
Abstract
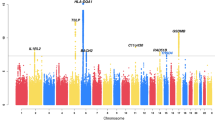
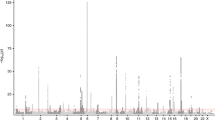
We examined common variation in asthma risk by conducting a meta-analysis of worldwide asthma genome-wide association studies (23,948 asthma cases, 118,538 controls) of individuals from ethnically diverse populations. We identified five new asthma loci, found two new associations at two known asthma loci, established asthma associations at two loci previously implicated in the comorbidity of asthma plus hay fever, and confirmed nine known loci. Investigation of pleiotropy showed large overlaps in genetic variants with autoimmune and inflammatory diseases. The enrichment in enhancer marks at asthma risk loci, especially in immune cells, suggested a major role of these loci in the regulation of immunologically related mechanisms.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Akinbami, L. J. et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. (U.S. Department of Health and Human Services, Washington, DC, 2012; 1–8. (NCHS Data Brief no. 94).
Duffy, D. L., Martin, N. G., Battistutta, D., Hopper, J. L. & Mathews, J. D. Genetics of asthma and hay fever in Australian twins. Am. Rev. Respir. Dis. 142, 1351–1358 (1990).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
Igartua, C. et al. Ethnic-specific associations of rare and low-frequency DNA sequence variants with asthma. Nat. Commun. 6, 5965 (2015).
Bouzigon, E. et al. Effect of 17q21 variants and smoking exposure in early-onset asthma. N. Engl. J. Med. 359, 1985–1994 (2008).
Galanter, J. M. et al. Genome-wide association study and admixture mapping identify different asthma-associated loci in Latinos: the Genes-environments & Admixture in Latino Americans study. J. Allergy Clin. Immunol. 134, 295–305 (2014).
Hirota, T. et al. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat. Genet. 43, 893–896 (2011).
Ferreira, M. A. et al. Genome-wide association analysis identifies 11 risk variants associated with the asthma with hay fever phenotype. J. Allergy Clin. Immunol. 133, 1564–1571 (2014).
Higgins, J. P. & Thompson, S. G. Quantifying heterogeneity in a meta-analysis. Stat. Med. 21, 1539–1558 (2002).
Grundberg, E. et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 44, 1084–1089 (2012).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Liang, L. et al. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res. 23, 716–726 (2013).
Hao, K. et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 8, e1003029 (2012).
Noguchi, E. et al. Genome-wide association study identifies HLA-DP as a susceptibility gene for pediatric asthma in Asian populations. PLoS Genet. 7, e1002170 (2011).
Ding, L. et al. Rank-based genome-wide analysis reveals the association of ryanodine receptor-2 gene variants with childhood asthma among human populations. Hum. Genomics 7, 16 (2013).
Sleiman, P. M. et al. Variants of DENND1B associated with asthma in children. N. Engl. J. Med. 362, 36–44 (2010).
Himes, B. E. et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am. J. Hum. Genet. 84, 581–593 (2009).
Torgerson, D. G. et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 43, 887–892 (2011).
Bønnelykke, K. et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 46, 51–55 (2014).
Ferreira, M. A. et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet 378, 1006–1014 (2011).
Moffatt, M. F. et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 363, 1211–1221 (2010).
Zeller, T. et al. Genetics and beyond: the transcriptome of human monocytes and disease susceptibility. PLoS One 5, e10693 (2010).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Raychaudhuri, S. et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 5, e1000534 (2009).
Hong, S. W., Kim, S. & Lee, D. K. The role of Bach2 in nucleic acid-triggered antiviral innate immune responses. Biochem. Biophys. Res. Commun. 365, 426–432 (2008).
Yang, M., He, R. L., Benovic, J. L. & Ye, R. D. Beta-Arrestin1 interacts with the G-protein subunits β1γ2 and promotes β1γ2-dependent Akt signalling for NF-kappaB activation. Biochem. J 417, 287–296 (2009).
Soler Artigas, M. et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet. 43, 1082–1090 (2011).
Goenka, S. & Kaplan, M. H. Transcriptional regulation by STAT6. Immunol. Res. 50, 87–96 (2011).
Qian, X., Gao, Y., Ye, X. & Lu, M. Association of STAT6 variants with asthma risk: a systematic review and meta-analysis. Hum. Immunol. 75, 847–853 (2014).
Wang, Y., Tong, X. & Ye, X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 189, 5304–5313 (2012).
Venuprasad, K., Zeng, M., Baughan, S. L. & Massoumi, R. Multifaceted role of the ubiquitin ligase Itch in immune regulation. Immunol. Cell Biol. 93, 452–460 (2015).
Javierre, B. M. et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369–1384 e19 (2016).
Barnes, P. J. Pathophysiology of allergic inflammation. Immunol. Rev. 242, 31–50 (2011).
Vicente, C. T. et al. Long-range modulation of PAG1 expression by 8q21 allergy risk variants. Am. J. Hum. Genet. 97, 329–336 (2015).
Davison, L. J. et al. Long-range DNA looping and gene expression analyses identify DEXI as an autoimmune disease candidate gene. Hum. Mol. Genet 21, 322–333 (2012).
Wang, L. et al. CPAG: software for leveraging pleiotropy in GWAS to reveal similarity between human traits links plasma fatty acids and intestinal inflammation. Genome Biol. 16, 190 (2015).
Rottem, M. & Shoenfeld, Y. Asthma as a paradigm for autoimmune disease. Int. Arch. Allergy Immunol. 132, 210–214 (2003).
Li, X. et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J. Allergy Clin. Immunol. 130, 861–868.e7 (2012).
Hayes, J. E. et al. Tissue-specific enrichment of lymphoma risk loci in regulatory elements. PLoS One 10, e0139360 (2015).
Liang, L. et al. An epigenome-wide association study of total serum immunoglobulin E concentration. Nature 520, 670–674 (2015).
Wenzel, S. E. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat. Med. 18, 716–725 (2012).
DerSimonian, R. & Laird, N. Meta-analysis in clinical trials. Control. Clin. Trials 7, 177–188 (1986).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Li, Y., Willer, C., Sanna, S. & Abecasis, G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 10, 387–406 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Raychaudhuri, S. VIZ-GRAIL: visualizing functional connections across disease loci. Bioinformatics 27, 1589–1590 (2011).
So, H. C., Gui, A. H., Cherny, S. S. & Sham, P. C. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet. Epidemiol. 35, 310–317 (2011).
Acknowledgements
We thank all participants who provided data for each study and also thank our valued colleagues who contributed to data collection and phenotypic characterization of clinical samples, genotyping, and analysis of individual datasets. Detailed acknowledgments and funding for individual studies can be found in the Supplementary Note.
Author information
Authors and Affiliations
Consortia
Contributions
TAGC study management: F.D., K.C.B., W.O.C.C., M.F.M., C.O., and D.L.N.
F.D. and D.L.N. designed the study and wrote the manuscript. F.D., D.L.N., and P.M.-J. designed and conducted the statistical analysis. K.C.B., W.O.C.C., M.F.M., and C.O. designed the study and wrote the manuscript. M.B., A.V., S. Letort, and H.M. carried out the quality control of the data and performed statistical analysis.
AAGC (Australia): study principal investigators (PIs), M.A.F., M.C.M., C.F.R., and P.J.T.; data collection or analysis, M.A.F., M.C.M., C.F.R., G.J., and P.J.T.
ALLERGEN Canadian Asthma Primary Prevention Study (CAPPS) and Study of Asthma, Genes and the Environment (SAGE): study PIs, A.B.B., M.C.-Y., D.D., and A.L.K.; data collection or analysis, D.D. and J.E.P.; study phenotyping, A.B.B. and M.C.-Y.
Saguenay‐Lac‐Saint‐Jean (SLSJ) Study: study PIs, C.L. and T.J.H.; study design and management, C.L.
Analysis in Population-based Cohorts of Asthma Traits (APCAT) Consortium: study PIs, J.N.H., M.-R.J., and V. Salomaa. Framingham Heart Study (FHS): study PI, G.T.O.; data collection or analysis, S.V. and Z.G. The European Prospective Investigation of Cancer (EPIC)-Norfolk: study PI, N.J.W.; data collection or analysis, J.H.Z. and R.S. Northern Finland Birth Cohort of 1966 (NFBC1966): study PI, M.-R.J.; data collection or analysis, A.C.A. and A.R. FINRISK: study PI, V. Salomaa; data collection or analysis, M. Kuokkanen and T. Laitinen. Health 2000 (H2000) Survey: study PIs, M.H. and P.J.; data collection or analysis, M. Kuokkanen and T.H. Helsinki Birth Cohort Study (HBCS): study PI, J.G.E.; data collection or analysis, E.W. and A. Palotie. Young Finns Study (YFS): study PI, O.T.R.; data collection or analysis, T. Lehtimäki and M. Kähönen.
African Ancestry Studies from the Candidate Gene Association Resource (CARe) Consortium: study PIs, J.N.H. and S.S.R.; data collection or analysis, C.D.P., D.B.K., L.J.S., R.K., K.M.B., and W.B.W.
Multi‐Ethnic Study of Atherosclerosis (MESA): study PIs, R.G.B. and S.S.R.; data collection or analysis, K.M.D. and A.M.
Atherosclerosis Risk in Communities Study (ARIC): study PI, S.J.L.; data collection or analysis, S.J.L. and L.R.L.
Cardiovascular Health Study (CHS): study PIs, S.A.G. and S.R.H.; data collection or analysis, G.L., S.A.G., and S.R.H.
deCode genetics: study PIs, K.S., I.J., D.F.G., U.T., and G.T.; data collection or analysis, I.J., D.F.G., and G.T.; study phenotyping, U.S.B.
Early Genetics and Lifecourse Epidemiology (EAGLE) Consortium: PI, H.B. Cophenhagen Prospective Study on Asthma in Childhood (COPSAC): study PIs, H.B. and K.B.; data analysis, E. Kreiner and J.W.; study phenotyping, K.B. Danish National Birth Cohort (DNBC): study PI, M.M.; data collection or analysis, B.F. and F. Geller. GENERATION R: study PI, J.C.d.J.; data collection or analysis, R.J.P.v.d.V., L.D., and V.W.V.J. GINIplus/LISAplus: study PI, J. Heinrich; genotyping, data collection or analysis, M. Standl and C.M.T.T.; study phenotyping, J. Heinrich. Manchester Asthma and Allergy Study (MAAS): study PIs, A.S. and A.C.; data collection or analysis, J.A.C. Western Australian Pregnancy Cohort Study (RAINE): study PI, P.H.; data collection or analysis, W.A. and C.E.P.
British 1958 Birth Cohort (B58C) Study: PI and statistical analysis, D.P.S.
EVE Consortium: study PIs, C.O., D.L.N., K.C.B., E. Bleecker, E. Burchard, J. Gauderman, F. Gilliland, S.J.L., F.J.M., D.M., I.R., S.T.W., L.K.W., and B.A.R.; data collection or analysis, D.L.N., J. Gauderman, S.J.L., D.M., D.G.T., B.A.R., B.E.H., P.E.G., M.T.S., C.E., B.E.D.-R.-N., J.J.Y., A.M.L., R.A. Myers, R.A. Mathias, and T.H.B.
Japanese Adult Asthma Research Consortium (JAARC): study PI, T.T.; data collection or analysis, T.T., A.T., and M. Kubo.
Japan Pediatric Asthma Consortium (JPAC): study PI, E.N.; data collection or analysis, H.H. and K.M.
GABRIEL Consortium: study PIs, W.O.C.C. and E.V.M.; genotyping, M.L.; data analysis, E.B., F.D., M.F., and D.P.S. Epidemiological study on the Genetics and Environment of Asthma (EGEA): study PIs, V. Siroux and F.D.; genotyping, data collection or analysis, M.L. and E. Bouzigon. Avon Longitudinal Study of Parents and Children (ALSPAC): study PI, J. Henderson; genotyping, data collection or analysis, W.L.M. and R.G.; study phenotyping, J. Henderson. European Community Respiratory Health Survey (ECRHS): study PI, D.J.; data collection or analysis, C.J. and J. Heinrich. Children, Allergy, Milieu, Stockholm, Epidemiology (BAMSE) study: study PIs, E.M., M.W., and G.P. Busselton Health Study: study PIs, A.W.M., A.J., and J.B.; genotyping, data collection or analysis, A.W.M., A.J., J. Hui, and J.B. GABRIEL Advanced Surveys: study PI, E.V.M.; data collection or analysis, M. Kabesch and J. Genuneit. Kursk State Medical University (KSMU) Study: study PI, A. Polonikov; data collection or analysis, M. Solodilova and V.I.; Medical Research Council-funded Collection of Nuclear Families with Asthma (MRCA-UKC): study PIs, W.O.C.C. and M.M.; data collection or analysis, L. Liang. Multicentre Asthma Genetics in Childhood Study (MAGICS): study PI, M. Kabesch; data collection or analysis, A.V.B. and S.M. German Multicentre Allergy Study (MAS): study PI, Y.-A.L.; data collection or analysis, S. Lau and I.M. Prevention and Incidence of Asthma and Mite Allergy (PIAMA) cohort : study PIs, G.H.K. and D.S.P.; data collection or analysis, G.H.K., D.S.P., and U.G. Swiss Cohort Study on Air Pollution and Lung and Heart Diseases in Adults (SAPALDIA): study PI, N.P.-H.; data collection or analysis, M.I. and A.K. Tomsk Study: study PIs, L.M.O. and V.P.P.; data collection or analysis, M.B.F. and P.A.S. UFA Study: study PI, E. Khusnutdinova; data collection or analysis, A.S.K. and Y.F. Industrial Cohorts Research Group (INDUSTRIAL): study PIs, D.H. and T.S.; data collection or analysis, I.M.W. and V. Schlünssen. Severe Asthma Cohorts (SEVERE): study PIs, A.B., K.F.C., and C.E.B.
Netherlands Twin Register (NTR) Study: study PI, D.I.B.; genotyping, data collection or analysis, J.J.H., H.M., and G.W.
Rotterdam Study: study PIs, A.H., B.H.S., and G.G.B.; genotyping, data collection or analysis, G.G.B., B.H.S., D.W.L., L. Lahousse, and A.G.U.
Dutch Asthma Genetics Consortium (DAGC): study PIs, G.H.K. and D.S.P.; genotyping, data collection or analysis, G.H.K., D.S.P., J.A., M.A.E.N., and J.M.V.
All authors provided critical review of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors affiliated with deCODE (D.F.G., I.J., K.S., U.T., and G.T.) are employees of deCODE genetics/Amgen. All other coauthors have no conflicts of interest to declare.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Supplementary Tables 3, 5–7, 9, 10, 12–17, 20 and 21, and Supplementary Note.
Supplementary Table 1
Description of TAGC studies included in the meta-analysis.
Supplementary Table 2
Information on genotyping methods, imputation, and statistical analysis by study. Details and references for each study are in the Supplementary Note.
Supplementary Table 4
Genome-wide significant SNPs (Prandom ≤ 5 × 10−8) in the European-ancestry meta-analysis.
Supplementary Table 8
Genome-wide significant SNPs (Prandom ≤ 5 × 10−8) in the multi-ancestry meta-analysis.
Supplementary Table 11
Association of 17q12-21 SNPs with asthma in multi-ancestry and pediatric meta-analyses.
Supplementary Table 18
Overlap between TAGC asthma-association signals (Prandom <10−3) and GWAS signals with diseases/traits in the GWAS catalog.
Supplementary Table 19
Enrichment of asthma risk loci in promoter and enhancer marks by cell type. The results presented in this table are for 16 out of the 18 asthma loci shown in Table 1. The 6p21.33 and 6p21.32 loci spanning the HLA complex were excluded because of high variability and LD in the region. Enhancer and promoter marks were defined using the ChromHMM 15-state model applied to 127 ROADMAP/ENCODE reference epigenomes (PMID 25693563).
Rights and permissions
About this article

Cite this article
Demenais, F., Margaritte-Jeannin, P., Barnes, K.C. et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet 50, 42–53 (2018). https://doi.org/10.1038/s41588-017-0014-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-017-0014-7
This article is cited by
-
A systematic two-sample and bidirectional MR process highlights a unidirectional genetic causal effect of allergic diseases on COVID-19 infection/severity
Journal of Translational Medicine (2024)
-
Genetics of chronic respiratory disease
Nature Reviews Genetics (2024)
-
The Rotterdam Study. Design update and major findings between 2020 and 2024
European Journal of Epidemiology (2024)
-
Identifying the potential causal role of insomnia symptoms on 11,409 health-related outcomes: a phenome-wide Mendelian randomisation analysis in UK Biobank
BMC Medicine (2023)
-
Childhood asthma phenotypes and endotypes: a glance into the mosaic
Molecular and Cellular Pediatrics (2023)
