
Abstract
Mutations that add, subtract, rearrange, or otherwise refashion genome structure often affect phenotypes, although the fragmented nature of most contemporary assemblies obscures them. To discover such mutations, we assembled the first new reference-quality genome of Drosophila melanogaster since its initial sequencing. By comparing this new genome to the existing D. melanogaster assembly, we created a structural variant map of unprecedented resolution and identified extensive genetic variation that has remained hidden until now. Many of these variants constitute candidates underlying phenotypic variation, including tandem duplications and a transposable element insertion that amplifies the expression of detoxification-related genes associated with nicotine resistance. The abundance of important genetic variation that still evades discovery highlights how crucial high-quality reference genomes are to deciphering phenotypes.
Similar content being viewed by others

Main
Mutations underlying phenotypic variation remain elusive in trait-mapping studies1 despite the exponential accumulation of genomic data, suggesting that many causal variants are invisible to current genotyping approaches2,3,4,5. In fact, mutations like duplications, deletions, and transpositions6,7 are systematically under-represented by standard methods7, even as a consensus emerges that such structural variants (SVs) are important factors in the genetics of complex traits2. Addressing this problem requires compiling an accurate and complete catalog of the genomic features that are relevant to phenotypic variation, a goal most readily achieved by comparing nearly complete high-quality genomes7. Although the development of high-throughput short-read sequencing led to a steep drop in cost and a commensurate increase in the pace of sequencing8, it also led to a focus on single-nucleotide changes and small indels3,9. Paradoxically, this has also resulted in deterioration of the contiguity and completeness of new genome assemblies, due primarily to read-length limitations10.
Here we present a reference-quality assembly of a second D. melanogaster strain called A4 and introduce a comprehensive map of SVs, which identifies a large amount of hidden variation exceeding that due to SNPs and small indels, and which includes strong candidates to explain complex traits. The A4 strain is a part of the Drosophila Synthetic Population Resource (DSPR)11, a resource for mapping phenotypically relevant variants. We assembled the new A4 genome using high-coverage (147×) long reads through single-molecule real-time sequencing of DNA extracted from females (Supplementary Fig. 1), following an approach that has been shown to yield complete and contiguous assemblies12. The A4 assembly is more contiguous than release 6 of the ISO1 strain13—which is arguably the best metazoan whole-genome sequence assembly—with 50% of the genome contained in contiguous sequences (contigs) 22.3 Mb in length or longer (Supplementary Figs. 2 and 3). As compared to the ISO1 assembly, the A4 assembly comprises far fewer sequences (161 scaffolds versus 1,857 non-Y-chromosome scaffolds14) while maintaining comparable completeness (Supplementary Table 1)15. The two genomes are collinear across all major chromosome arms, making large-scale misassembly unlikely (Fig. 1a). An optical map of the A4 genome also supported the accuracy of the assembly (Supplementary Figs. 4 and 5).

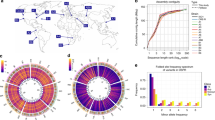
a, Dot plot between the D. melanogaster reference (ISO1) and A4 assemblies. The A4 assembly is as contiguous as the ISO1 assembly (scaffold N50 = 25.4 Mb versus 25.2 Mb; Supplementary Table 1). Repeats and TEs were masked to highlight the correspondence of the two genomes. b, The proportions of large (>100-bp) SVs in the A4 chromosome 2L assembly relative to the ISO1 2L assembly that were identified (visible) or missed (invisible) by short-read methods (Methods). c, Relationship between the lengths of TEs in ISO1 (median 5.1 kb) and the lengths of the introns into which they are inserted. Nearly equal intron and TE lengths indicate that many introns comprise mainly TEs. d, Distribution of SVs (>100 bp) across chromosome arms in the A4 genome. Track 1 shows pericentric heterochromatin (black). Tracks 2–4 show TEs, duplicate CNVs (relative to ISO1), and non-TE indels >100 bp in length, respectively. CNVs and TEs are present in higher densities in heterochromatin as compared to euchromatin, whereas non-TE indels are less numerous in heterochromatin.
We identified putative SVs by classifying regions of disagreement in a genome-wide pairwise alignment of the A4 and ISO1 assemblies as indels, copy number variants (CNVs), or inversions (Table 1). Reads spanning SVs showed that genotyping error was rare (<2.5%; Supplementary Table 2). However, because extremely long repeats are common in heterochromatin and require specialized approaches for assembly and validation16, we focused on euchromatin (Supplementary Table 3). We discovered 1,890 large (>100-bp) indels (Supplementary Fig. 6 and Supplementary Table 4), which affected more than 7 Mb. In contrast, mutations <100 bp in length affected only 1.4 Mb (indels, 722 kb; SNPs, 687 kb). Among large indels, 79% (1,486/1,890) were transposable element (TE) insertions (Supplementary Figs. 7–17). A previously published catalog of TE insertions in A4 based on 70× short-read coverage17 failed to find 38% of the TE insertions in A4 reported here (Fig. 1b, Supplementary Fig. 18, and Supplementary Table 5). These insertions, which are invisible to short-read approaches, often occur (in 34% of instances) when a TE is inserted near another TE, resulting in complex, non-uniquely mapping reads that are difficult to interpret. One such insertion was found in the A4 allele of the MRP gene (encoding multidrug-resistance-like protein 1), which is a candidate gene for resistance to the chemotherapy drug carboplatin18 (Supplementary Fig. 17).
We found that many TE insertions affected introns (395/718 in ISO1, 435/768 in A4), often greatly lengthening them (Fig. 1c and Supplementary Fig. 19). Additionally, TEs inserted into exons can be spliced out, effectively becoming new introns. We saw evidence of this in cDNA from ISO119 and in RNA-seq reads in A4 that showed exon junctions flankng TE insertions (Supplementary Figs. 20–22 and Supplementary Table 6), which represents a genome-wide view of TE-derived introns segregating in a population. TE insertions within introns are associated with decreased transcription20, possibly caused by a phenomenon called intron delay, which slows transcription in long introns21. TE insertions can affect phenotype directly22, perhaps by modulating or disrupting the expression of important genes. Because most TEs are rare in D. melanogaster 23, they are poorly tagged by common variants, complicating genome-wide association study (GWAS) approaches for mapping traits; this mirrors similar complications in human GWAS24.
Non-TE insertions represented 20% of ISO1 and 23% of A4 insertions, and they accounted for 170 kb of sequence variation (Fig. 1d and Table 1). Although these mutations were much smaller than TEs (median 213 bp versus 4.7 kb), they often affected genes, and 23% even escaped detection by short reads (Fig. 1b). For example, among both hidden and visible deletions, there were 18 genes that were present in ISO1 and partially or completely absent in A4 (Supplementary Table 7), including Cyp6a17 (Fig. 2a and Supplementary Fig. 23). Knockout of Cyp6a17 in a previous study increased cold preference25. Indeed, A4 flies preferred colder temperatures than flies from a strain carrying an intact copy of Cyp6a17 (Fig. 2b and Supplementary Fig. 24). Furthermore, this mutation was more common than expected for a deleterious allele (Fig. 2c), suggesting that it has a role in regulating how flies respond to temperature in the wild. One deletion missed by short-read genotyping removed the second exon of Mur18B (and 41 amino acids of the encoded chitin-binding protein that confers resistance to high-temperature stress26) (Supplementary Fig. 25), likely rendering the A4 Mur18B allele defective.

a, Cyp6a17 is deleted in the A4 genome relative to the ISO1 genome. Alignment between annotated ISO1 and A4 assemblies on chromosome arm 2R shows a large ISO1 region (red) missing in A4. Gene models are shown (gray indicates noncoding sequences, and yellow indicates coding sequences). b, Temperature preference of strains A4 (∆Cyp6a17) and w 1118 (intact Cyp6a17 23). Preference was measured by recording the position of 100 flies along a linear 8 °C–30 °C temperature gradient after an adjustment period (Methods). Each dot represents the position of a fly along the gradient. Each experiment number is an independent pairwise trial. A4 flies occupy colder regions of the gradient than w 1118 flies (Fisher’s method on Wilcoxson rank-sum tests, meta P value << 10−16). Upper and lower hinges of the box plots represent 25% and 75% quantiles, respectively; the upper whisker indicates the largest observation less than or equal to the upper hinge + 1.5 times the interquartile range (IQR); the lower whisker indicates the smallest observation greater than or equal to the lower hinge – 1.5 times the IQR; and the middle horizontal bar indicates the median, 50% quantile. c, Frequency of the Cyp6a17 deletion in African (DPGP2) and North American (DGRP) populations.
We discovered 27 inversions, ranging from 100 bp to 21 kb in length (Supplementary Table 4), that affected 60 kb of sequence, only 4 of which were detected by paired-end methods (Fig. 1b and Supplementary Table 5). These inversions often (in 21/27 instances) affected regions harboring genes, including a 21-kb region that spanned five genes encoding gustatory receptors: Gr22a, Gr22b, Gr22c, Gr22d, and Gr22e (Supplementary Table 4). Although such clusters of related sequences may obscure the read-mapping information used to detect inversions, we could not find genomic features that might explain why the other inversions were missed. The A4 optical map identified a putative inversion occupying 300 kb of the proximal end of the X-chromosome scaffold that was not resolved by the A4 assembly (Supplementary Figs. 4 and 5). Failure to resolve this inversion is not unexpected because assembly methods tuned for euchromatin perform poorly in heterochromatic regions16.
We discovered 390 CNVs (209 in A4 and 181 in ISO1) that affected ~600 kb (Fig. 1d, Supplementary Figs. 26–36, and Supplementary Table 4). Although some CNVs were missed by paired-end methods owing to spacer sequences between copies that were longer than the library fragments (Fig. 3a,d), most (~90%) of the CNVs were missed because they occurred in complex tandem repeats (Supplementary Fig. 37). Unlike indels, most CNVs (64%) affected exons. Additionally, short-read CNV genotyping methods missed 13 of 34 protein-coding genes that were duplicated in A4. In total, only ~40% of CNVs were discoverable with high-specificity split-read and read-orientation methods27,28 (Fig. 1b and Supplementary Fig. 38). Consistent with previous observations29, coverage-based methods were extremely nonspecific (Supplementary Fig. 38) and were therefore excluded from analysis. We next compared published gene expression data from larvae of A4 to expression data for a DSPR strain called A330 and identified 17 A4 duplicate genes that are single copy in ISO1 with increased expression (Supplementary Table 8), including genes previously identified as candidates for cold adaptation, olfactory response, and toxin resistance, among others (Fig. 3a,d and Supplementary Tables 8 and 9). Notably, eight of these CNVs were invisible to short-read methods (Supplementary Table 8).

Shaded parallelograms (light blue, distal copy; dark blue, proximal copy) correspond to the single and duplicated regions in ISO1 and A4. a, Schematic showing duplication of Cyp28d1 and CG7742 in A4. ISO1 and strain A3 possess one copy of Cyp28d1, whereas A4 has two copies. A 1.5-kb Accord fragment (pink) containing an LTR (blue) is located between the proximal Cyp28d1 and the distal CG7742. Gene models are shown with gray (noncoding) and orange (coding) rectangles. b, Paralog-specific expression of candidate QTL genes at Q1 in A4 and A3 in the presence and absence (control) of nicotine in the food. CG7742 and Cyp28d1 copies located nearer the Accord element are transcribed at higher levels than those that are more distal. FPKM, fragments per kilobase of transcript per million mapped reads. c, Combined frequency of four Cyp28d duplicate alleles in African (DPGP2 and DPGP3) and North American populations. d, Schematic showing that tandem duplication of Ugt86Dh in A4 created Ugt86Dh-d. e, Paralog-specific expression of candidate QTL gene Ugt86Dh in A4 and A3 in the presence and absence (control) of nicotine in the food. In contrast to Cyp28d1 duplicates, the two copies of Ugt86Dh are expressed at similar levels, and their expression nearly doubles in the presence of nicotine. f, Frequency of the Ugt86Dh duplicate in African (DPGP2 and DPGP3) and North American populations.
A longstanding concern in trait-mapping studies is failure to genotype candidate mutations2. Because A4 is a parental line of the DSPR trait-mapping panel11, we could confront this problem directly. Among the eight duplicate genes with increased expression in A4 that escaped detection, Cyp28d1 and Ugt86Dh fell under quantitative trait loci (QTLs) for resistance to nicotine, a plant defense toxin30,31. One QTL (Q1) contains two genes, Cyp28d1 and Cyp28d2, that encode cytochrome P450 enzymes, both of which were upregulated30. The other candidate region that showed a major effect contains the Ugt86D gene cluster, which includes several differentially regulated genes, including Ugt86Dh (Fig. 3d,e). Candidate mutations like these are of obvious interest to researchers trying to dissect any trait, and yet they were not visible in the initial study30.
In the A4 assembly, Q1 contains a 3,755-bp tandem duplication in which the duplicated regions are separated by a 1.5-kb spacer, resulting in two copies of Cyp28d1 (Fig. 3a and Supplementary Figs. 39–41). We compared paralog-specific expression levels of the Cyp28d1 copies in A4 to expression of the single copy in A3. In the absence of nicotine, the proximal and distal copies in A4 exhibited ~41-fold and ~6.3-fold higher expression, respectively, than the single copy in A3 (Fig. 3b). The intervening spacer sequence proved to be the 5′ end of Accord, a long terminal repeat (LTR) retrotransposon (Fig. 3a). Insertion of Accord upstream of another gene called Cyp6g1 has been linked to upregulation of the encoded cytochrome P450 enzyme32, suggesting that the retrotransposon may be responsible for the upregulated expression rather than the tandem duplication of the Cyp28d gene. The second nicotine-resistance QTL contains several Ugt genes, including Ugt86Dh, which have previously been implicated in increased resistance to the pesticide DDT33. Of note, we found that Ugt86Dh was duplicated in A4 (Fig. 3d and Supplementary Figs. 42 and 43); this mutation escaped detection by paired-end short reads (Supplementary Table 5). Although several Ugt genes in the Q4 QTL showed higher expression in nicotine-resistant A4 larvae than in sensitive A3 larvae30 (Fig. 3e), candidate variants that explain these differences have yet to be identified.
Because nicotine analogs are widely used pesticides, we predict that resistance-conferring mutations are common, mirroring observations for DDT. Indeed, we found that four duplicate alleles spanning Cyp28d1 and Cyp28d2 segregated at intermediate to high frequencies in multiple populations (Fig. 3c) in a 25-kb region where we expected duplicate heterozygosity to be less than 0.1. Similarly, the single duplicate allele of Ugt86Dh segregated at high or intermediate frequency in nearly all of the populations we examined6 (Fig. 3f). Finally, patterns of SNP variation surrounding both Cyp28d1 and Ugt86Dh are consistent with recent bouts of natural selection (Supplementary Figs. 44 and 45), suggesting recent adaptation to nicotinoids.
Although we focus on genetic variation in A4 relative to ISO1, there is no biologically meaningful sense in which any individual of a species is a more appropriate reference than another. Yet, despite the prevalence of heritable phenotypic variation, functional work often describes results derived from individuals with diverse genotypes as applying to an entire species34. Approaches like RNA interference (RNAi) or gene editing with CRISPR require precise sequence information about their targets and can be easily misled by hidden structural variation. One study on the origin of new genes in D. melanogaster argues that new genes rapidly become essential, and the authors even report a new gene called p24-2 that is so young that it is present in only D. melanogaster 35. Experiments targeting p24-2 using RNAi constructs suggested that, although new, p24-2 is essential. However, p24-2 was absent in eight of the ten strains we examined, including A4 and Oregon-R (Supplementary Figs. 46 and 47), which calls into question its essential nature in D. melanogaster. Because the original construct actually targeted both p24-2 and its essential paralog eca 36,37 (Supplementary Note), we tested two other constructs targeting p24-2, neither of which resulted in any reduction in viability (Supplementary Table 10), thus bolstering the suggestion that p24-2 is not essential.
The ubiquity of hidden variation in genome structure is merely an indication of the extent of the underlying genetic variation governing phenotypes. Together with careful phenotypic measurements, a new generation of high-quality genomes will identify previously invisible heritable phenotypic variation. Our results show that popular genotyping approaches miss a significant fraction of SVs (Fig. 1b, Supplementary Figs. 18 and 38, and Supplementary Table 5), including ones that affect gene expression and organismal phenotype (Supplementary Tables 8 and 9), suggesting that previous estimates of the contribution of SVs to regulatory38 and phenotypic variation are misleading39. The extensive hidden variation we observe segregates in D. melanogaster, a species that likely harbors fewer complex structural features than humans or livestock, as well as crop species like wheat and maize. Consequently, we suggest that the true medical and agricultural impact of structural variation is likely to be much greater than the already considerable estimates made without recourse to multiple reference-grade assemblies29.
Methods
DNA sequencing and genome assembly
A4 DNA was extracted from females and used in SMRTbell library preparation as described previously12. We sequenced this library on 30 SMRTcells using P6-C4 chemistry on a Pacific Biosciences RSII platform at the University of California High-Throughput Genomics Facility, yielding 18.7 Gb of sequence. We then followed the method described previously12 to assemble the A4 genome. We assembled a draft genome using PBcR-MHAP40 in wgs 8.3rc1 and PacBio reads (NG50 = 13.9 Mb, 147 Mb in total; NG50 is the contig length such that 50% of an assumed assembly size is contained within contigs of this length or longer) and then generated a hybrid assembly with DBG2OLC41 using the longest 30× PacBio reads and 75× paired-end Illumina reads from ref. 42 (assuming a genome size of 130 Mb; NG50 = 4.23 Mb, 129 Mb in total). We merged the two assemblies using quickmerge v0.1 with default settings, except hco = 5, c = 1.5, and l = 2 Mb. The merge yielded an assembly (NG50 = 21.3 Mb, 130 Mb in total) that was both smaller than expected42 and smaller than the PacBio-only assembly. Therefore, we added contigs that were unique to the PacBio assembly to the hybrid assembly using quickmerge as described above but with I = 5 Mb. Finally, we generated the final assembly by running finisherSC43 with default settings, polishing the assembly twice with quiver (SMRT Analysis v2.3), and with Pilon v1.344 (using A4 reads from ref. 42). This yielded a final assembly of 144 Mb with N50 = 22.3 Mb (Supplementary Table 1).
Bionano data
A4 embryos less than 12 h old were collected on Petri dishes containing apple juice and agar, dechorionated using 50% bleach, rinsed with water, and stored at –80 °C. DNA was extracted from frozen embryos using the Animal Tissue DNA Isolation kit (Bionano Genomics). Bionano Irys optical data were generated and assembled with IrysSolve 2.1 at Bionano Genomics. We then merged the Bionano assembly with the final assembly contigs (described in “DNA sequencing and genome assembly”) using IrysSolve, retaining Bionano assembly features when the two assemblies disagreed.
Comparative scaffolding
The scaffold for the A4 assembly was prepared with the software mscaffolder (see URLs) using the release 6 D. melanogaster genome (r6.09) assembly13 as the reference. Prior to scaffolding, TEs and repeats in both assemblies were masked using default settings for RepeatMasker (v4.0.6). The repeat-masked A4 assembly was aligned to the repeat-masked major chromosome arms (X, 2L, 2R, 3L, 3R, and 4) of the D. melanogaster ISO1 assembly using MUMmer45. Alignments were further filtered using the delta-filter utility with the -m option, and the contigs were assigned to specific chromosome arms on the basis of the mutually best alignment. Contigs showing less than 40% of the total alignment for any chromosome arms could not be assigned a chromosomal location and therefore were not scaffolded. The mapped contigs were ordered on the basis of the starting coordinate of their alignment that did not overlap with the preceding reference chromosome–contig alignment. Finally, the mapped contigs were joined with 100 Ns, a convention representing assembly gaps. The unscaffolded sequences were named with a ‘U’ prefix.
Benchmarking universal single-copy orthologs (BUSCO) analysis
We used BUSCO (v1.22)15 to evaluate the completeness and accuracy of the A4 and ISO1 release 6 assemblies. ISO1 contains five BUSCOs (BUSCOaEOG75R3J9, BUSCOaEOG7SJRJ9, BUSCOaEOG7SJRK2, BUSCOaEOG7WMR0H, and BUSCOaEOG71S8ZH) that are missing from the A4 assembly. To validate the absence of these five BUSCOs in the A4 assembly, the full-length sequences of the ISO1 genes (Ftz-f1, CG7627, Raw, Maf1, and Cv-c) were downloaded from FlyBase14 and queried against the A4 assembly with MUMmer. MUMmer found all five ‘missing’ BUSCOs in the A4 assembly in single copies. The BUSCO counts for A4 were adjusted accordingly.
Structural variant detection
Detection of CNVs via whole-genome alignment. We aligned the ISO1 and A4 assemblies using MUMmer45 (mummer -mumreference -l 20 -b) and then clustered maximal exact matches (MEMs) between the two mgaps (mgaps -C -s 200 -f 0.12 -l 100). The l parameter in mgaps was set to 100 to detect duplicates that were 100 bp or longer. We used a pipeline called svmu (structural variants from MUMmer; see URLs) to automate CNV detection from overlapping mgaps clusters. When reference sequence regions in two separate alignment clusters overlapped, the overlapping segment of the reference sequence regions was inferred to be duplicated in the query sequence. This approach can also identify (i) a duplicated sequence that is present in both the genomes but has diverged owing to the presence of repeats or indels and (ii) CNVs containing TE sequences. We filtered the latter using RepeatMasker (v4.0.6). We identified false-positive duplication calls by aligning the putatively duplicated reference sequences back to the ISO1 and A4 genomes using nucmer (nucmer --maxmatch --g 200) and then counting the copy number using checkCNV, which is also included in the svmu pipeline. svmu was run with the default parameters; checkCNV was run with c = 500 (max copy number 500), qco = 10,000 (10 kb of insertion or deletion allowed within a copy), and rco = 0.2 (unaligned length of up to 20% of the sequence length between reference and query copies allowed). CNVs occurring within 2 kb of each other were designated as ‘complex events’ and combined (bedtools merge --d 2000)46 for the purpose of counting the total number of CNVs present in the genome (Supplementary Table 11). However, the total sequence affected by CNVs was counted before merging. Functional annotation of CNVs was based on gene annotation of ISO1 release 6.
Detection of indels via whole-genome alignment
Insertions (>100 bp) in the A4 genome appear as alignment gaps between two adjacent syntenic blocks when ISO1 is aligned to A4 (and vice versa). We aligned the A4 sequence to the ISO1 sequence using nucmer (default parameters) and then identified adjacent syntenic blocks with gaps >100 bp in length between them in the A4 assembly but <10% the gap length in the ISO1 assembly. Indel detection was carried out with the svmu utility findInDel. A deletion was inferred for a specific gene (e.g., Cyp6a17) when an ortholog of the gene was present in the closely related species Drosophila simulans.
Detection of inversions via whole-genome alignment
We identified inversions in the A4 genome by aligning it to the ISO1 genome using nucmer (-mumreference) and then processing the outputted delta file using findInDel. A4 regions that ran in the reverse direction with respect to the ISO1 sequence were recorded as inversions. TEs were removed from this list using RepeatMasker annotations for ISO1.
Genotyping CNVs, indels, and inversions using Illumina reads
Three common, complementary strategies are typically used to discover CNVs with paired-end Illumina reads: read depth, read-pair mapping orientation, and split-read mapping7. We identified duplications (100 bp to 25 kb long) in the A4 genome using 70× paired-end reads11 with CNVnator47 for the read depth approach, pecnv28 for the read-pair orientation approach, and Pindel27 for the split-read mapping approach. We mapped reads to ISO1 release 6 using bwa-mem for CNVnator and pindel and bwa-aln for pecnv48. We required at least three supporting read pairs for pecnv calls28 and used a bin size of 100 for CNVnator because of the data’s high coverage. Furthermore, we used CNVnator and Pindel to identify large (>100-bp) indels and Pindel to identify inversions. We manually compared these short-read-based calls to our alignment-based CNV calls for all of chromosome arm 2L.
TE insertion coordinates for A4 were obtained from DSPR (http://wfitch.bio.uci.edu/~dspr/). We manually compared our TE insertion calls and those from ref. 17 for all of chromosome arm 2L.
SNP and small indel detection
SNPs and small (<100-bp) indels in the A4 assembly were identified using the show-snps utility from MUMmer45. We aligned A4 scaffolds to ISO1 scaffolds using nucmer (-mumreference) and then filtered repeats using delta-filter in conjunction with the --r and --q options. SNPs and small indels were called from the filtered data using show-snps with --Clr options.
Validation of duplicates and indels
Dot plots between A4 and ISO1 for all SV loci on chromosome arm 2L were manually inspected to confirm the accuracy of the MUMmer-based genotyping. All manually inspected loci corresponded to the automated genotype calls. To quantify the effect of assembly errors in A4 on SV calls, we required that unassembled, corrected long reads from A4 agree with the A4 assembly in the region spanning the entire mutation. To do this, we mapped the PBcR-MHAP-corrected long reads to the A4 assembly using blasr v1.3.1.142244 (-bestn 1 --sam) and identified all of the reads that spanned the mutation-containing region with anchors in the flanking sequence of at least 250 bp on each side. For our stringent validation criteria, we required at least two fully spanning reads to overlap each SV (Supplementary Fig. 48a). These fully spanning reads were required to have at least 99.5% alignment coverage (P aligned) and less than a ratio of 0.005 of gaps to read length (R gaps; Supplementary Fig. 48a). For our standard validation criteria, we permitted validation under the following relaxed criteria: (i) overlap-spanning reads (at least two on each side) that otherwise fit the stringent criteria above and (ii) fully spanning reads with at least 97.5% alignment coverage (P aligned) and less than a ratio of 0.025 of gaps to read length (R gaps; Supplementary Fig. 48b).
Half of our sequencing data were present in reads that were 17,885 bp or longer, which was enough to achieve more than 60-fold coverage across the entirety of the euchromatin and more than 10-fold coverage of the genome in reads that were 30 kb or longer. Such long reads contained unique sequences flanking each side of the mutation, as well as the mutation breakpoints and the mutation itself, making this a powerful approach to validating SV calls.
PCR validation
We assayed for the presence and absence of Cyp28d1 and p24-2 copies using PCR (Supplementary Figs. 41 and 47, and Supplementary Table 12). We extracted DNA from 25 flies from each strain using the Magattract HMW DNA kit (Qiagen), and we used Phusion (New England Biolabs) for PCRs that had an amplification time of 15 s for the Cyp28d1 reactions and 30 s for the p24-2 reactions.
Temperature-preference assay
We created a linear temperature gradient on a solid aluminum bar (total dimensions: 24 inches × 4 inches × 4 inches) by placing 4 inches of one end of the bar inside a reservoir containing ice water (0 °C) and 4 inches of the other end inside a reservoir containing warm water (35 °C) (Supplementary Fig. 24). This left ~40 cm of aluminum bar exposed between the baths. Temperatures along the bar were measured by 11 temperature sensors (Tmp36 analog temperature sensors from Adafruit) that were evenly spaced at 4-cm intervals and sealed into holes drilled into the bar after being secured with thermal epoxy (OMEGABOND 101 Two-Part Epoxy). The probes were connected to three four-channel 16-bit analog-to-digital converters (ADS1115 from Adafruit), which were in turn calibrated and monitored by a Raspberry Pi 3 single-board computer. Automated temperatures were recorded every second using a custom Python script (see URLs) during the experiment to verify the stability of the gradient. The temperature measurements at the end of the experiment were used in assigning temperatures to individual flies. The temperature gradient on the aluminum bar ranged from 9 °C to 30 °C (Fig. 2b). We compared the preference of A4 flies, which lack the Cyp6a17 gene, to that of w 1118 flies (BDSC stock 5905), which have an intact copy of Cyp6a17 25. We collected groups of 100 1- to 3-d-old flies of mixed sex and kept them at 25 °C for 24 h. Before the assay, flies were immobilized with light anesthesia and placed between a thin aluminum sheet cut into the shape of the aluminum bar surface and an acrylic lid possessing a partition to create two ‘lanes’ for the flies to behave without interacting with each other. Quinine sulfate was applied to the roof and walls of each channel in the lid so that the flies would avoid these surfaces and be constantly in contact with the aluminum surface. Flies were allowed to recover on the aluminum sheet in a 25 °C incubator for 40 min after being anesthetized. The aluminum sheet was then placed on top of the aluminum bar and left for 40 min in the dark. A photo was taken to record the positions of the flies on the block after 40 min. We recorded fly positions and interpolated their temperatures using linear regression based on temperature-probe readings.
Statistical analyses
We replicated the temperature preference assay experiment six times. Three replicates were conducted with A4 flies in lane 1 and w 1118 flies in lane 2, and three replicates were conducted with the lane assignments reversed. We performed a nonparametric Wilcoxon rank-sum test, which does not assume a particular distribution for the data, on each of these six replicates to test for a difference in temperature preference between the two strains. These six individual tests produced P values of 2.12 × 10–10, 6.76 × 10–10, 1.89 × 10–6, 9.21 × 10–14, 1.96 × 10–6, and 1.25 × 10–24. To obtain a combined P value, we performed a meta-analysis using Fisher’s method, which gave a very low meta P value (P << 10−16).
RNAi strain construction and screening
Strain 60100 (Vienna Drosophila Resource Center) contains two attP sites at 2L: 22,019,296 (near tiptop; VIE260B) and 2L: 9,437,482 (VIE260B-2). Activation of RNAi constructs inserted into VIE260B results in ectopic activation of tiptop and phenotypes independent of the RNAi target49. PCR screening showed that KK109179 contained insertions at both sites and likely caused the lethal phenotype observed in ref. 35 (Supplementary Fig. 49). We removed the insertion at VIE260B following the crossing scheme outlined by ref. 49 and kept two of the resulting lines with insertions only at VIE260B-2 (Supplementary Fig. 49).
We generated a new p24-2 RNAi line as previously described50. We designed the RNAi construct CG33105_RNAi using the E-RNAi server (see URLs). CG33105_RNAi was the only possible construct >50 bp in length with 100% of the possible 19-mers uniquely matching p24-2. CG33105_RNAi was cloned into pKC26 and then injected into flies from strain 60100 at 250 ng/μl. We isolated transformants using Bloomington Drosophila Stock Center (BDSC) balancer stock 9325 to ensure that the RNAi construct was inserted only at VIE260B-2 using PCR54. NV-CG33105-2 and NV-CG33105-6 are derived from different transformants, but carry the same CG33105_RNAi construct. We drove RNAi expression using lines that constitutively expressed GAL4 under the control of the Act5C or αTub84B promoter (BDSC lines 4414 and 5138, respectively). Five males and five virgin driver females were allowed to cross for 9 d at 25 °C and a 12-h light/12-h dark cycle; they were then removed from the vials. F1 progeny flies were counted 19 d after crossing. The proportion of wild-type (RNAi-active) F1 flies was compared to the proportion of wild-type F1 flies from control crosses between 60,100 males and the driver strains. We confirmed presence of the p24-2 duplicate in each of these lines using PCR (Supplementary Table 12) and Sanger sequencing.
Expression analysis
Genome-wide gene expression differences between A3 and A4 larvae were analyzed as described previously30. Sequences of the genes from A3 larvae were obtained from an A3 genome assembly constructed with publicly available A3 Illumina paired-end reads. To compare the expression levels of Cyp28d1, CG7742, and Ugt86Dh gene copies, we aligned publicly available 100-bp RNA-seq reads30 to A4 mRNA sequences using Bowtie251 (with --score-min L,0,0 to ensure that only perfectly aligned unique (i.e., copy-specific) reads were kept for FPKM calculations). We adjusted transcript length by subtracting the length of regions to which no SNP-covering read aligned because only reads overlapping the SNPs could be included in FPKM calculations. For example, Cyp28d1 gene copies are distinguishable by 15 SNPs. When regions that cannot be spanned by perfectly aligned unique reads are removed from the effective transcript length, 310 bp is subtracted from the total 1,509-bp transcript length, leaving an effective transcript length of 1,199 bp. Similarly, for Ugt86Dh and CG7742, transcript lengths of 1,065 bp and 755 bp were used to calculate FPKM values, respectively. No such adjustments were made for the single-copy genes not segregating for duplications. The total number of reads aligned to the genomes was calculated based on alignment of the single-end RNA-seq reads aligned to the A4 and A3 genomes using TopHat52.
Testing for selective sweeps
We used the composite likelihood ratio (CLR) statistic of SweepFinder2 v1.0 to test for recent selective sweeps53,54. CLR values were calculated using the frequency of SNPs present in each sample over a grid with 250-bp increments. Sites were polarized using D. simulans, Drosophila yakuba, and Drosophila erecta. Invariant sites that differed from the inferred ancestral state (substitutions) were included in the analysis, thus improving power and robustness to bottlenecks53,55. The significance of the results was evaluated by comparing the CLR values to 100 coalescent neutral simulations generated using ms56. Estimates of the effective population size, neutral mutation rate, and recombination rate were taken from previous publications57. The 95% confidence intervals were computed using the largest CLR values from each neutral simulation.
Estimating frequencies of duplicate alleles
The frequency of duplicate alleles was estimated from next-generation Illumina data (Supplementary Note) by analyzing the density of divergently mapped read pairs. Reads were mapped against the release 6 ISO1 reference genome using bwa-mem48. Divergent read pairs were selected by taking the complement of paired reads in the BAM file that mapped with proper orientation, defined as pairs of reads that mapped to the same chromosome on opposite strands and were flagged by the aligner as being properly aligned with respect to each other. Duplications were called for samples that showed a clear peak and high signal-to-noise ratio in the coverage density for divergent read pairs at breakpoints surrounding genes that were found to be duplicated in the A4 sequence. The divergent read pair signals for several duplicate alleles for Cyp28d1 from various populations are shown in Supplementary Fig. 50. Samples with low genomic coverage (<10 Mb over the chromosome containing the duplication) or inferred to be identical by descent to other samples over a region containing the duplication, using estimates of homozygous coverage and identity by descent from ref. 58, were excluded from analysis. Populations were excluded from this analysis if they contained fewer than ten samples.
URLs
All codes used for variant calling and scaffolding have been deposited to GitHub (https://github.com/mahulchak). Codes used in the temperature-gradient experiment have been deposited to GitHub (https://github.com/jjemerson/TemperatureGradient). RNAi was designed using the E-RNAi server at http://www.dkfz.de/signaling/e-rnai3/.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
All single-molecule sequence data have been deposited to the NCBI Sequence Read Archive (SRA) and can be found under accession SRX2729308. The A4 scaffolded assembly has been deposited in the NCBI Assembly database under accession GCA_002300595.1. All the variant calls are provided in the supplementary files.
References
Rockman, M. V. The QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution 66, 1–17 (2012).
Eichler, E. E. et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450 (2010).
Wray, N. R. et al. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 14, 507–515 (2013).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
McCarthy, M. I. et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369 (2008).
Emerson, J. J., Cardoso-Moreira, M., Borevitz, J. O. & Long, M. Natural selection shapes genome-wide patterns of copy number polymorphism in Drosophila melanogaster. Science 320, 1629–1631 (2008).
Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011).
Anonymous. The human genome at ten. Nature 464, 649–650 (2010).
Frazer, K. A., Murray, S. S., Schork, N. J. & Topol, E. J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251 (2009).
Alkan, C., Sajjadian, S. & Eichler, E. E. Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65 (2011).
King, E. G. et al. Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 22, 1558–1566 (2012).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44, e147 (2016).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458 (2015).
dos Santos, G. et al. FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 43, D690–D697 (2015).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Khost, D. E., Eickbush, D. G. & Larracuente, A. M. Single molecule long read sequencing resolves the detailed structure of complex satellite DNA loci in Drosophila melanogaster. Preprint at bioRxiv https://doi.org/10.1101/054155 (2016).
Cridland, J. M., Macdonald, S. J., Long, A. D. & Thornton, K. R. Abundance and distribution of transposable elements in two Drosophila QTL mapping resources. Mol. Biol. Evol. 30, 2311–2327 (2013).
King, E. G., Kislukhin, G., Walters, K. N. & Long, A. D. Using Drosophila melanogaster to identify chemotherapy toxicity genes. Genetics 198, 31–43 (2014).
Stapleton, M. et al. The Drosophila gene collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 12, 1294–1300 (2002).
Cridland, J. M., Thornton, K. R. & Long, A. D. Gene expression variation in Drosophila melanogaster due to rare transposable element insertion alleles of large effect. Genetics 199, 85–93 (2015).
Swinburne, I. A. & Silver, P. A. Intron delays and transcriptional timing during development. Dev. Cell 14, 324–330 (2008).
Long, A. D., Lyman, R. F., Morgan, A. H., Langley, C. H. & Mackay, T. F. C. Both naturally occurring insertions of transposable elements and intermediate frequency polymorphisms at the achaete–scute complex are associated with variation in bristle number in Drosophila melanogaster. Genetics 154, 1255–1269 (2000).
Petrov, D. A., Fiston-Lavier, A.-S., Lipatov, M., Lenkov, K. & González, J. Population genomics of transposable elements in Drosophila melanogaster. Mol. Biol. Evol. 28, 1633–1644 (2011).
Lohmueller, K. E. et al. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am. J. Hum. Genet. 93, 1072–1086 (2013).
Kang, J., Kim, J. & Choi, K. W. Novel cytochrome P450, cyp6a17, is required for temperature preference behavior in Drosophila. PLoS One 6, e29800 (2011).
MacMillan, H. A. et al. Cold acclimation wholly reorganizes the Drosophila melanogaster transcriptome and metabolome. Sci. Rep. 6, 28999 (2016).
Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Rogers, R. L. et al. Landscape of standing variation for tandem duplications in Drosophila yakuba and Drosophila simulans. Mol. Biol. Evol. 31, 1750–1766 (2014).
Huddleston, J. & Eichler, E. E. An incomplete understanding of human genetic variation. Genetics 202, 1251–1254 (2016).
Marriage, T. N., King, E. G., Long, A. D. & Macdonald, S. J. Fine-mapping nicotine resistance loci in Drosophila using a multiparent advanced generation inter-cross population. Genetics 198, 45–57 (2014).
Glendinning, J. I. How do herbivorous insects cope with noxious secondary plant compounds in their diet? Entomol. Exp. Appl. 104, 15–25 (2002).
Chung, H. et al. Cis-regulatory elements in the Accord retrotransposon result in tissue-specific expression of the Drosophila melanogaster insecticide resistance gene Cyp6g1. Genetics 175, 1071–1077 (2007).
Pedra, J. H. F., McIntyre, L. M., Scharf, M. E. & Pittendrigh, B. R. Genome-wide transcription profile of field- and laboratory-selected dichlorodiphenyltrichloroethane (DDT)-resistant Drosophila. Proc. Natl. Acad. Sci. USA 101, 7034–7039 (2004).
modENCODE Consortium. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797 (2010).
Chen, S., Zhang, Y. E. & Long, M. New genes in Drosophila quickly become essential. Science 330, 1682–1685 (2010).
Saleem, S. et al. Drosophila melanogaster p24 trafficking proteins have vital roles in development and reproduction. Mech. Dev. 129, 177–191 (2012).
Bartoszewski, S., Luschnig, S., Desjeux, I., Grosshans, J. & Nüsslein-Volhard, C. Drosophila p24 homologues eclair and baiser are necessary for the activity of the maternally expressed Tkv receptor during early embryogenesis. Mech. Dev. 121, 1259–1273 (2004).
Stranger, B. E. et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853 (2007).
Gamazon, E. R., Nicolae, D. L. & Cox, N. J. A study of CNVs as trait-associated polymorphisms and as expression quantitative trait loci. PLoS Genet. 7, e1001292 (2011).
Berlin, K. et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630 (2015).
Ye, C., Hill, C. M., Wu, S., Ruan, J. & Ma, Z. S. DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third-generation sequencing technologies. Sci. Rep. 6, 31900 (2016).
Hoskins, R. A. et al. Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol. 3, RESEARCH0085.1–RESEARCH0085.16 (2002).
Lam, K. K., LaButti, K., Khalak, A. & Tse, D. FinisherSC: a repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 31, 3207–3209 (2015).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Quinlan, A. R. BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34 (2014).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Li, H. & Durbin, R. Fast and accurate short-read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Green, E. W., Fedele, G., Giorgini, F. & Kyriacou, C. P. A Drosophila RNAi collection is subject to dominant phenotypic effects. Nat. Methods 11, 222–223 (2014).
Dietzl, G. et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156 (2007).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Nielsen, R. et al. Genomic scans for selective sweeps using SNP data. Genome Res. 15, 1566–1575 (2005).
DeGiorgio, M., Huber, C. D., Hubisz, M. J., Hellmann, I. & Nielsen, R. SweepFinder2: increased sensitivity, robustness, and flexibility. Bioinformatics 32, 1895–1897 (2016).
Huber, C. D., DeGiorgio, M., Hellmann, I. & Nielsen, R. Detecting recent selective sweeps while controlling for mutation rate and background selection. Mol. Ecol. 25, 142–156 (2016).
Hudson, R. R. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18, 337–338 (2002).
Fiston-Lavier, A. S., Singh, N. D., Lipatov, M. & Petrov, D. A. Drosophila melanogaster recombination rate calculator. Gene 463, 18–20 (2010).
Lack, J. B. et al. The Drosophila genome nexus: a population genomic resource of 623 Drosophila melanogaster genomes, including 197 from a single ancestral range population. Genetics 199, 1229–1241 (2015).
Acknowledgements
We thank L. T. Ngo, J. Yan, and A. Yue for help with fly maintenance and SV analysis, and J. Mohammed for providing the multiple-sequence alignment of the Drosophila species group. We thank A. Carrillo, E. Azizi, D. German, and M. McHenry for assistance in assembling the temperature-gradient instrument, N. Nirale for uploading the sequencing data and assembly, B. Gaut, G. C. G. Lee, A. Long, and K. Thornton for thoughtful comments on the manuscript, and M. Long for discussion and for permission to use the RNAi data. The work was supported by US National Institutes of Health (NIH) grant R01GM123303-1 (J.J.E.), University of California, Irvine setup funds (J.J.E), National Science Foundation (NSF) Graduate Research Fellowships (DGE-1321846 to R.Z. and DGE-1144082 to N.W.V.), and NIH Genetics and Regulation Training Grant T32-GM007197 (N.W.V.). This work was made possible, in part, through access to the Genomics High-Throughput Facility Shared Resource of the Cancer Center Support Grant CA-62203 at the University of California, Irvine, and NIH shared-instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01.
Author information
Authors and Affiliations
Contributions
M.C. and J.J.E. conceived the project, designed the experiments, and wrote the paper. M.C. collected the sequencing data, assembled the A4 genome, designed the pipelines for calling SVs, and genotyped variants from genome alignment. N.W.V. conceived and performed the RNAi experiments. R.Z. performed the selective sweep analysis. R.Z. and J.J.E. conceived and analyzed CNV genotypes based on paired-end Illumina reads, and R.Z. analyzed the frequencies of Cyp6a17, Cyp28d1, and Ugt86Dh. X.Z. and M.C. measured the paralog-specific expression patterns. S.K. generated the DNA for the Bionano optical data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note:Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–50, Supplementary Tables 1, 3, 9, 10, and 12, and Supplementary Note
Supplementary Table 2
Validation summary of the genomic intervals containing SVs
Supplementary Table 4
Coordinates of the CNVs, indels, and inversions in A4 and ISO1
Supplementary Table 5
CNVs and indels for chromosome arms 2L as called by various CNV-calling software
Supplementary Table 6
Summary of putative TE introns
Supplementary Table 7
Genes mutated by non-TE indels in A4
Supplementary Table 8
Expression changes in genes in A4 with increased copy number
Supplementary Table 11
The unmerged CNV calls from A4–ISO1 genome alignment
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, M., VanKuren, N.W., Zhao, R. et al. Hidden genetic variation shapes the structure of functional elements in Drosophila . Nat Genet 50, 20–25 (2018). https://doi.org/10.1038/s41588-017-0010-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-017-0010-y
