accurately reflects the structure of P. marinus chromosomes. For many vertebrates, it is possible to independently assess long-range contiguity by measuring conservation of gene orders with closely related species. Highly contiguous assemblies are not yet available for any other jawless vertebrate, although an unanchored draft assembly does exist for the Arctic lamprey (Lethenteron camtschaticum: syn. Lethenteron japonicum)17. To provide perspective on the chromosomal structure of a closely related species, we developed a meiotic map for the Pacific lamprey (Entosphenus tridentatus). The species is a representative of a clade of lampreys (genera Entosphenus, Lethenteron and Lampetra) that diverged from the lineage represented by Petromyzon ~40 MYA18, and embryos of known parentage are available through ongoing hatchery efforts aimed at restoring the species to its native waterways in the US Pacific Northwest19. Meiotic mapping was performed using restriction site–associated DNA (RAD) sequencing of 94 F1 siblings generated from a controlled cross between two wild-captured individuals. The resulting meiotic map provides dense coverage of the genome and represents 83 linkage groups, covering 9,956 cM with an average intermarker distance of 3.4 cM (Supplementary Table 1). Alignment of RAD markers to the sea lamprey genome identified 1,733 homologous sequences, which show strong conservation of synteny and gene order (Fig. 3, Supplementary Table 1). This broad conservation of gene order is considered strong evidence that the sea lamprey assembly and Pacific lamprey meiotic map accurately reflect the chromosomal structure of their respective species.

The relative position of homologous sequences is shown for sea lamprey (y axis) and Pacific lamprey (x axis). A single homologous site (aligning RAD-seq read, Supplementary Table 1) is marked by a single dot. Chromosomes and linkage groups (LGs) are ordered from longest to shortest within species, and individual chromosomes and LGs are highlighted by alternating dark and light shading. Groups of adjacent dots (regions showing conservation of synteny and gene order) appear as diagonal lines.
The repetitive nature of the lamprey genome presents challenges not only to its assembly but also the identification of genes within assembled contigs. This is largely attributable to the interspersion of transposable coding sequences within and among the coding sequences of low-copy genes. To circumvent these issues, we used a two-tiered approach to gene prediction. Annotation and identification of repetitive elements was performed using RepeatModeler and RepeatMasker20,21. The entire set of annotated repeats, published gene models and transcriptomic data sets10,13 were integrated to generate a conservative set of 18,205 gene predictions using MAKER22. After generating initial gene calls, a second round of gene predictions was generated that permitted extraction of gene models that include low-copy repetitive sequences, yielding another 2,745 gene models, for a total of 20,950 MAKER gene models. In total, Maker was able to assign 18,367 of these gene models to a likely vertebrate homolog on the basis of multispecies BLAST alignments, which included the vast majority of single-copy orthologs expected for lamprey (Supplementary Note)23,24. An additional 2,583 genes (12%) could not be immediately assigned a homolog on the basis of multispecies alignments. Although these may represent lamprey-specific genes, careful manual curation is likely to be necessary to define their precise evolutionary origins. Such efforts will be enabled through the publicly available genome browser (see URLs). This annotation set was subsequently used to identify the location of 35,382 long noncoding RNA (lncRNA) transcripts in 18,857 lncRNA gene bodies (Supplementary Note, Supplementary Table 2 and Supplementary Fig. 1). These and other annotation sets, including RNA sequencing and genome re-sequencing tracks, are available through SIMRbase (see URLs).
Vertebrate genome evolution
Lamprey occupies a critical phylogenetic position with respect to reconstructing ancestral karyotypes and inferring the timing and mode of duplication events that occurred in ancestral vertebrate and gnathostome lineages. Alignment to the chicken25 and gar26 genomes (Supplementary Tables 3–5) permits reconstruction of ancestral orthology groups that are highly consistent with previous reconstructions based on the lamprey meiotic map4. Because these comparisons require resolution of homologies that are the product of duplication (i.e., 1:1 orthology is not expected) our operational definition of “orthology groups” is expanded to include higher-order relationships (see ref. 4 for more detail). Inclusion of comparative mapping data from the recently published gar genome assembly provides further support for the observation that the majority of ancestral vertebrate chromosomes experienced a single large-scale duplication event in the ancestral vertebrate lineage (Fig. 4, Supplementary Fig. 2). Most ancestral orthology groups correspond to two derived chicken chromosomes (6/11 chicken–lamprey orthology groups identified here). Three other orthology groups possess four derived chromosomes, suggesting that these groups have experienced an additional large-scale duplication: these include well-defined fourfold orthology regions harboring the HOX and MHC clusters in one orthology group, the NPYR locus and ParaHOX cluster in a second, and the RAR and ALDH1 loci in a third4 (Fig. 4). Two remaining orthology groups present more complex ratios of ancestral to derived chromosomes. Notably, though, comparative mapping with gar shows that chicken chromosome 26 and a portion of chicken chromosome 1 were likely fused in the bony vertebrate (Euteleostome) ancestor approximately 450 MYA and subsequently experienced a derived fission in the chicken lineage. Other deviations from 1:2 or 1:4 are interpreted as the product of derived fission/fusion events that occurred during the first 150 MY following divergence of basal lamprey and gnathostome lineages, derived fission/fusion events in the lamprey lineage, or misassembled regions of the lamprey genome. Although it is possible that the observed genome-wide patterns of conserved synteny could have arisen through two whole-genome duplication events (the 2R hypothesis)27,28 accompanied by large numbers of chromosome losses29,30, a previously proposed alternative scenario involving one whole-genome duplication preceded by three distinct chromosome-scale duplication events requires fewer evolutionary steps and is consistent with the data underlying all previous reconstructions4.

These patterns are consistent with those from the lamprey somatic genome assembly and show both chromosomal or segmental and whole-genome duplications. Lamprey super-scaffolds are oriented along the y axis and chicken chromosomes along the x axis. Circles reflect counts of syntenic orthologs on the corresponding lamprey and chicken chromosomes, with the size of each circle being proportional to the number of orthologs on that pair. The color of each circle represents the degree to which the number of observed orthologs deviates from null expectations under a uniform distribution across an identical number of lamprey and chicken chromosomes with identical numbers of orthology-informative genes. Shaded regions of the plot designate homology groups that correspond to presumptive ancestral chromosomes. Syntenic groups that are linked by lines marked EA are predicted to correspond to a presumptive single chromosome in the Euteleostome ancestor, on the basis of conserved synteny with spotted gar (Lepisosteus oculatus). The three largest super-scaffolds are each marked with an arrowhead along the y axis. The ordering of lamprey super-scaffolds along the y axis is provided in Supplementary Table 4.
Lamprey HOX clusters: duplication and divergence
Historically, descriptions of genome duplications have relied heavily on the HOX gene clusters. This is partially due to their highly conserved organization with respect to gene order and orientation, which contributes to the generation of coordinated patterns of axial expression (collinearity) associated with their roles in embryonic development. Assembly of the Arctic lamprey genome led to the tentative prediction of (at least) six, and possibly eight, HOX clusters, suggesting that the duplication history of at least the lamprey HOX-bearing chromosomes differs from that in the jawed vertebrates17. We identify 42 HOX genes in the sea lamprey, which all fall within six HOX clusters that are highly similar in content to the HOX clusters predicted in the Arctic lamprey (Fig. 5a, Supplementary Figs. 3 and 4). Additionally, we are able to place these in their broader chromosomal context, showing that these six HOX clusters are embedded in larger chromosomal regions that share conserved synteny with the presumptive ancestral HOX-bearing chromosome (Fig. 4).

a, Six HOX clusters can be identified within the sea lamprey genome assembly. Lamprey cluster designations α through ζ follow the convention of Mehta et al.17. HOX genes are represented as boxes, with the direction of their transcription indicated by the black arrow. Flanking non-HOX genes are depicted as arrowheads, which indicate their direction of transcription. The positions of known microRNAs are indicated. The four human HOX loci and the inferred ancestral vertebrate HOX locus40 are shown for comparison. The white arrow downstream of the lamprey HOX-γ cluster represents PMZ_0048273, an uncharacterized non-HOX gene. b, The evolutionary history was inferred using the neighbor-joining method41. The optimal tree with the sum of branch length = 9.68 is shown. The percentages of replicate trees in which the associated taxa clustered together (bootstrap test with 100 replicates) are shown next to the branches. c, Tests for enrichment of two-copy duplicates among all pairs of HOX-bearing chromosomes (super-scaffolds). Colors correspond to the degree to which the counts of shared duplicates on each pair of chromosomes deviates from the expected value given an identical number of chromosomes and paralogs retained on each chromosome (probability estimates were generated using two-tailed χ2 tests and a total of n = 200 independent pairs of duplicated genes: see Supplementary Table 6). Plus and minus symbols indicate the direction of deviation from expected for chromosome pairs with P < 0.01.
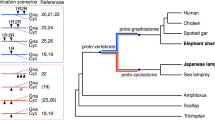
In principle, a number of duplication scenarios could potentially explain the existence of six paralogous HOX-bearing chromosomes. These include: (1) whole-genome duplication followed by triplication, or vice versa; (2) a gnathostome-like duplication history (either 2R accompanied by large numbers of chromosome losses29,30 or one whole-genome duplication preceded by three chromosome-scale duplication events4) followed by a further round of whole-genome duplication (yielding eight ancestral HOX clusters) and loss of two entire paralogous chromosomes; (3) a gnathostome-like duplication history followed by duplication of two individual chromosomes. Initial synteny comparisons between lamprey and gnathostome HOX loci showed no clear orthology relationships, but show substantial similarities in the gene content of lamprey HOX-ε and HOX-β clusters. Notably, phylogenetic analyses of paralogy groups with ≥4 retained copies (HOX4, HOX8, HOX9, HOX11 and HOX13) also show no clear orthology between lamprey and gnathostome clusters, but they reproducibly place members of HOX-ε and HOX-β clusters in sister clades with high bootstrap support (Fig. 5b, Supplementary Figs. 5–9). Taken at face value, this would seem to suggest that the ε and β clusters diverged from one another more recently than other paralogous clusters, apparently lending support to scenario 3. Alternatively, this might also reflect greater functional constraint with respect to the membership of these clusters.
To gain further perspective on the duplication history of lamprey HOX clusters, we extended the analyses to compare the chromosome-wide distribution of two-copy paralogs on all HOX-bearing chromosomes. Because post-duplication patterns of conserved synteny are strongly driven by paralog loss, we reasoned that more recent duplication events should yield pairs of chromosomes that share more two-copy duplications, exclusive of all other paralogous chromosomes (the latter of which would have experienced more extensive loss of redundant paralogs over time). Two pairs of chromosomes were observed to share more duplicates relative to all other pairwise combinations of HOX-bearing chromosomes. The strongest enrichment of two-copy paralogs was observed between super-scaffolds 5 and 16 (χ2 = 14.22, P = 1.6 × 10−4, d.f. = 1, Fig. 5, Supplementary Table 6), which carry the HOX-ε and HOX-β clusters. In conjunction with the internal structure of HOX clusters and consistent phylogenetic clustering of ε and β HOX members, we interpret this as indicating that the ε- and β-bearing chromosomes trace their ancestry to a chromosome-scale duplication event that occurred substantially more recently than the genome- and chromosome-scale duplication events that define all other pairwise contrasts, perhaps within the last 200–300 MY. Only one other pair of chromosomes shows significant enrichment of two-copy paralogs relative to all other contrasts. The chromosomes bearing HOX-α and HOX-δ clusters are enriched in shared two-copy paralogs (χ2 = 8.41, P = 3.7 × 10−3, d.f. = 1, Fig. 5, Supplementary Table 6), although α and δ HOX members show no consistent pattern of clustering within gene trees. This difference could be interpreted as indicating that these two chromosomes are the product of a slightly older duplication event, or alternatively it might reflect differential constraints relative to the retention of duplicates by individual pairs of paralogous chromosomes. However, it is unclear what processes might constrain the evolution of one pair of paralogous chromosomes relative to all others.
Programmed genome rearrangement
Identification of eliminated DNA
In lampreys approximately 20% of zygotically inherited DNA is eliminated from somatic cell lineages during early embryogenesis, being retained only by the germline8,10,31. To identify germline-enriched (i.e., somatically eliminated) regions, we generated whole-genome shotgun sequence data for both sperm (73× coverage) and blood (80× coverage) DNAs that were isolated from the same individual. Analysis of read counts identified 1,077 super-scaffolds with enrichment scores (log2(standardized sperm coverage/blood coverage)) exceeding 2, over more than 80% of the scaffold (Fig. 6, Supplementary Table 7). These presumptively germline-specific regions cover ~13 Mb of the genome assembly and contain 356 annotated protein coding genes. The distribution of enrichment scores also suggests that other regions with lower enrichment scores are likely to be affected by PGR. To further evaluate our predictions, we designed primers for the 96 longest super-scaffolds with enrichment scores of 2 or higher. In total, primers from 90 (94%) of these scaffolds yielded specific amplification in testes relative to blood, confirming that they are deleted during PGR (Supplementary Table 8).

Comparative sequencing reveals germline enrichment of several single- and low-copy intervals. The distribution of coverage ratios reveals a long tail corresponding to segments with higher sequence coverage in sperm relative to blood. This excess is highlighted in red, assuming a symmetrical distribution of enrichment scores for non-eliminated regions and an absence of somatic-specific sequence.
Notably, the estimates above only account for single-copy DNA of sufficient complexity to yield unique alignments. Eliminated sequences with retained paralogs or that contain low-copy repetitive elements are expected to show relatively lower enrichment scores. To gain further insight into elimination of repetitive DNA, we performed similar analyses targeting repetitive sequences (Supplementary Note). These analyses identify an additional 102 Mb of eliminated sequence that can be directly assigned to assembly-amenable repetitive sequences and indicate that remaining fractions of the germline-specific subgenome likely consist of arrays of short or incomplex/simple repetitive sequence that are less amenable to sequencing, mapping or assembly (Supplementary Note and Supplementary Fig. 10).
Function of PGR
It has been proposed that PGR serves to prevent the expression of genes with beneficial functions in the germline and deleterious functions in soma (such as oncogenesis and aging)8,10,12. To gain further insight into the functions of eliminated genes and the underlying evolutionary logic of PGR, we asked whether human homologs of eliminated genes are enriched for defined functional categories. In interpreting these ontology enrichment studies, it is important to recognize that these analyses define a single human or mouse ortholog for each lamprey gene. While this scenario does not accurately reflect duplication events that have structured lamprey and gnathostomes, or divergence in gene functions over more than 500 MY of independent evolution, such analyses are expected to provide some (albeit conservative) perspective on the likely function of lamprey genes. Despite this deep divergence, ontology analyses showed enrichment for several categories, including pathways related to oncogenesis, including regulation of cell division, epithelial migration, adhesion and cell fate commitment (Supplementary Table 9, Supplementary Note).
While ontology analyses provide some insight into the likely functions of eliminated genes, it is important to recognize that curated ontology databases do not capture all of the biological functions that are encoded in the genome. To gain additional insight into the functional consequences of PGR, we searched for enrichment of eliminated orthologs among 645 chromatin immunoprecipitation (ChIP) experiments (ChEA 2016)32,33 (Supplementary Table 10). To identify subcategories of enriched ChIP data sets, we performed two-way hierarchical clustering of presence–absence calls from the top 50 enriched ChIP data sets. These analyses showed two distinct categories of lamprey genes and ChIP experiments (Fig. 7). One cluster (denoted C1; Fig. 7) corresponds to the binding sites of PRC genes in mouse embryonic stem cells, apparently indicating that that these genes may be marked by bivalent promoters in embryonic stem cells (ESCs) and then presumably released from silencing in germline at later developmental stages. To test this idea, we more closely examined a cluster of genes (denoted GS3) that was highly enriched within C1 ChIP experiments. Notably, all of these genes were previously found to be marked by bivalent (poised) promoters in murine ESCs and primordial germ cells34 (bivalent in ESCs: 16/16, χ2 = 77.0, P = 8.8 × 10−19, d.f. = 1; bivalent in primordial germ cells (PGCs): 15/16, χ2 = 47.3, P = 3.1 × 10−12, d.f. = 1). A second cluster of eliminated genes (denoted GS1) also showed strong enrichment for these two functional categories (bivalent in ESCs: 14/22, χ2 = 34.6, P = 2.0 × 10−9, d.f. = 1; bivalent in PGCs: 14/22, χ2 = 23.2, P = 7.5 × 10−7, d.f. = 1).

Homologs of eliminated genes show strong overlap for the binding targets of polycomb repressive complexes in mouse ESCs and the binding sites of transcription factors in multipotent progenitor lineages and cancer cells (from ChEA 2016)32. Red cells denote ChIP experiments |(x axis) that identify peaks overlapping orthologs of lamprey genes (y axis). ChIP enrichment statistics and ordering along the x axis are provided in Supplementary Table 9. Labels GS1, GS2 and GS3 denote three primary clusters of germline-specific genes; C1 and C2 denote two primary clusters of ChIP experiments.
Other enriched ChIP experiments (C2) correspond primarily to the binding targets of transcriptional modifiers in embryonic stem cells (N = 7), embryonic progenitor lineages (N = 7) and transcriptional activators in cancer (N = 15; Fig. 7). Notably, all but one (PCDHGB5) of the genes detected in C1 are present in one or more experiments in C2. Overall, comparisons with ChIP analyses performed in non-eliminating species lends further support to the idea that PGR acts to prevent misexpression of ‘germline’ genes and suggests that misexpression of orthologous genes may directly contribute to oncogenesis in a diverse range of cancers. Moreover, these comparative analyses provide new insight into the regulatory functions of PGR by finding overlap between early gene-silencing events that are achieved by PGR and those that are mediated by the PRC during differentiation of germline and soma.
Discussion
The lamprey genome presents an interesting target for sequencing because of its phylogenetic position and unique genome biology, yet a particularly challenging target given its high repeat content and divergence from other species with highly contiguous assemblies. In an attempt to resolve this complexity, we leveraged several complementary technologies to generate a highly contiguous assembly that approaches the scale of entire chromosomes. Moreover, we were able to validate the chromosome-scale contiguity of our assembly by generating a dense meiotic map for a related species. The high contiguity of our assembly provides critical context for understanding the evolution of gene content and genome structure in vertebrates. Here we highlighted the utility of this assembly in addressing fundamental questions related to understanding changes in large-scale structure of vertebrate genomes, specifically reconstructing the deep evolutionary origins of vertebrate chromosomes and understanding how PGR mediates genetic conflicts between germline and somatic tissues.
Our improved assembly permits robust resolution of a complement of ancestral chromosomes that existed before the divergence of ancestral gnathostome and agnathan lineages and prior to whole-genome duplication(s) within the shared ancestral lineage of all extant vertebrates. These reconstructions largely validate previous analyses that were performed using meiotic mapping data, but they provide improved resolution of ancestral homology groups. Analyses also lend further support to the idea that chromosome-scale duplication events may have been more common over the course of vertebrate ancestry than has been appreciated from the analysis of bony vertebrate genomes. Parallel lines of evidence supporting a relatively recent duplication having given raise to lamprey HOX-ε- and HOX-β-bearing chromosomes further highlights the potential for large-scale duplication outside the context of whole-genome duplication. It appears that two features of lamprey biology might favor the fixation of chromosomal duplications. First, lampreys possess a large number of small chromosomes, and consequently chromosomal duplications will generally impact fewer genes than similar events in human. Duplication events (in addition to a single presumptive whole-genome duplication) appear to have affected other groups of lamprey chromosomes, though not all (Supplementary Fig. 11). Second, individuals are highly fecund (~100,000 eggs per female), and therefore a single mutant can introduce thousands of carriers (including stable carriers) into a population4,35,36,37. While it is likely that the reproductive biology and distribution of chromosome sizes has fluctuated over the course of vertebrate evolution, available evidence suggests that lampreys have possessed similar karyotypes and reproductive biologies for hundreds of millions of years. As such, extant lampreys may represent a better model for conceptualizing phases of evolution during which ancestral vertebrates were characterized by higher fecundity and larger numbers of relatively gene-poor microchromosomes, in addition to providing phylogenetic perspective on early stages of vertebrate genome evolution.
The assembly also identifies a large number of genes that are reproducibly eliminated via PGR. Enrichment analyses reveal a strong overlap in the targets of PGR-mediated elimination and the targets of silencing via PRC proteins in embryonic stem cells. The PRC is a deeply conserved complex that plays roles in gene silencing related to the maintenance of stem cell identity, silencing of oncogene expression and X-chromosome inactivation, among other functions38,39. These well-defined functions of the PRC mirror several aspects of PGR, particularly in that both act to achieve strong transcriptional silencing and both appear to target an overlapping subset of proto-oncogenes. It is interesting to speculate that the overlapping targets of PGR and the PRC may indicate that these two modes of silencing share common underlying mechanisms. However, it is notable that PRC repression is strongly associated with the deposition and binding to trimethylated lysine 27 of histone H3 (H3K27me3), whereas previous studies have shown that this mark is absent prior to the onset of PGR in lamprey embryos11. It therefore appears that PGR acts to (in part) regulate a subset of germline-expressed targets of the PRC and that it may work upstream of the PRC in lamprey embryos.
The analyses presented here address a focused set of topics that are specifically related to understanding the evolution and development of genome structure in lamprey and other vertebrates. We anticipate that this assembly will substantially improve our ability to use lamprey as a comparative evolutionary model. Because sequences are anchored to their broader chromosomal structure, the current assembly should enhance the ability to reconstruct the deep evolutionary history of the vast majority of genes within vertebrate genomes and perform robust tests of hypotheses related to historical patterns of duplication and divergence. Moreover, the availability of a highly contiguous assembly for an agnathan species should aid in the development and analysis of other genome assemblies from this highly informative vertebrate lineage.
URLs
SIMRbase/Lamprey Genome Browser, https://genomes.stowers.org/organism/Petromyzon/marinus; DifCover, https://github.com/timnat/DifCover; RepeatMasker, http://www.repeatmasker.org. Original data pertaining to the Chicago assembly (Dovetail) and HOX cluster curation can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/LIBPB-1215.
Methods
Research animals
This study complied with all relevant ethical guidelines and was performed under protocol number 2011-0848 (University of Kentucky Institutional Animal Care and Use Committee).
Sequencing
Fragment libraries were prepared by Covaris shearing of sperm genomic DNA isolated from a single individual and size selected to achieve average insert sizes of ~205 and 231 bp. These libraries were sequenced on the Illumina HiSeq2000 platform. Two separate 4-kb mate pair libraries were generated. One 4-kb library was prepared and sequenced by the Genomic Services Laboratory at HudsonAlpha (Huntsville, AL) and another was prepared and sequenced using the standard Illumina mate-pair kit. Two 4-kb libraries were prepared and sequenced by Lucigen (Middleton, WI). Long reads were prepared by the University of Florida Interdisciplinary Center for Biotechnology Research (Gainesville, FL) and sequenced using Pacific Biosciences (Menlo Park, CA) XL/C2 chemistry on a Single Molecule, Real-Time (SMRT) Sequencing platform.
Hybrid assembly
Hybrid assembly of Illumina fragment reads and Pacific Biosciences single-molecule reads was performed using the programs SparseAssembler42 and DBG2OLC15. The first 159 Gb of the high-quality paired-end reads were used to construct short but accurate de Bruijn graph contigs using the program SparseAssembler42 with k-mer size 51 and a skip length of 15. The program DBG2OLC15 was then used to map short contigs to PacBio SMRT sequencing reads and generate a hybrid assembly. Each PacBio read was compressed using high-quality short-read contigs and aligned to all other reads for structural error correction wherein chimeric PacBio reads are identified and trimmed. A read-overlap-based assembly graph was generated and unbranched linear regions of the graph were output as the initial assembly backbones. Consensus sequences for the backbones were generated by joining overlapped raw sequencing reads and short-read contigs. In practice, many regions of the initial consensus sequences can be erroneous due to the high error rates of the PacBio reads. In order to polish each backbone, all related PacBio reads and contigs are first collected and realigned using Sparc43 to calculate the most likely consensus sequence for the genome.
Scaffolding
Scaffolding of the hybrid assembly was performed using SSPACE 2.044 to incorporate mate pair data, followed by ALLMAPS version 0.5.316 to incorporate optical mapping (BioNano), linked-read (Dovetail) and previously published meiotic mapping data4. Scaffolding by SSPACE imposed a stringent scaffolding threshold requiring 5 or more consistent linkages to support scaffolding of any pair of contigs. Scaffolding via ALLMAPS was implemented with default parameters and with equal weights assigned to all three types of mapping data with initial anchoring to meiotic maps. For scaffolds without linkage mapping data, additional ALLMAPS runs were performed using the remaining data sets. Conflicts among the three mapping methods were resolved by majority rule or by manually breaking contigs that could not be placed by majority rule.
Meiotic mapping of E. tridentatus
A meiotic map was generated for E. tridentatus using a single outbred adult pair collected from Willamette Falls (Oregon City, OR, USA) and from which larvae were artificially propagated in May 2013 at the USGS Columbia River Cook Laboratory (Cook, WA, USA) and reared for 2 weeks until they were sacrificed after hatching at around ~10 mm in total length. Restriction site–associated DNA sequencing (RAD-seq45) Illumina sequencing libraries were prepared using a modified version of a previously published protocol46). A total of 250 ng of DNA from each sample was added to a 100 μL restriction digest using the Sbf1 restriction enzyme (New England Biolabs, Ipswich, MA, USA). Each sample was then tagged by ligation of one of 96 unique barcoded adapters (P1 adaptor) to the Sbf1 site. Once barcoded, the samples were mixed together into three libraries of 96 individuals per library, and approximately 4 μg of each was sheared using a Bioruptor UCD-300 instrument (Diagenode, Denville, NJ, USA). Following sonication, each library was concentrated using the Qiagen MinElute PCR purification kit (Qiagen) in preparation for size selection by agarose gel electrophoresis. Prior to sequencing RAD-seq libraries were quantified by qPCR and Illumina library quantification standards (Kappa Biosystems Inc, Woburn, MA, USA) on an ABI 7900HT Sequence Detection System (Life Technologies). Libraries were sequenced with single-end 100-bp reads on an Illumina HiSeq2000 sequencer (Illumina Inc., San Diego, CA, USA). Genotypes from 94 individuals with the greatest marker densities were used to reconstruct a consensus meiotic map from maternal and paternal meiosis. Maximum-likelihood mapping and manual curation were performed using the Join Map software package with default parameters for an outbred crossing design, except that the number of optimization rounds was increased to ten in order to better optimize the internal ordering of markers47,48.
Annotation
Identification of repetitive elements
Repeats were identified within assembled scaffolds using RepeatModeler20 and annotated using RepeatMasker version open-4.0.521 (see URLs) and a library of vertebrate repeats from repbase (repeatmaskerlibraries-20140131).
Identification of coding sequences
Genome annotations were produced using the MAKER22,49,50 genome annotation pipeline, which supports re-annotation using pre-existing gene models as input. Previous Petromyzon marinus gene models (WUGSC 7.0/petMar2 assembly)51 were mapped against the new genome assembly into GFF3 format and were used as prior model input to MAKER for re-annotation. Snap52 and Augustus53,54 were also used with MAKER and were trained using the pre-existing lamprey gene models. Additional input to MAKER included previously published mRNA-seq reads derived from lamprey embryos and testes10,12,13 and assembled using Trinity55, as well as mRNA-seq reads (NexSeq 75–100 bp paired-end) that were derived from whole embryos and dissected heads at Tahara stage 20 and dissected embryonic dorsal neural tubes at Tahara stage 18, 20 and 21. The following protein data sets were also used: Ciona intestinalis (sea squirt)56, Lottia gigantea (limpet)57, Nematostella vectensis (sea anemone)58, Takifugu rubripes (pufferfish)59, Branchiostoma floridae (lancelet)60, Callorhinchus milii (elephant shark)61, Xenopus tropicalis (western clawed frog)62, Drosophila melanogaster (fruit fly)63, Homo sapiens (human)64,65, Mus musculus (mouse)66, Danio rerio (zebrafish)67, Hydra magnipapillata68, Trichoplax adhaerens69, and the Uniprot/Swiss-Prot protein database70,71. Protein domains were identified in final gene models using the InterProScan domain identification pipeline72,73,74, and putative gene functions were assigned using BLASTP75 identified homology to the Uniprot/Swiss-Prot protein database.
lncRNA annotation
Putative lncRNAs were predicted from RNA-seq reads obtained from brain, heart, kidney, and ovary/testis sampled from two ripe adult individuals (one female, one male). In total, 8 libraries were produced using the Illumina stranded TruSeq mRNA kit (Illumina Inc.). Sequencing (single-end, directional 100 bp) was performed on a HiSeq 2000. The resulting reads were mapped to the germline genome assembly using GSNAP (v2017-04-24)76; the resulting bam files were then assembled into transcript models using StringTie (v1.3.3b)77. The following parameters were optimized in order to maximize the number of predicted lncRNAs and reduce the number of assembly artifacts: (1) minimum isoform abundance of the predicted transcripts as a fraction of the most abundant transcript assembled at a given locus: lower-abundance transcripts are often artifacts of incompletely spliced precursor of processed transcripts; (2) minimum read coverage allowed for the predicted transcripts; (3) minimum locus gap separation value: reads that are mapped closer than 10 bp distance are merged together in the same processing bundle; (4) smallest anchor length: junctions that do not have spliced reads that align across them with at least 10 bases on both sides are filtered out; (5) minimum length allowed for the predicted transcripts (200 bp); (6) minimum number of spliced reads that align across a junction (i.e. junction coverage); (7) removal of monoexonic transcripts. The resulting transcriptomes from each library were then merged into a single GTF file (–merge option in StringTie).
Transcripts overlapping (in sense) exons of the protein coding annotated genes were removed using the script FEELnc_filter.pl78. The filtered gene models file was then used to compute the Coding Potential Score (CPS) for each of the candidate non-coding transcript with the script FEELnc_codpot.pl78. In the absence of a species-specific lncRNA set, as is the case for P. marinus, the implemented machine-learning strategy requires to simulate non-coding RNA sequences to train the model by shuffling the set of mRNAs while preserving their 7-mer frequencies. This approach is based on the hypothesis that at least some lncRNAs are derived from “debris” of protein-coding genes79. The simulated data were then used to calculate the CPS cutoff separating coding (mRNAs) from non-coding (lncRNAs) using 10-fold cross-validation on the input training files in order to extract the CPS that maximizes both sensitivity and specificity.
Analysis of conserved synteny
Analyses of conserved synteny were performed as previously described4. Briefly, predicted protein sequences from the lamprey genome were aligned to proteins from the Gar (LepOcu1: GCA_000242695.1) and Chicken (Galgal4: GCA_000002315.2) genome assemblies80. All alignments with bitscore ≥100 and ≥90% of the best match (within a species) were considered putative orthologs of each lamprey, chicken or gar gene. Groups of orthologs were filtered to remove those with more than 6 members in any given species. Enrichment of orthologs on chromosomes or chromosomal segments was assessed using χ2 tests, incorporating Yates’ correction for continuity and Bonferroni corrections for multiple testing as previously described4.
Identification and characterization of germline-specific/enriched sequences
Single-copy genes
To identify germline-specific regions, we separately aligned paired-end reads from blood and sperm DNA to the germline genome assembly using BWA-MEM (v.0.7.10)81 with default parameters and filtered to exclude unmapped reads and supplementary alignments (samtools v.1.2 with option: view -F2308)82. Initial coverage analyses was implemented using bedtools v2.23.083 and revealed that the modal coverage of reads from sperm DNA was slightly lower than the coverage of reads from blood, ~73× and ~80×, respectively, but contained a larger amount of low-copy DNA (Supplementary Fig. 12). To identify germline-enriched intervals, data were filtered to remove regions with coverage both from sperm and blood of <10 (underrepresented regions: computed with genomecov -bga, bedtools v2.23.0) and also regions with coverage exceeding three times the modal value in sperm or blood (high-copy regions). The remaining data were processed to generate coverage ratios for discreet intervals containing 1,000 bp (or >500 bp at contig ends) of approximately single-copy sequence. Identification of contiguous intervals and re-estimation of coverage ratios was performed using DNAcopy version 1.42.084 after removing trailing windows that were <500 bp in length. Ontology analyses used naming assignments that were generated using multispecies BLAST alignments via MAKER22,49,50 and were performed using Enrichr33.
Repetitive sequences
High-identity repetitive elements were assembled de novo from k-mers (k = 31) that were abundant in sperm and blood reads, with k-mer counting via Jellyfish version 2.2.485 and assembly using Velvet version 1.2.1086. Copy-number thresholds for abundant k-mers set at 3× modal copy numbers for 31-mers: 165 for sperm and 180 for blood. Abundant k-mers from sperm and blood were combined and used as a single-end reads for Velvet running with 29-mers. These analyses resulted in a de novo repeat library with 130,632 sequences (overall length ~11 Mb with individual contigs lengths range from 57 bases to 15.5 kb). These repeats were annotated using RepeatMasker version open-4.0.521 (see URLs) and repeat libraries generated for the germline assembly and from Repbase (repeatmaskerlibraries-20140131: “vertebrate repeats”).
For downstream analyses we used a set of model repeats representing the union of de novo repeats, those identified within assembled genomic sequences via RepeatModeler20 and an updated assembly of the previously identified Germ1 element8. Enrichment analyses were performed by separately aligning paired-end reads from blood and sperm DNA to the repeat data set. As with single-copy sequence, alignments were pre-filtered to exclude unmapped reads and supplementary alignments. The remaining data were processed to generate average coverage ratios for intervals of ~100 bp.
Manual curation of HOX clusters
Manual curation of gene models was carried out using Apollo87 implemented in JBrowse88. Indels in the assembly were identified and corrected by comparison with RNA-seq and genomic DNA re-sequencing data. Gene predictions from Maker were refined based on whole-embryo RNA-seq data from multiple developmental stages and homology with gene sequences from other vertebrates.
In addition to the 42 clustered HOX genes in the genome assembly, 6 further HOX genes were predicted that did not fall within the 6 HOX clusters. To investigate these genes further, the genomic scaffolds harboring these gene loci were extracted and used as queries for alignment against the assembly by BLAST89. Five of these gene loci (homologs of hoxA3, D8, C9, B13 and B13a) were found to align with high sequence similarity (>97% identity) across long stretches of their sequence (>4 kb, containing predicted HOX coding sequence and flanking, non-coding sequence) to loci of individual members of the 42 clustered lamprey HOX genes (Supplementary Table 13). These loci either could represent recent duplications of HOX loci or could be assembly artifacts arising from the relatively high heterozygosity of the lamprey genome. Based on their exceptionally high levels of coding and non-coding sequence similarity to clustered HOX loci, we infer that these 5 loci are assembly artifacts due to polymorphism and that they do not represent additional singleton HOX genes in the lamprey genome. The 6th predicted singleton HOX gene shows equal levels of homology to ANTP-class homeobox genes of both HOX and non-HOX families, suggesting it is a derived ANTP-class homeobox gene and not necessarily a HOX gene.
Phylogenetic analysis of HOX genes
Phylogenetic analysis was performed on HOX paralog groups with 4 or more members in sea lamprey: groups 4, 8, 9, 11 and 13. For each paralog group, predicted sea lamprey HOX protein sequences were aligned against homologs from other vertebrate species and amphioxus, retrieved from GenBank. Our approach was informed by the experiences detailed by Kuraku et al.90, Qiu et al.91, Mehta et al.17 and Manousaki et al.92. In selecting jawed vertebrate taxa for these analyses, we avoided teleost fish and Xenopus laevis as these lineages have undergone additional genome duplication events, which can lead to their co-orthologous genes/proteins being more derived than those from non-duplicated lineages. Thus, we opted for elephant shark (C. milii) and coelacanth (L. menadoensis) as Chondrichthian and ‘basal’ Sarcopterygian representatives, respectively; both have slowly evolving protein-coding genes and well characterized HOX gene complements93,94. Urochordates are the sister group of vertebrates, but the divergent nature of their HOX genes led us to favor the cephalochordate amphioxus as a source for outgroup sequences in our analyses. We chose to perform protein alignments rather than DNA alignments due to the high coding GC content in lamprey, which can result in artifactual clustering of lamprey genes in DNA trees. Nevertheless, the unique pattern of amino-acid composition in lamprey proteins is an unavoidable complicating factor that impinges on their phylogenetic analysis and can lead to artifactual clustering of lamprey proteins, as described in Qiu et al.91. The MEGA741 software suite was used for sequence alignment, best-fit substitution model evaluation and phylogeny reconstruction. Protein alignments were performed with full available length protein sequences using MUSCLE41. Best-fit substitution models were evaluated and chosen for each alignment. Maximum likelihood, neighbor joining and maximum parsimony approaches were used for phylogenetic analysis, with 100 bootstrap replicates generated for node support. For each method, all positions in the alignment containing gaps and missing data were eliminated.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Code availability
Custom code (DifCover) is available on GitHub (see URLs).
Data availability
Accession numbers
Genome Assembly: NCBI Genome PIZI00000000.1; BioProject PRJNA357048. Raw sequence data used for genome assembly: NCBI SRA SRR5503831–SRR5503843. Re-sequencing data for detection of eliminated segments: NCBI SRA SRR5535434–SRR5535435. Previously published RNA-seq data were used for annotation: NCBI SRA SRX110029.2–SRX110035.2 (ref. 13), NCBI SRA SRX1483277–SRX1483282 (ref. 12) and NCBI SRA SRX104180 (ref. 10). Source data is available online for Figs. 3, 4, 6 and 7.
Change history
17 September 2018
When published, this article did not initially appear open access. This error has been corrected, and the open access status of the paper is noted in all versions of the paper. Additionally, affiliation 16 denoting equal contribution was missing from author Robb Krumlauf in the PDF originally published. This error has also been corrected.
19 April 2018
In the version of this article initially published, the present addresses for authors Dorit Hockman and Chris Amemiya were switched. The error has been corrected in the HTML and PDF versions of the article.
References
Parker, H. J., Bronner, M. E. & Krumlauf, R. A Hox regulatory network of hindbrain segmentation is conserved to the base of vertebrates. Nature 514, 490–493 (2014).
Green, S. A., Simoes-Costa, M. & Bronner, M. E. Evolution of vertebrates as viewed from the crest. Nature 520, 474–482 (2015).
Sower, S. A. et al. Emergence of an ancestral glycoprotein hormone in the pituitary of the sea lamprey, a basal vertebrate. Endocrinology 156, 3026–3037 (2015).
Smith, J. J. & Keinath, M. C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 25, 1081–1090 (2015).
Das, S. et al. Evolution of two prototypic T cell lineages. Cell. Immunol. 296, 87–94 (2015).
Doolittle, R. F. Bioinformatic characterization of genes and proteins involved in blood clotting in lampreys. J. Mol. Evol. 81, 121–130 (2015).
McCauley, D. W., Docker, M. F., Whyard, S. & Li, W. Lampreys as diverse model organisms in the genomics era. Bioscience 65, 1046–1056 (2015).
Smith, J. J., Antonacci, F., Eichler, E. E. & Amemiya, C. T. Programmed loss of millions of base pairs from a vertebrate genome. Proc. Natl. Acad. Sci. USA 106, 11212–11217 (2009).
Smith, J. J., Stuart, A. B., Sauka-Spengler, T., Clifton, S. W. & Amemiya, C. T. Development and analysis of a germline BAC resource for the sea lamprey, a vertebrate that undergoes substantial chromatin diminution. Chromosoma 119, 381–389 (2010).
Smith, J. J., Baker, C., Eichler, E. E. & Amemiya, C. T. Genetic consequences of programmed genome rearrangement. Curr. Biol. 22, 1524–1529 (2012).
Timoshevskiy, V. A., Herdy, J. R., Keinath, M. C. & Smith, J. J. Cellular and molecular features of developmentally programmed genome rearrangement in a vertebrate (sea lamprey: Petromyzon marinus). PLoS Genet. 12, e1006103 (2016).
Bryant, S. A., Herdy, J. R., Amemiya, C. T. & Smith, J. J. characterization of somatically-eliminated genes during development: lamprey (Petromyzon marinus). Mol. Biol. Evol. 33, 2337–2344 (2016).
Smith, J. J. et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 45, 415–421 (2013).
Speir, M. L. et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 44 D1, D717–D725 (2016).
Ye, C., Hill, C. M., Wu, S., Ruan, J. & Ma, Z. S. DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 6, 31900 (2016).
Tang, H. et al. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 16, 3 (2015).
Mehta, T. K. et al. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc. Natl. Acad. Sci. USA 110, 16044–16049 (2013).
Kuraku, S. & Kuratani, S. Time scale for cyclostome evolution inferred with a phylogenetic diagnosis of hagfish and lamprey cDNA sequences. Zool. Sci. 23, 1053–1064 (2006).
Lampman, R. et al. in Jawless Fishes of the World Vol. 2 (eds. A. Orlov & R. Beamish) Ch. 22, 160–195 (Cambridge Scholars Publishing, Newcastle-upon-Tyne, UK, 2016).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0 (2015).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0 (2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Hara, Y. et al. Optimizing and benchmarking de novo transcriptome sequencing: from library preparation to assembly evaluation. BMC Genomics 16, 977 (2015).
Warren, W. C. et al. A new chicken genome assembly provides insight into avian genome structure. G3 (Bethesda) 7, 109–117 (2017).
Braasch, I. et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 48, 427–437 (2016).
Ohno, S. Evolution by Gene Duplication (Springer-Verlag, New York, 1970).
Holland, P. W., Garcia-Fernàndez, J., Williams, N. A. & Sidow, A. Gene duplications and the origins of vertebrate development. Dev. Suppl. 1994, 125–133 (1994).
Nakatani, Y., Takeda, H., Kohara, Y. & Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 17, 1254–1265 (2007).
Murat, F., Van de Peer, Y. & Salse, J. Decoding plant and animal genome plasticity from differential paleo-evolutionary patterns and processes. Genome Biol. Evol. 4, 917–928 (2012).
Timoshevskiy, V. A., Lampman, R. T., Hess, J. E., Porter, L. L. & Smith, J. J. Deep ancestry of programmed genome rearrangement in lampreys. Dev. Biol. 429, 31–34 (2017).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013).
Sachs, M. et al. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Reports 3, 1777–1784 (2013).
Moore, C. M. & Best, R. G. Chromosomal Genetic Disease: Structural Aberrations (eLS, Chichester, UK, 2001).
Hardisty, M. W. Fecundity and speciation in lampreys. Evolution 17, 17–22 (1963).
Hardisty, M. W. & Cosh, J. Primordial germ cells and fecundity. Nature 210, 1370–1371 (1966).
Grossniklaus, U. & Paro, R. Transcriptional silencing by polycomb-group proteins. Cold Spring Harb. Perspect. Biol. 6, a019331 (2014).
Aloia, L., Di Stefano, B. & Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 140, 2525–2534 (2013).
Acemel, R. D. et al. A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat. Genet. 48, 336–341 (2016).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Ye, C., Ma, Z. S., Cannon, C. H., Pop, M. & Yu, D. W. Exploiting sparseness in de novo genome assembly. BMC Bioinformatics 13 (Suppl. 6), S1 (2012).
Ye, C. & Ma, Z. S. Sparc: a sparsity-based consensus algorithm for long erroneous sequencing reads. PeerJ 4, e2016 (2016).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A. & Johnson, E. A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248 (2007).
Miller, M. R. et al. A conserved haplotype controls parallel adaptation in geographically distant salmonid populations. Mol. Ecol. 21, 237–249 (2012).
Stam, P. Construction of integrated genetic-linkage maps by means of a new computer package: Join Map. Plant J. 3, 739–744 (1993).
Van Ooijen, J. W. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 93, 343–349 (2011).
Campbell, M. S. et al. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524 (2014).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Hwang, J. Y., Smith, S. & Myung, K. The Rad1-Rad10 complex promotes the production of gross chromosomal rearrangements from spontaneous DNA damage in Saccharomyces cerevisiae. Genetics 169, 1927–1937 (2005).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 (Suppl. 2), ii215–ii225 (2003).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Dehal, P. et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298, 2157–2167 (2002).
Simakov, O. et al. Insights into bilaterian evolution from three spiralian genomes. Nature 493, 526–531 (2013).
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007).
Aparicio, S. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297, 1301–1310 (2002).
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071 (2008).
Venkatesh, B. et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 505, 174–179 (2014).
Hellsten, U. et al. The genome of the Western clawed frog Xenopus tropicalis. Science 328, 633–636 (2010).
Adams, M. D. et al. The genome sequence of Drosophila melanogaster. Science 287, 2185–2195 (2000).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Waterston, R. H. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002).
Howe, K. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 (2013).
Chapman, J. A. et al. The dynamic genome of Hydra. Nature 464, 592–596 (2010).
Srivastava, M. et al. The Trichoplax genome and the nature of placozoans. Nature 454, 955–960 (2008).
The UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 39, D214–D219 (2011).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Mulder, N. J. et al. InterPro: an integrated documentation resource for protein families, domains and functional sites. Brief. Bioinform. 3, 225–235 (2002).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Wucher, V. et al. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 45, e57 (2017).
Duret, L., Chureau, C., Samain, S., Weissenbach, J. & Avner, P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312, 1653–1655 (2006).
Yates, A. et al. Ensembl 2016. Nucleic Acids Res. 44 D1, D710–D716 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Venkatraman, E. S. & Olshen, A. B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23, 657–663 (2007).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Lee, E. et al. Web Apollo: a web-based genomic annotation editing platform. Genome Biol. 14, R93 (2013).
Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J. & Holmes, I. H. JBrowse: a next-generation genome browser. Genome Res. 19, 1630–1638 (2009).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Kuraku, S., Meyer, A. & Kuratani, S. Timing of genome duplications relative to the origin of the vertebrates: did cyclostomes diverge before or after? Mol. Biol. Evol. 26, 47–59 (2009).
Qiu, H., Hildebrand, F., Kuraku, S. & Meyer, A. Unresolved orthology and peculiar coding sequence properties of lamprey genes: the KCNA gene family as test case. BMC Genomics 12, 325 (2011).
Manousaki, T. et al. in Jawless Fishes of the World Vol. 1 (eds. Orlov, A. & Beamish, R.) 2–16 (Cambridge Scholars Publishing, Newcastle-upon-Tyne, UK, 2016).
Ravi, V. et al. Elephant shark (Callorhinchus milii) provides insights into the evolution of Hox gene clusters in gnathostomes. Proc. Natl. Acad. Sci. USA 106, 16327–16332 (2009).
Amemiya, C. T. et al. Complete HOX cluster characterization of the coelacanth provides further evidence for slow evolution of its genome. Proc. Natl. Acad. Sci. USA 107, 3622–3627 (2010).
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the US National Institutes of Health under award number R01GM104123 to J.J.S., the Stowers Institute under award number SIMR-1001 to H.J.P., M.E.C., L.M.W., S.M.C.R. and R.K., and the Bonneville Power Administration to J.E.H. and S.R.N. E.E.E. is an investigator of the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged. Additional computational support was provided by The University of Kentucky High Performance Computing complex.
Author information
Authors and Affiliations
Contributions
J.J.S., R.K., C.T.A. and G.E. conceived of the study. J.J.S., N.T., C.Y., C.H., M.C.K., H.J.P., M.E.C., J.E.H., S.R.N., V.A.T., C.K.M.W., C.S., H.K., F.L., L.M.W., S.M.C.R., C.B., E.E.E., D.H., T.S.-S., M.Y. and R.K. contributed analyses. J.J.S., N.T., M.C.K., H.J.P. and R.K. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 lncRNA subclasses identified in the sea lamprey genome.
a) Categories of annotated intergenic lncRNAs based on their location and transcriptional direction, relative to their closest neighboring gene. b) Categories of annotated genic lncRNAs based on their location and transcriptional direction, relative an overlapping gene. c) Counts of genic and intergenic lncRNAs. d) Counts for subcategories of intergenic lncRNAs segregated on the basis of location and direction of transcription. e) Counts for subcategories of genic lncRNAs segregated on the basis of exon/intron overlaps. f) Counts for subcategories of genic lncRNAs segregated on the basis of location and direction of transcription relative to their overlapping gene.
Supplementary Figure 2 The distribution of conserved syntenies in lamprey and gar reveals patterns of ancient large-scale duplication.
These patterns are consistent with those from chicken/lamprey comparisons and reveal both chromosomal/segmental and whole genome duplications. Lamprey super-scaffolds are oriented along the y-axis and gar chromosomes (linkage groups) are oriented along the x-axis. Circles reflect counts of syntenic orthologs on the corresponding lamprey and gar chromosomes, with the size of each circle being proportional to the number of orthologs on that pair. The color of each circle represents the degree to which the number of observed orthologs deviates from null expectations under a uniform distribution across an identical number of lamprey and gar chromosomes with identical numbers of orthology-informative genes. Shaded regions of the plot designate homology groups that correspond to presumptive ancestral chromosomes and mark sets of lamprey chromosomes that are identical to those depicted in Fig. 4 in order to facilitate direct comparison. The ordering of lamprey super-scaffolds along the y-axis is provided in Supplementary Table 4. Altering the ordering of gar chromosomes or lamprey chromosomes within orthology groups or the relative ordering of orthology groups would not change the interpretation of patterns depicted herein.
Supplementary Figure 3 Hox gene loci in the sea lamprey showing the order of assembled scaffolds and gaps.
Hox gene exons and non-Hox flanking genes are represented by boxes. Loci are not drawn to scale. The six Hox clusters fall within separate super-scaffolds with the constituent scaffolds and gaps indicated as lines beneath each cluster. Positions of known micro-RNAs are shown. All Hox genes are transcribed from left to right unless indicated (black arrows). The assembled Hox-ε cluster contains a scaffold arrangement that results in a non-canonical order of Hox genes, with Hox-ε13 and Hox-ε14 exon2-3 being positioned outside of the cluster with respect to evx. Hox-ε14 exon2-3 displays an opposite direction of transcription to the majority of the genes in the cluster, including its first exon. Given that homologues of Hox-ε11, 13 and 14 show a canonical arrangement in the closely related L. camtschaticum Hox-ε cluster, which is based on contiguous BAC sequences, we tentatively infer that these scaffolds have been mis-ordered in the P. marinus assembly and their true positions are between Hox-ε14exon1 and Hox-ε11 as indicated (red arrows). In the current assembly, a homeodomain-containing fragment of a PG8 Hox gene (8e2), with very high similarity to Hox-ε8, falls within the gap-spanning intron of Hox-ε7 and has an opposite direction of transcription to that of the majority of the genes in the cluster. We tentatively infer this to be an assembly error.
Supplementary Figure 4 A comparison between assembled Hox loci of the sea lamprey and the Japanese lamprey.
Orthologous sea lamprey (P.m.) and Japanese lamprey (L.c.) Hox loci are shown side-by-side. Hox genes are represented as boxes. Flanking non-Hox genes are depicted as arrowheads, which indicate their direction of transcription. The sea lamprey assembly supports the ε and ζ clusters that were tentatively predicted in the Japanese lamprey assembly, reveals an additional Hox gene (Hox-ζ1), an additional micro-RNA (miR-196ε), and expands these loci to reveal the genes flanking the ε and ζ clusters. Hox-containing loci homologous to L.c. Hox-η and Hox-θ were not identifiable in the sea lamprey assembly. The white arrow downstream of the lamprey hox-γ cluster represents PMZ_0048273, an uncharacterized non-Hox gene.
Supplementary Figure 5 Phylogenetic analysis of sea lamprey Hox4 genes.
Six predicted full available length sea lamprey Hox4 protein sequences were aligned with selected vertebrate Hox4 homologues, with amphioxus Hox4 as an outgroup. Trees generated by Maximum Likelihood (ML), Maximum Parsimony (MP) and Neighbor Joining (NJ) approaches are shown, based on 190 positions in the alignment. Support for each node is shown as a percentage from 100 bootstrap replicates. ML and NJ trees are drawn to scale, with the unit for evolutionary distance being the number of amino acid substitutions per site. Lamprey genes are highlighted (blue shading). Pm, Petromyzon marinus (Sea lamprey); Lc, Lethenteron camtschaticum (Japanese lamprey); Hs, Homo sapiens (Human); Cm, Callorhinchus milii (Elephant shark); Lm, Latimeria menadoensis (Coelocanth); Gg, Gallus gallus (Chicken); Bf, Branchiostoma floridae (Amphioxus).
Supplementary Figure 6 Phylogenetic analysis of sea lamprey Hox8 genes
Four predicted full available length sea lamprey Hox8 protein sequences were aligned with selected vertebrate Hox8 homologues, with amphioxus Hox8 as an outgroup. Trees generated by Maximum Likelihood (ML), Maximum Parsimony (MP) and Neighbor Joining (NJ) approaches are shown, based on 124 positions in the alignment. Support for each node is shown as a percentage from 100 bootstrap replicates. ML and NJ trees are drawn to scale, with the unit for evolutionary distance being the number of amino acid substitutions per site. Lamprey genes are highlighted (blue shading). Pm, Petromyzon marinus (Sea lamprey); Lc, Lethenteron camtschaticum (Japanese lamprey); Hs, Homo sapiens (Human); Cm, Callorhinchus milii (Elephant shark); Lm, Latimeria menadoensis (Coelocanth); Gg, Gallus gallus (Chicken); Bf, Branchiostoma floridae (Amphioxus).
Supplementary Figure 7 Phylogenetic analysis of sea lamprey Hox9 genes
Five predicted full available length sea lamprey Hox9 protein sequences were aligned with selected vertebrate Hox9 homologues, with amphioxus Hox9 as an outgroup. Trees generated by Maximum Likelihood (ML), Maximum Parsimony (MP) and Neighbor Joining (NJ) approaches are shown, based on 74 positions in the alignment. Support for each node is shown as a percentage from 100 bootstrap replicates. ML and NJ trees are drawn to scale, with the unit for evolutionary distance being the number of amino acid substitutions per site. Lamprey genes are highlighted (blue shading). Pm, Petromyzon marinus (Sea lamprey); Lc, Lethenteron camtschaticum (Japanese lamprey); Hs, Homo sapiens (Human); Cm, Callorhinchus milii (Elephant shark); Lm, Latimeria menadoensis (Coelocanth); Gg, Gallus gallus (Chicken); Bf, Branchiostoma floridae (Amphioxus).
Supplementary Figure 8 Phylogenetic analysis of sea lamprey Hox11 genes
Four predicted full available length sea lamprey Hox11 protein sequences were aligned with selected vertebrate Hox11 homologues. Amphioxus Hox11 was not used as an outgroup due to its relatively short sequence impinging on the alignment. Hox-β11 from the Japanese lamprey was omitted from the alignment as full-length sequence is not available. Trees generated by Maximum Likelihood (ML), Maximum Parsimony (MP) and Neighbor Joining (NJ) approaches are shown, based on 229 positions in the alignment. Support for each node is shown as a percentage from 100 bootstrap replicates. ML and NJ trees are drawn to scale, with the unit for evolutionary distance being the number of amino acid substitutions per site. Lamprey proteins are highlighted (blue shading). Pm, Petromyzon marinus (Sea lamprey); Lc, Lethenteron camtschaticum (Japanese lamprey); Hs, Homo sapiens (Human); Cm, Callorhinchus milii (Elephant shark); Lm, Latimeria menadoensis (Coelocanth); Gg, Gallus gallus (Chicken).
Supplementary Figure 9 Phylogenetic analysis of sea lamprey Hox13 genes.
Five predicted full available length sea lamprey Hox13 protein sequences were aligned with selected vertebrate Hox13 homologues, with amphioxus Hox13 as an outgroup. Trees generated by Maximum Likelihood (ML), Maximum Parsimony (MP) and Neighbor Joining (NJ) approaches are shown, based on 74 positions in the alignment. Support for each node is shown as a percentage from 100 bootstrap replicates. ML and NJ trees are drawn to scale, with the unit for evolutionary distance being the number of amino acid substitutions per site. Lamprey proteins are highlighted (blue shading). Pm, Petromyzon marinus (Sea lamprey); Lc, Lethenteron camtschaticum (Japanese lamprey); Hs, Homo sapiens (Human); Cm, Callorhinchus milii (Elephant shark); Lm, Latimeria menadoensis (Coelocanth); Gg, Gallus gallus (Chicken); Bf, Branchiostoma floridae (Amphioxus).
Supplementary Figure 10 Diversity and abundance of eliminated repeats.
Assembly of repeats and comparative sequencing reveals germline enrichment of several high-copy sequences. The distribution of coverage ratios reveals a long tail corresponding to repetitive sequences with higher sequence coverage in sperm relative to blood. This excess is highlighted in red, assuming a symmetrical distribution of enrichment scores for non-eliminated regions and an absence of somatic-specific sequence. B) Competitive genomic hybridization of labeled CoT2 repetitive DNA from soma (liver: green) and germline (sperm: red) to elimination anaphases. Germline-enriched repeats are nearly absent from somatically retained chromatin (migrating toward the spindle poles) and highly enriched in lagging chromatin that is slated for elimination. DNA is counterstained with DAPI (blue). A similar pattern was observed on 22 additional anaphases across two slides.
Supplementary Figure 11 The relative locations of duplicated genes on lamprey chromosomes derived from four distinct orthology groups.
The locations of duplicate genes on lamprey super-scaffolds are shown for a subset of ancestral linkage orthology groups. a) Lamprey duplications on super-scaffolds corresponding to an ancestral orthology group that encompasses chicken chromosomes 2, 7, 27 and 29. b) Lamprey duplications on super-scaffolds corresponding to an ancestral orthology group that encompasses chicken chromosomes 14 and 18. c) Lamprey duplications on super-scaffolds corresponding to an ancestral orthology group that encompasses chicken chromosomes 17 and 8. Lamprey super-scaffolds 105 and 107 are not shown, these contain no duplicates on any of the other lamprey super-scaffolds that were assigned to this orthology group. d) Lamprey duplications on super-scaffolds corresponding to an ancestral orthology group that encompasses chicken chromosomes 15 and 19. Two-copy duplicates (referenced in Fig. 5 and Supplemental Table 6) are shown in bold. Higher-copy relationships are shown with thinner lines.
Supplementary Figure 12 Distribution of read depths from germline and somatic re-sequencing data
Sequence reads from sperm and blood were mapped separately to the germline genome assembly. Values represent the number of base pairs at a given depth of coverage. These distributions reveal that the modal coverage of reads from sperm DNA was slightly lower than the coverage of reads from blood, ~73X and ~80X, respectively, and sperm sequences map to a larger proportion of the approximately single copy sequence within the germline genome.
Supplementary information
Supplementary Figures, Text and table
Supplementary Figures 1–12, Supplementary Note and Supplementary Table 13.
Supplementary Table 1
The Pacific lamprey (Entosphenus tridentatus) meiotic map and alignment of RADseq markers to sea lamprey (Petromyzon marinus) super-scaffolds.
Supplementary Table 2
Noncoding RNAs detected in the germline genome assembly.
Supplementary Table 3
A sea lamprey/chicken comparative map.
Supplementary Table 4
Ordering of lamprey super-scaffolds along the y-axis of Fig. 4 (ordered from bottom to top).
Supplementary Table 5
A sea lamprey/gar comparative map.
Supplementary Table 6
Statistics for all pairwise comparisons of numbers of shared twocopy paralogs on all lamprey derivatives of the ancestral hoxbearing chromosome. Probability estimates were generated using two-tailed tests and a total of n = 200 independent pairs of duplicated genes.
Supplementary Table 7
Depth of coverage ratios for all genomic intervals.
Supplementary Table 8
Primers used for PCR validation of candidate PGR targets and assay results.
Supplementary Table 9
Enrichment statistics for overlap between PGR-orthologs and curated "biological process" ontology terms. These comparisons reflect enrichment statistics derived from n = 102 independent lamprey/human orthologs. Statistics are as described in reference 25. P-value is calculated using the Fisher’s exact test. Adjusted P-value is calculated using the Benjamini-Hochberg method for correction for multiple hypothesis testing. Z-score is calculated by comparing the observed ranking of Fisher’s exact tests relative to those derived from random sets of genes. Combined score is the product of the log P-value and the Z-score.
Supplementary Table 10
Enrichment statistics for overlap between PGR-orthologs and published chromatin immunoprecipitation (ChIP) experiments (ChEA 2016). These comparisons reflect enrichment statistics derived from n = 102 independent lamprey/human orthologs. Statistics are as described in reference 25. P-value is calculated using the Fisher’s exact test. Adjusted P-value is calculated using the Benjamini-Hochberg method for correction for multiple hypothesis testing. Z-score is calculated by comparing the observed ranking of Fisher’s exact tests relative to those derived from random sets of genes. Combined score is the product of the log P-value and the Z-score.
Supplementary Table 11
Copy number estimates and depth of coverage ratios for intervals within all characterized repetitive elements.
Supplementary Table 12
Repeat classification for all repetitive elements with matches in RepBase. The best match for each element is shown here. Elements with no match to RepBase or RepeatModeler repeats are not shown here.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smith, J.J., Timoshevskaya, N., Ye, C. et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat Genet 50, 270–277 (2018). https://doi.org/10.1038/s41588-017-0036-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-017-0036-1
This article is cited by
-
Genome sequencing and analysis of black flounder (Paralichthys orbignyanus) reveals new insights into Pleuronectiformes genomic size and structure
BMC Genomics (2024)
-
Diversity and evolution of the vertebrate chemoreceptor gene repertoire
Nature Communications (2024)
-
The hagfish genome and the evolution of vertebrates
Nature (2024)
-
Sea lamprey enlightens the origin of the coupling of retinoic acid signaling to vertebrate hindbrain segmentation
Nature Communications (2024)
-
Shared features of blastula and neural crest stem cells evolved at the base of vertebrates
Nature Ecology & Evolution (2024)
