
Abstract
Cell and gene therapies using haematopoietic stem cells (HSCs) epitomize the transformative potential of regenerative medicine. Recent clinical successes for gene therapies involving autologous HSC transplantation (HSCT) demonstrate the potential of genetic engineering in this stem cell type for curing disease. With recent advances in CRISPR gene-editing technologies, methodologies for the ex vivo expansion of HSCs and non-genotoxic conditioning protocols, the range of clinical indications for HSC-based gene therapies is expected to significantly expand. However, substantial immunological challenges need to be overcome. These include pre-existing immunity to gene-therapy reagents, immune responses to neoantigens introduced into HSCs by genetic engineering, and unique challenges associated with next-generation and off-the-shelf HSC products. By synthesizing these factors in this Review, we hope to encourage more research to address the immunological issues associated with current and next-generation HSC-based gene therapies to help realize the full potential of this field.
Similar content being viewed by others
Introduction
A rare population of bone marrow cells known as haematopoietic stem cells (HSCs) are responsible for supporting lifelong homeostasis of the blood and immune systems. HSCs achieve this through their unique abilities for both self-renewal and multipotent differentiation1,2,3. As a result, genetic mutations in HSCs (either hereditary or somatic) manifest in a wide range of disease phenotypes, some of which directly affect HSC function but the majority of which affect their differentiated progeny. Thus, as mutations in HSCs are the root cause of many genetic haematological disorders, the correction of these disease-causing mutations in HSCs can be curative.
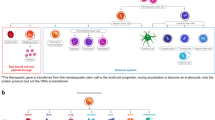
This curative potential of HSC-based therapies is exemplified by HSC transplantation (HSCT), whereby genetically healthy HSCs are transplanted into a patient to reconstitute a new, healthy haematopoietic system. Pioneered in 1957, HSCT has been widely used in the treatment of haematological malignancies4. HSCT has also been used to successfully treat a broad range of non-malignant genetic haematological disorders, such as X-linked severe combined immunodeficiency (SCID-X1), Fanconi anaemia and sickle cell disease (Table 1). For example, SCID-X1 is caused by mutations in the IL-2 receptor-γ (IL2RG) gene that prevent lymphoid progenitors (derived from HSCs) from responding to IL-2, which leads to a complete lack of B cells, T cells and natural killer cells. Transplantation of HSCs with a functional copy of IL2RG into patients with SCID-X1 leads to the generation of lymphoid progenitors that can differentiate into T cells, B cells and natural killer cells, and results in complete correction of the disease5 (Fig. 1). However, despite decades of clinical success, HSCT has long been hampered by the need to identify a source of allogeneic HSCs from a donor whose human leukocyte antigen (HLA; MHC) profile matches that of the patient to avoid rejection of the transplanted HSCs6,7,8. In addition, there is a potential for graft-versus-host-disease to occur after HSCT, a life-threatening disorder whereby alloreactive T cells from the donor attack the patient’s cells and organs7,9.

a–d | Current paradigms for gene therapy of haematopoietic stem cells (HSCs) follow a four-step process: isolation of haematopoietic stem and progenitor cells (HSPCs) from the patient, which contain a population of long-term HSCs (panel a); their ex vivo genetic engineering (for example, using retroviral transduction or CRISPR-based platforms) (panel b); genotoxic conditioning of the patient (typically using the chemotherapeutic busulfan) to create space for transplanted HSCs to engraft (panel c); and transplantation of genetically modified HSPCs back into the patient (panel d). Gene correction and transplantation of multipotent self-renewing HSCs result in the stable reconstitution of a healthy haematopoietic system within the patient.
To avoid these immunological barriers to HSCT, significant investment has been made in developing methods to genetically engineer autologous HSCs so that a patient’s own modified HSCs can be used as a genetically healthy source of cells for transplantation10 (Fig. 1). Furthermore, HSCs are a tractable model for exploring the potential of cell and gene therapies in general, owing to the relative ease with which HSCs can be isolated and transplanted. Until recently, however, ex vivo culture of functional HSCs remained challenging1, which limited the ability to maintain and genetically manipulate HSCs ex vivo. Decades of research have now resulted in the development of viral vectors and gene-editing platforms (Box 1) for ex vivo use that have been successfully implemented in clinical settings to treat genetic haematological diseases11,12,13,14 (Figs 2 and 3; Table 1).

Timeline highlighting major developments in the fields of haematopoietic stem cell (HSC) transplantation (HSCT) and gene therapy, including both major successes and major clinical challenges arising from immune responses against gene-therapy reagents4,11,36,44,140,146,147,148,149,150,151,152,153. ADA–SCID, adenosine deaminase deficiency–severe combined immunodeficiency.

Several platforms have been developed that can be used to engineer the genome of haematopoietic stem cells (HSCs). a | Lentiviral vectors allow for semi-random insertion of transgenes into the genome. b | Site-specific nucleases such as zinc finger nucleases (not shown) and the CRISPR–Cas9 system can mutate regions of the genome through creation of a double-strand break in DNA and its repair through the non-homologous end joining pathway, which creates insertions and/or deletions. In the CRISPR–Cas9 system, a single guide RNA (sgRNA) guides Cas9 to create a double-strand break at a specific target DNA sequence. c | Methods based on homology directed repair rely on the creation of a double-strand break in the genome using a site-specific nuclease (as shown in part b), followed by homology directed repair of the double-strand break using an exogenously supplied DNA donor, from an adeno-associated virus (AAV) vector or a single-stranded oligodeoxynucleotide (ssODN), that has homology to the break site. d | Next-generation gene-editing platforms (such as base editors and prime editing) allow for manipulation of the genome without use of a double-strand break. Cytidine base editors (shown) consist of a catalytically dead Cas9 fused to cytidine deaminase that is guided to the sequence of interest by sgRNA. Cytidine deamination (C → U) followed by mismatch repair can convert a G:C base pair to an A:T base pair. Adenosine base editors (not shown) convert an A:T base pair to a G:C base pair. Prime editors consist of a catalytically impaired (nickase) Cas9 fused to a reverse transcriptase, and a prime editing guide RNA (pegRNA) that contains a sgRNA sequence and a reverse transcriptase template sequence. Cas9 nickase generates a single-stranded break in target DNA, and reverse transcriptase then reverse transcribes pegRNA from the free 3′ DNA end. This initially generates a branched intermediate with the endogenous DNA strand as a 5′ flap. This is then cleaved by endogenous nuclease activity, and ligation repair incorporates the edit into the genome.
As our understanding of HSCs and genetic engineering approaches matures, novel strategies for engineering HSCs have sparked substantial interest in using HSC-based gene therapies for a wide range of diseases. For example, novel techniques for transplanting HSCs without the need for genotoxic conditioning regimens that damage DNA (Box 2) will expand the use of HSC-based gene therapy beyond the most severe life-threatening genetic diseases to become a potential long-term treatment for clinically manageable diseases such as autoimmune haemolytic anaemia15. In addition, improvements in ex vivo culture of HSCs are opening the door to more complex genetic engineering and selection protocols, as well as scalable off-the-shelf HSC products, thus expanding patient access to this therapy16,17,18. Furthermore, advances in genetic engineering should also allow for novel treatment strategies such as in vivo gene therapy of HSCs.
However, to achieve the full potential of HSC-based therapies for a broader range of disease indications, numerous immunological challenges must be addressed. Innate and adaptive immune responses to gene-therapy reagents remain a significant potential barrier to the efficacy of these platforms. Furthermore, the possibility of in vivo immune reactions to neoantigens introduced into HSCs through gene-editing procedures has yet to be addressed in clinical studies. In this Review, we synthesize recent advances in HSC-based gene therapy and highlight the immunological challenges that need to be overcome for the success of both current and next-generation technologies.
Immunity to gene-therapy reagents
The development of tools to efficiently genetically manipulate HSCs must take into consideration both intracellular signalling pathways of the innate immune system and responses of the adaptive immune system (Fig. 4). For ex vivo gene-therapy approaches (Box 1), innate immune signalling pathways within HSCs have been the primary barrier to efficient gene correction; however, as the field moves closer to realizing in vivo HSC gene therapies, adaptive immune responses against gene-therapy reagents must also be considered.

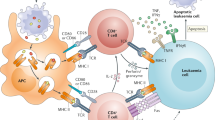
Gene therapy of haematopoietic stem cells (HSCs) is challenged by various immune barriers of both innate and adaptive immune systems. a | Innate immune pathways that detect gene-therapy reagents include double-stranded DNA (dsDNA)-sensing pathways such as those mediated by Toll-like receptor 9 (TLR9) (not shown) and the cGAS–STING pathway; DNA damage response pathways such as through the kinase ATM and p53, which allow for detection of viral episomal DNA in the nucleus; RNA-sensing pathways such as through TLR3, TLR7, TLR8 and TLR13, or through retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), that allow for detection of synthetic or viral RNA; and pathways that allow for the detection of xenogeneic contaminants that can be present when gene-editing reagents such as Cas9 are produced in xenogeneic hosts, such as lipopolysaccharide (LPS) which can be detected by TLR4. The ability of HSCs to digest proteins via the proteasome and to present peptides from these proteins on MHC class I molecules is also shown. b | The adaptive immune system can also present significant challenges to the genetic engineering of HSCs, either when genetically edited cells are transplanted back into a patient or, potentially, if genetic engineering reagents were delivered to cells in vivo. T cells can recognize cells containing foreign proteins (either neoantigens introduced into HSCs or proteins used to genetically engineer them) through interaction with antigen-presenting MHC class I molecules, which leads to T cell activation and destruction of the genetically modified cell. Antibodies produced by B cells can neutralize viral vectors or gene-therapy proteins present in the bloodstream. AAV, adeno-associated virus; IFNγ, interferon-γ; TCR, T cell receptor; TNF, tumour necrosis factor.
Innate immunity to lentiviruses
HSCs express various pattern recognition receptors (PRRs) that enable them to detect and respond to foreign pathogens or endogenous damage through the sensing of conserved pathogen-associated molecular patterns or damage-associated molecular patterns, respectively19. These PRRs include Toll-like receptors (TLRs) that sense foreign molecules at the cell membrane and in endosomes and lysosomes, retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) that detect foreign RNA, and DNA sensors such as the cGAS–STING pathway that detect foreign DNA. As a result, any gene-therapy reagents must be designed and delivered to HSCs in a way that minimizes the activation of PRR-mediated signalling pathways. These pathways and related challenges to genetic engineering have been discussed in depth elsewhere19,20, and so are only summarized briefly here.
Lentiviruses, a type of retrovirus, have made ideal vectors for HSC-based gene therapy because retroviruses have evolved numerous mechanisms to avoid innate immune detection within cells (Figs 3 and 4). For example, 5′ capping of viral RNA prevents the activation of RLR signalling, and direct nuclear import of the viral genome minimizes activation of cGAS–STING in the cytoplasm21,22. Nevertheless, lentiviruses do not completely evade intracellular immune responses. For example, the presence of lentiviral DNA in the nucleus has been shown to trigger activation of the kinase ATM, which functions in the detection of free double-stranded DNA (dsDNA) ends or short single-stranded DNA overhangs. Lentivirus-mediated activation of ATM leads to the activation of p53 in HSCs, thus resulting in the upregulation of p53 targets such as the cyclin-dependent kinase inhibitor p21 and in HSC cell cycle arrest or apoptosis, which has been shown to reduce the engraftment potential of transduced HSCs by at least 2-fold23. In addition, HSCs and other stem cells are highly refractory to lentiviral transduction, partly because they constitutively express large numbers of restriction factors compared with more differentiated cell types; for example, HSCs express high levels of the interferon-induced antiviral protein IFITM1 (ref.24). As such, a better understanding of the innate immune pathways triggered by lentiviruses and how to inhibit them will be important to developing more efficacious lentiviral gene-therapy protocols through both increased engraftment and increased transduction. For example, inhibition of p53 has been shown to increase the engraftment of lentivirus-transduced HSCs by blocking ATM-mediated signalling, and cyclosporine H has been shown to increase lentiviral transduction of HSCs by 10-fold through a reduction in IFITM1 expression23,25. Further investigation into lentiviral transduction pathways will likely yield even more promising solutions to improve the efficacy of lentivirus-mediated genetic engineering of HSCs.
Innate immunity to gene-editing enzymes
In contrast to lentivirus-based gene therapies, which take advantage of the natural life cycle of a virus to enter and genetically engineer a cell, gene editing requires the introduction of reagents into HSCs that cannot naturally penetrate them. Electroporation has emerged as a simple method of introducing foreign gene-editing reagents into HSCs11. DNA vectors are commonly used to genetically engineer cell lines, but their use in primary cells leads to the activation of PRR pathways such as the cGAS–STING pathway by dsDNA in the cytoplasm. To avoid the activation of DNA-sensing pathways in HSCs, current gene-editing methods avoid DNA components and, instead, rely on the delivery of RNA, protein or ribonucleoprotein complexes in the case of CRISPR-based platforms (Cas9 protein complexed to a synthetic single guide RNA (sgRNA)). However, the synthetic or xenogeneic origin of these reagents can lead to the activation of PRR-mediated signalling in HSCs. For example, synthetic mRNA has been shown to trigger an innate cellular immune response in HSCs, leading to the upregulation of cytokines such as interferon-γ (IFNγ). Innate immune detection of mRNA can be mitigated by incorporating pseudouridine rather than uridine, by 5-methylcytosine modification or by uridine depletion26,27,28,29. Similarly, in vitro transcribed sgRNA has been shown to cause significant innate immune responses in HSCs that can be mitigated by chemical synthesis and 2′-O-methyl 3′-phosphorothioate modification of the sgRNA30,31. The delivery of gene-editing enzymes as protein has not been shown to trigger a significant innate immune response within cells; however, stringent purification is required if the gene-editing protein is produced in a xenogeneic host, to minimize activation of TLR4 signalling by contaminants such as lipopolysaccharide (LPS)18. In addition, the potential effects of novel gene-editing platforms, such as base editors and prime editing, on innate immune pathways have yet to be addressed in HSCs32,33.
Innate immunity to DNA donors
Compared with the delivery of gene-editing enzymes, the delivery of a DNA donor to stimulate homology directed repair in HSCs has proven to be a much greater immunological challenge because naked dsDNA moieties can trigger PRR pathways such as the cGAS–STING pathway in HSCs, resulting in cell death34 (Box 1; Fig. 4). Alternative methods of delivering DNA donor templates have therefore been investigated, primarily focusing on viral vectors because they have evolved to evade intracellular innate immune responses (Fig. 3). Clinically relevant levels of homology directed repair in HSCs were first achieved through the use of non-integrating lentiviral vectors, and subsequently through the use of adeno-associated virus (AAV) vectors35,36,37. In HSCs, AAV6 has proven to be particularly effective for stimulating homology directed repair, and clinical trials using AAV6 to correct mutations in the haemoglobin-β (HBB) gene in patients with sickle cell disease are expected to begin later this year (ClinicalTrials.gov NCT04819841). However, AAV vectors have been shown to activate TLR2 and/or TLR9 signalling in cells, as well as DNA damage responses involving ATM signalling and p53 activation in HSCs (also seen with lentiviral transduction), leading to reduced HSC engraftment after viral transduction23,38,39,40. Alternative sources of DNA donor templates are single-stranded oligodeoxynucleotides (ssODNs), which have limited immunogenicity but can, currently, only deliver relatively short DNA sequences (≤200 bp)41. Further investigation into the innate immune pathways triggered by AAV vectors and ssODNs will likely result in improvements in gene-therapy efficacy.
Adaptive immune challenges
As many of the reagents that are used for genetic engineering of HSCs are of xenogeneic origin, they have the potential to be recognized by our adaptive immune system. The current paradigm of HSC gene therapy has centred on the delivery of gene-therapy reagents to autologous HSCs ex vivo, followed by their transplantation back into a patient who has received genotoxic conditioning. As these reagents are delivered ex vivo, their exposure to the adaptive immune system is minimized and adaptive immune responses to gene-therapy reagents have, thus far, not proved to be a major barrier to the clinical efficacy of ex vivo HSC gene therapies11,12,13,14. However, as the field of HSC gene therapy evolves, efforts to develop in vivo HSC gene therapies have gained significant research interest (as discussed later). For in vivo HSC gene therapy to be successful, potential adverse responses to gene-therapy reagents delivered in vivo must be carefully considered.
Delivery of gene-editing reagents in vivo, both in animal models and in humans, can lead to their neutralization in the extracellular environment by antibodies. In addition, any reagents delivered intracellularly as protein can be broken down into peptides and presented on MHC class I molecules to cytotoxic T cells, potentially leading to T cell-mediated cytotoxicity against genetically modified cells42,43,44,45 (Fig. 4). If there is no pre-existing immunity to gene-editing reagents, evidence from animal models and clinical experience of in vivo gene-therapy trials (not in HSCs) indicate that transient exposure to these reagents is not likely to result in an adverse immune response owing to the time needed to mount a robust adaptive immune response against a foreign antigen45,46,47,48. However, once an adaptive immune memory of gene-editing reagents has developed, the adaptive immune system can respond rapidly to their subsequent use44,46,49.
Pre-existing immunity has, thus far, proven to be a major challenge to the application of in vivo gene therapy in preclinical and clinical studies, leading to complete inhibition of therapeutic effect44,45,48,49. Whereas pre-existing immunity to lentiviral vectors is uncommon, pre-existing immunity to AAV vectors (present in ~30–60% of adults) and to the CRISPR–Cas9 system (present in ~60% of adults) is common in the human population owing to exposure to the pathogens from which these platforms are derived, with the frequency and level of immunogenicity depending on the reagent serotype or species of origin50,51,52,53. Even in cases where a patient has no pre-existing immunity to gene-editing reagents, exposure of the adaptive immune system to these reagents as part of the gene therapy can lead to the development of immune memory responses that prevent the possibility of re-dosing or future applications of gene-therapy reagents to the same patient44,54,55. Notably, the problem of pre-existing immunity has been observed in clinical trials of autologous ex vivo HSC gene therapy. Recent clinical trials of HSCs engineered ex vivo with VSV-G pseudo-typed lentiviral vectors detected the development of adaptive immune responses against VSV-G after HSCT in humans, indicating that the adaptive immune system can recognize and respond to gene-editing reagents in vivo even when they have been applied to cells in an ex vivo setting56. In addition, contaminants that arise during the manufacture of gene-therapy reagents can also lead to undesired adaptive immune responses. For example, lentiviral vectors can incorporate HLA molecules into their viral coat when produced in a cell line, unless an alloantigen-free cell line is used57. These contaminating HLA molecules can lead to neutralization of the vector by antibodies or clearance of transduced cells by T cells.
Immunity to HSC neoantigens
Even if genetic engineering reagents can be applied to HSCs without triggering immune responses against them, the successful transplantation and/or survival in vivo of modified HSCs presents an immunological challenge for successful gene therapy. The modification of an HSC genome, either through the correction of disease-causing mutations or by therapies that involve the introduction of transgenes, can lead to the expression of neoantigens, which introduces the risk of adaptive immune responses against genetically engineered cells and their clearance from the body (Fig. 4).
The potential for neoantigens introduced as part of gene therapy to induce adaptive immune responses against genetically engineered cells has been demonstrated in numerous preclinical studies in both HSCs and other organ systems, as well as in clinical studies (although not yet in HSC-based clinical trials)58,59,60,61,62,63,64,65. However, one unique aspect of HSC gene therapy is that lymphoablation can be carried out before HSCT to facilitate tolerance to neoantigens introduced through gene therapy; in this case, the immune system that is reconstituted from transplanted HSCs undergoes negative selection in vivo for reactivity against self-antigens, including neoantigens, to ensure central tolerance66,67,68. Nevertheless, lymphoablation has significant limitations, including the complete loss of immune memory, which makes patients transiently immunosuppressed and exposes them to opportunistic or latent infections69,70. Furthermore, lymphoablation becomes less feasible with increasing age owing to fibrosis of the thymus and a significantly reduced ability to reconstitute the immune system following lymphoablation71,72. These constraints have prompted research into non-lymphoablative genotoxic conditioning, which raises the possibility of antigen-specific immune responses against genetically modified HSCs when HSCT is carried out in the absence of lymphoablation.
Green fluorescent protein (GFP) is often used as a model antigen to study the effect of xenogene introduction into HSCs. Studies have shown the development of B cells and T cells specific for GFP following HSCT in mouse and non-human primate models after non-lymphoablative genotoxic conditioning, which ultimately led to the rejection of transplanted cells in many studies61,62,63,64. Notably, some groups have reported that transient post-transplant immunosuppression (with drugs such as rapamycin, cyclosporine and abatacept) can mitigate the development of adaptive immune responses and induce tolerance against introduced antigens, thus allowing for the introduction of xenogene-expressing HSCs without immune rejection in mice, dogs and non-human primates61,73,74. Furthermore, long-term engraftment of GFP-expressing HSCs has been achieved without any lymphoablation or immunosuppression in some studies75. These mixed findings regarding tolerance to xenogenes highlight the need for further investigation into the immune response against introduced transgenes in different settings of HSC conditioning, particularly as newer non-genotoxic methods for conditioning begin to reach clinical use (as discussed later).
Allogeneic or autologous HSCT can be curative for autoimmune diseases when carried out in the context of lymphoablation, presumably owing to lymphoablation resetting the immune memory and clearing autoreactive B cells and T cells from the body76. Furthermore, the introduction and expression of antigens in HSCs or professional antigen-presenting cells (APCs) that differentiate from HSCs has also been shown to induce tolerance in mouse models of autoimmunity, even without lymphoablation before HSCT77,78. Notably, the presentation of antigens by APCs on MHC class II molecules in the absence of inflammation has an important role in maintaining peripheral tolerance and may explain why the introduction of antigens into HSCs and their presentation by APCs that differentiate from HSCs can be tolerizing79. These studies of HSC gene therapy to treat autoimmune diseases highlight the potential for HSC gene therapy to specifically tolerize the immune system to foreign antigens and may explain some of the conflicting results regarding tolerance to xenogenes after HSCT. However, it is also worth noting that genotoxic conditioning has been shown to induce significant inflammation, which may prime the immune system to reject transplanted cells that express xenogenes80,81,82. This may help explain why the use of immunosuppression aids in the induction of tolerance to xenogenes introduced into HSCs, as it provides time for inflammation to reduce after HSCT and for APCs expressing xenogenes to differentiate from the transplanted HSCs. However, a clear answer regarding immunity or tolerance to xenogenes expressed by HSCs in vivo requires further investigation.
Compared with xenogenes, the risk of adaptive immune responses against endogenous genes that are reintroduced (via gene correction) into HSCs is lower61,83,84. This is likely owing to the healthy endogenous gene product and the mutated gene product having identical or mostly identical antigens. However, the more prominent the mutation in the endogenous gene, the stronger the adaptive immune response to the corrected gene product is expected to be. For example, the prevalence of adaptive immune responses against factor VIII, which is used in enzyme replacement therapy (ERT) by intravenous infusion for patients with haemophilia, has been shown to correlate strongly with the location of genetic mutations in the patients being treated; patients with genetic mutations leading to complete abrogation of factor VIII expression are seven to ten times more likely to develop inhibitory antibodies to ERT than are patients with milder genetic defects (such as small deletions or splice site mutations)85. Notably, patients often develop adaptive immune responses to proteins given through ERT, such as the lysosomal enzyme α-l-iduronidase (IDUA) that is used to treat mucopolysaccharidosis type 1 (MPS1)85,86. Pre-existing adaptive immunity to IDUA (owing to ERT) has led to the clearance of genetically modified HSCs in mouse and dog models of MPS1, and lymphoablation is the only known method to ensure engraftment of HSCs in the setting of pre-existing immunity against an antigen introduced as part of HSC gene therapy83,87.
Fortunately, no adverse immune reactions to neoantigens have been reported so far in clinical trials of HSC gene therapy. However, clinical application of HSC gene therapy has, thus far, centred on the correction of immunodeficiencies, metabolic disorders or β-haemoglobinopathies, in which patients have weakened immune systems or have received a functional copy of an endogenous gene. Furthermore, in cases where patients have pre-existing immunity to the introduced transgene, such as in patients with MPS1 owing to prior ERT, chemotherapeutic lymphoablation has been included as part of the conditioning regimen to prevent undesired immune responses against introduced transgenes88. As the applications of gene therapy expand beyond the most life-threatening diseases, the potential of immune responses against introduced neoantigens must be carefully considered to ensure the successful engraftment of genetically engineered HSCs. Novel methods to avoid neoantigen presentation by APCs or to induce neoantigen-specific tolerance should therefore be investigated. The ability of HSC gene therapy itself to induce tolerance to antigens through transient post-transplant immunosuppression or through restricting expression of neoantigens to APCs offers one attractive approach to ensure tolerance without the use of lymphoablation.
Immunity to next-generation therapies
Having successfully used HSC gene therapy to clinically treat multiple diseases (Table 1), it is now possible to envision new ways in which HSCs may be genetically engineered beyond the correction of severe life-threatening mutations. One area that has exceptional potential is the introduction of new immune functions into the haematopoietic system. For example, work is currently underway to use HSC gene therapy to confer resistance to HIV-1 infection through knockout of CCR5 (a co-receptor for HIV-1) in the HSC genome and by the introduction of restriction factors, small interfering RNAs or the CRISPR–Cas13 system (to inhibit the viral life cycle directly or to knockdown genes involved in HIV-1 replication)89,90,91. As these approaches become more refined, their application may be expanded to other infections that are major causes of disease burden such as Epstein–Barr virus, cytomegalovirus or malaria. HSC gene therapy has also been suggested as a way to improve the safety of chimeric antigen receptor T cell (CAR T cell) therapies for leukaemia. By knocking out the antigen that is targeted by CAR T cells (such as CD33) within donor HSCs, unwanted ablation of donor HSCs by CAR T cells can be prevented92.
Other future modifications of HSCs could include insertion of sequences encoding pathogen-specific antibodies, T cell receptors (TCRs) or CARs as part of gene therapies to create HSC-based vaccines. By introducing desired pathogen-specific sequences into HSCs, mature immune cells that differentiate from them may provide lifelong protection from disease without the need for immunization93,94,95,96. Furthermore, recent epidemics of SARS-CoV, Ebola virus and SARS-CoV-2, which have proved highly dangerous in human populations, have been shown to infect members of the Chiroptera order (bats) without significant pathogenicity97; therefore, a better understanding of the immune systems of other species may offer the potential to re-engineer our own immune system through HSC gene therapy to provide greater protection from infection and to improve human health generally.
Looking beyond the current HSC gene-therapy pipeline, recent advances in our ability to expand HSCs ex vivo, carry out non-genotoxic conditioning for HSCT and deliver gene-editing enzymes in vivo now offer the potential to develop novel HSC gene therapy paradigms in a more scalable, affordable and safe manner. However, the successful development and clinical implementation of these next-generation HSC gene-therapy platforms requires overcoming several immunological barriers, as discussed below.
Off-the-shelf HSC gene therapy
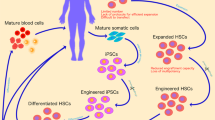
With advances in human HSC expansion protocols and genetic engineering, a single genetically engineered HSC population may eventually be mass-produced for use in off-the-shelf HSC therapies for numerous patients18,98,99,100. In addition, significant research efforts are currently underway to differentiate HSCs from human pluripotent stem cells or to trans-differentiate other haematopoietic cell types into HSCs101,102,103. Although methods to expand and/or generate HSCs ex vivo will markedly improve the availability of HSCs, the immunological challenges associated with transplantation of these cells have yet to be fully addressed.
In order for off-the-shelf allogeneic HSC gene therapy to be successful, HSCs must be engrafted without rejection by the recipient’s immune system. Any given off-the-shelf HSC product derived from a pluripotent stem cell or human donor is highly unlikely to be a complete immunological match to the patient, owing to the highly variable nature of HLA alleles (with more than 15,000 different known HLA alleles present in the human population)6,104,105. One potential solution would be to genetically engineer ex vivo-expanded HSCs to be a complete match for any patient they are transplanted into through the knockout of mismatched HLA alleles; however, this would need to be carried out on a patient by patient basis106. Instead, a more feasible and universal approach is to genetically modify HSCs to become hypoimmune, such that they are not recognized by the immune system. Notably, the creation of hypoimmune cells has been demonstrated in pluripotent stem cells through knockout of the β2-microglobulin locus (a component of MHC class I molecules) and knock in of either a synthetic HLA-E molecule or overexpression of the anti-phagocytic signal CD47 (refs107,108). Knockout of β2-microglobulin prevents the presentation of allogeneic HLA molecules on the cell surface, thus preventing the development of antibodies to HLA and T cell-mediated lysis of cells expressing mismatched HLA; and expression of a synthetic HLA-E molecule or CD47 prevents attack by natural killer cells.
Genetically engineered hypoimmune mouse cells have proven successful for allogeneic transplantation of embryonic stem cells and cardiovascular cells between MHC-mismatched hosts without rejection107,109. However, these approaches have not yet been applied to HSCT. MHC (HLA) molecules have important roles in the development and functions of immune cells, and their lack of expression through knockout approaches may have significant detrimental effects on the overall function of the immune system110,111,112. Furthermore, the overexpression of negative regulators of immune activation such as CD47 could have serious detrimental effects on overall haematopoiesis, such as by inhibiting the normal turnover of red blood cells113. Therefore, the generation of clinically translatable hypoimmune HSCs remains an open challenge.
Off-the-shelf HSCs from allogeneic donors would ideally be transplanted without any need for lymphoablation, which leads to loss of immune memory, or for the modification of HLA loci, which affects antigen presentation. One promising solution could be the induction of tolerance to foreign HLA genes using regulatory T cells or CAR regulatory T cells, for which clinical trials are currently underway108,114,115,116. Alternatively, the liver has been shown to be a highly tolerogenic environment and targeted overexpression of both xenogenes and allogeneic HLAs in this environment (through the use of liver-specific promoters and microRNAs to prevent expression in off-target tissues) has been shown to induce peripheral tolerance to them117,118,119. It is thus possible that these approaches could be applied to facilitate the transplantation of allogeneic HLA-mismatched HSCs into patients without additional negative modification of the immune system.
Non-genotoxic conditioning for HSCT
Current approaches to HSC gene therapy generally rely on use of the chemotherapeutic busulfan, an alkylating agent that creates DNA cross links to induce genotoxic stress. HSCs are significantly more sensitive to such genotoxic stress as compared with other cell types, such as cells of the adaptive immune system. Busulfan therefore clears endogenous HSCs from the HSC niche so that space is made available for transplanted genetically modified HSCs to engraft, without any lymphoablation120. However, the use of such genotoxic conditioning is also associated with significant toxicities to patients including infertility, organ toxicity and risk of secondary malignancy, thus limiting the application of HSC gene therapy to only the most severe forms of genetic disease121. The negative side effects of genotoxic conditioning regimens have spurred significant investigation into alternative methods to facilitate the engraftment of transplanted HSCs into a patient.
Many of these efforts have centred on clearing endogenous HSCs from the niche through the use of antibody-based drugs that target cell surface antigens restricted in expression primarily to HSCs and their early progenitors. Of the various antibodies developed thus far, those targeting CD117 have generated the most interest. Anti-CD117 conditioning has now reached the clinic for allogeneic HSCT, but has not yet been used for genetically engineered HSCs122. Several optimized strategies to clear endogenous HSC niches involving anti-CD117 have been developed, including a combined anti-CD117 and anti-CD47 approach to facilitate macrophage-mediated phagocytosis; anti-CD117 conjugated with the protein synthesis inhibitor saporin to facilitate drug-mediated cytotoxicity; and CAR T cells that target CD117-expressing cells to facilitate T cell-mediated cytotoxicity15,123,124. As these approaches specifically deplete HSCs, they should allow for transplantation of genetically engineered HSCs without harm to the immune system.
More recently, the use of antibody-mediated conditioning for HSCT has been further refined to facilitate antibody-mediated lymphoablation through a combination of antibodies to CD117, CD47, CD4, CD8, CD40L and CD122, thus allowing for transplantation of HSCs from completely immunologically mismatched donors without graft rejection125. There is exciting potential synergy here with the off-the-shelf allogeneic HSCs described above. However, it will be important to further understand how the use of such non-genotoxic methods alters the immune response to transplanted HSCs and how immune memory is altered following antibody-mediated immune depletion.
Alternative non-genotoxic conditioning strategies have also been proposed. For example, our group has shown in mice that depletion of the amino acid valine leads to loss of HSCs in the bone marrow and facilitates high levels of engraftment of transplanted HSCs126. Although complete removal of valine from the diet may not be translatable to clinical settings, this study highlights that further investigation into HSC metabolism may open up new avenues for non-genotoxic conditioning for HSCT. Furthermore, recent studies have also shown that robust (>10%) engraftment of HSCs without any form of conditioning is feasible when supraphysiological numbers of HSCs (>400% of the total number of HSCs) are transplanted intravenously into mice18,127,128.
Proof of concept for non-conditioned HSC gene therapies has also recently been demonstrated; a clinical trial found that gene-corrected HSCs could engraft without conditioning in patients with Fanconi anaemia, albeit at low levels129. These patients have defects in DNA damage repair, which leads to progressive bone marrow failure. It is thought that this allows for HSC engraftment without conditioning and that gene therapy confers a strong selective advantage to the corrected HSCs following engraftment. Unfortunately, this approach is currently difficult to replicate in other disease settings where gene correction does not provide such a strong selective advantage without boosting the number of donor HSCs. Towards this goal, our laboratory has successfully demonstrated a protocol allowing for 900-fold ex vivo expansion of mouse HSCs over the course of a month and has shown that these ex vivo-expanded HSCs are amenable to non-conditioned transplantation as well as genetic engineering18,127,130. Further investigation into the ex vivo expansion of human HSCs will open up the possibility of non-conditioned transplants in clinical settings. However, questions remain to be addressed regarding the clonality of engrafted cells, the location of HSC engraftment, the durability of engraftment and the potential for adverse events owing to low levels of engraftment of corrected cells in different disease settings. Non-conditioned transplantation of haematopoietic stem and progenitor cells (HSPCs) has been shown to cause leukaemia in mouse models of SCID-X1 owing to the replicative stress that progenitors face following low levels of HSC engraftment or progenitor-only reconstitution. These studies demonstrate the potential for unexpected adverse events when carrying out non-conditioned HSCT in a clinical setting; of note, however, sub-physiological numbers of HSPCs were transplanted into mice in these studies, leading to low or no long-term engraftment in the bone marrow29,131.
Further investigations are warranted into how the type of conditioning regimen determines the immune response in autologous HSC gene therapies, as the level of innate and adaptive immune activity in the patient may affect the extent to which the neoantigen-expressing HSC is tolerated or rejected. For example, myeloablative conditioning regimens such as the use of total body irradiation or busulfan are highly genotoxic and induce significant amounts of inflammation, thus creating conditions that are more likely to induce immune reactivity to neoantigens rather than tolerance80,81,82. By contrast, non-genotoxic conditioning regimens induce far less cell death, which should reduce the level of inflammation in the HSC bone marrow microenvironment. Therefore, transplantation of neoantigen-expressing HSCs into a non-inflammatory environment may be more amenable for inducing tolerance to neoantigens than transplantation into a traditional myeloablative conditioned environment, although this remains to be investigated.
In vivo gene therapy of HSCs
As an alternative to transplanting gene-edited HSCs into a patient, it may be possible to genetically engineer HSCs in vivo. In vivo HSC gene therapies would avoid the need for HSC isolation, editing and HSCT, and could revolutionize how we treat and cure haematological diseases. Although in vivo gene therapy has been well developed for targeting other organ systems such as the liver, muscle and the eye132,133, its application to HSCs has been relatively underdeveloped owing to the existence of established protocols for ex vivo HSC gene therapy; thus, the best evidence we have regarding potential immune complications comes from these other systems. Currently, two major approaches have been suggested for in vivo gene therapy: use of viral vectors designed to express the desired genes or gene-editing reagents in cells in vivo; or delivery of mRNA or protein encapsulated in nanoparticles to introduce gene-editing reagents into cells in vivo.
Viral-vectored gene therapy is the most clinically well developed of the platforms for in vivo gene therapy and has proved efficacious in the treatment of genetic diseases of the eye and liver132,133. Several viral platforms (including adenoviral, AAV and lentiviral vectors) can transduce HSCs in vivo at varying levels of efficacy16,134,135. Adenoviral vectors have been used to successfully transduce HSCs in vivo and provide phenotypic improvement of disease in mouse models of sickle cell disease16. AAV vectors have also been shown to be capable of gene correction in vivo at curative rates in mouse HSCs (5–40% of HSCs being corrected)134.
In contrast to viral vectors, which often reside within cells long term and use potentially oncogenic promoters to drive transgene expression, the use of nanoparticles to deliver gene-editing reagents to HSCs as mRNA or protein is an attractive alternative because the transient expression of these reagents reduces the risk of oncogenesis. For example, lipid nanoparticles containing mRNA or protein have proven highly effective for gene editing of liver cells in vivo both in animal studies and in human clinical trials, and recent preclinical data suggest that human HSCs can also be targeted by nanoparticles in vivo, at least within immunodeficient NSG mice (NOD-scid Il2rgnull mice)17,136. In addition, several groups have also been working on delivery of the CRISPR–Cas9 system in vivo as a ribonucleoprotein by adding multiple nuclear localization sequences to the Cas9 protein or through the use of cell-penetrating peptides137,138.
However, the successful implementation of in vivo gene therapy is complicated by the potential for innate and adaptive immune responses against gene-therapy reagents (see earlier discussion of these issues). For example, adenoviral vectors and AAV vectors have previously led to the deaths of patients in clinical trials owing to systemic innate inflammatory responses and innate immune toxicity, respectively139,140. In addition, as described above, the delivery of gene-editing reagents in vivo can lead to undesired adaptive immune responses against gene-therapy reagents (such as neutralizing antibodies or T cell-mediated toxicity of transduced cells), which can negate any therapeutic effect44,46 (Fig. 4). Notably, this problem persists even for nanoparticle-based vectors. For example, in a macaque monkey model, the use of base editors in the liver was shown not to improve gene-editing efficiency upon re-dosing, despite their transient delivery as mRNA using lipid nanoparticles, presumably owing to the development of antigen-specific T cells following the primary dose and clearance of transduced cells by these T cells following the second dose141. These investigations into viral and nanoparticle-based genetic engineering platforms highlight the challenge that the adaptive immune system poses to the delivery of gene-editing reagents in vivo.
Furthermore, whereas ex vivo gene therapy allows for specific targeting of HSCs through their isolation from the rest of the body, delivery of genetic engineering reagents to HSCs in vivo introduces the risk of undesirable off-target tissue transduction. This off-target transduction can increase the risk of potential complications from innate immune toxicity and could lead to undesired genetic modifications of off-target cells that may be detrimental to patient health. In addition, off-target gene expression has been shown to impact the risk of adaptive immune responses against gene-therapy reagents and transgenes, as different tissues vary in their inflammatory environment. For example, successful long-term transgene expression in the liver from lentiviral vectors delivered in vivo has required the use of cell type-specific promoters and microRNAs to restrict gene expression to on-target tissues142. These were required to prevent undesired adaptive immune responses against the introduced transgene and resulting elimination of transduced cells by antigen-specific T cells.
Conclusions
It is an exciting time in the field of HSC gene therapy. Recent clinical successes have highlighted the transformative potential of HSC gene therapy in disease treatment. As technical challenges to the development of efficacious HSC gene therapies are resolved, new challenges to the implementation of the next generation of HSC gene therapies present themselves. The current paradigm of ex vivo autologous HSC gene therapy is highly personalized and extremely technically challenging to implement, thus making HSC gene therapy extremely expensive. This is exemplified by betibeglogene autotemcel (brand name Zynteglo) gene therapy for β-thalassaemia, which despite achieving clinical approval has been pulled from European markets owing to its cost of US $1.8 million per patient143. For some diseases, however, the high cost of a curative HSC gene therapy may, ultimately, prove to be less than the total costs of chronic use of conventional therapies for the disease across a patient’s lifetime. Unfortunately, for some of the major diseases for which HSC gene therapies are being developed, such as sickle cell disease and HIV-1, the majority of patients live in low-income countries and will likely not have access to these therapies unless the economic issues regarding their implementation are resolved. As autologous HSC gene therapy becomes more streamlined and closed loop, automated systems reduce its technical challenges as well as cost, and we hope that these economic challenges to autologous HSC gene therapy may be overcome. Alternatively, the development of next-generation, scalable off-the-shelf allogeneic or in vivo HSC gene therapies may offer alternative approaches to address these economic challenges, if the immunological challenges to their implementation can be addressed.
As the field of HSC gene therapy advances, challenges from both innate immunity and adaptive immunity have arisen. These challenges must be overcome to apply current and next-generation HSC gene therapies successfully and safely. We believe that in the near future, the progress of HSC gene therapy will be defined by our ability to address these immunological challenges. The adaptive immune system, in particular, poses a major challenge to the next generation of HSC gene therapies as it can lead to rejection of neoantigens introduced into HSCs, genetic engineering reagents delivered in vivo to HSCs or allogeneic off-the-shelf HSCs. The haematopoietic system is unique in that lymphoablation offers one potential avenue to address most of these challenges; however, lymphoablation has significant drawbacks in terms of patient health and other avenues to address these challenges should also be pursued. Some potential solutions to tolerize the immune system to neoantigens are engineering cells to become hypoimmune, as well as using HSC-derived APCs to tolerize the immune system. However, none of these strategies has been tested clinically and more research to incorporate these methods as part of HSC gene therapy is required. By synthesizing these immunological challenges within this Review, we hope to encourage more immunologists to investigate and address the immunological issues in current and next-generation HSC gene therapies in order to realize the full potential of this approach for the improvement of human health.
References
Wilkinson, A. C., Igarashi, K. J. & Nakauchi, H. Haematopoietic stem cell self-renewal in vivo and ex vivo. Nat. Rev. Genet. 21, 541–554 (2020).
Eaves, C. J. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood 125, 2605–2613 (2015).
Morrison, S. J. & Scadden, D. T. The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014).
Thomas, E. D., Lochte, H. L. Jr., Lu, W. C. & Ferrebee, J. W. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N. Engl. J. Med. 257, 491–496 (1957).
Wahlstrom, J. T., Dvorak, C. C. & Cowan, M. J. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Curr. Pediatr. Rep. 3, 1–10 (2015).
Tiercy, J. M. How to select the best available related or unrelated donor of hematopoietic stem cells? Haematologica 101, 680–687 (2016).
Granot, N. & Storb, R. History of hematopoietic cell transplantation: challenges and progress. Haematologica 105, 2716–2729 (2020).
Gragert, L. et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N. Engl. J. Med. 371, 339–348 (2014).
Hill, G. R., Betts, B. C., Tkachev, V., Kean, L. S. & Blazar, B. R. Current concepts and advances in graft-versus-host disease immunology. Annu. Rev. Immunol. 39, 19–49 (2021).
Morgan, R. A., Gray, D., Lomova, A. & Kohn, D. B. Hematopoietic stem cell gene therapy: progress and lessons learned. Cell Stem Cell 21, 574–590 (2017).
Frangoul, H. et al. CRISPR–Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2021). This work is the first clinical application of the CRISPR–Cas9 system for ex vivo HSC gene therapy.
Thompson, A. A. et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N. Engl. J. Med. 378, 1479–1493 (2018). This paper reports clinical trial results from lentiviral-based gene therapy for β-thalassaemia.
Boztug, K. et al. Stem-cell gene therapy for the Wiskott–Aldrich syndrome. N. Engl. J. Med. 363, 1918–1927 (2010).
Aiuti, A. et al. Correction of ADA–SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296, 2410–2413 (2002).
Czechowicz, A. et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 10, 617 (2019).
Li, C. et al. In vivo HSC gene therapy using a bi-modular HDAd5/35++ vector cures sickle cell disease in a mouse model. Mol. Ther. 29, 822–837 (2021).
Burns, S. In vivo gene editing of hematopoietic stem and progenitor cells. Intellia https://3o5c4w3neipl16yvhj3nfqam-wpengine.netdna-ssl.com/wp-content/uploads/Keystone_2021_BoneMarrow_10Mar2021-1.pdf (2021). This preclinical data release from Intellia demonstrates in vivo editing of human HSCs in xenografted mice.
Wilkinson, A. C. et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 571, 117–121 (2019). This paper is the first to show robust (>100-fold) expansion of HSCs ex vivo.
Piras, F. & Kajaste-Rudnitski, A. Antiviral immunity and nucleic acid sensing in haematopoietic stem cell gene engineering. Gene Ther. 28, 16–28 (2021).
Dudek, A. M. & Porteus, M. H. Answered and unanswered questions in early-stage viral vector transduction biology and innate primary cell toxicity for ex-vivo gene editing. Front. Immunol. 12, 660302 (2021).
Rasaiyaah, J. et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405 (2013).
Chiu, Y. L. et al. Tat stimulates cotranscriptional capping of HIV mRNA. Mol. Cell 10, 585–597 (2002).
Piras, F. et al. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 9, 1198–1211 (2017).
Wu, X. et al. Intrinsic immunity shapes viral resistance of stem cells. Cell 172, 423–438.e25 (2018).
Petrillo, C. et al. Cyclosporine H overcomes innate immune restrictions to improve lentiviral transduction and gene editing in human hematopoietic stem cells. Cell Stem Cell 23, 820–832.e9 (2018).
Vaidyanathan, S. et al. Uridine depletion and chemical modification increase Cas9 mRNA activity and reduce immunogenicity without HPLC purification. Mol. Ther. Nucleic Acids 12, 530–542 (2018).
Dowdy, S. F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 35, 222–229 (2017).
Kariko, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Schiroli, G. et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 9, eaan0820 (2017).
Wienert, B., Shin, J., Zelin, E., Pestal, K. & Corn, J. E. In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLoS Biol. 16, e2005840 (2018).
Hendel, A. et al. Chemically modified guide RNAs enhance CRISPR–Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015). This paper describes the development of chemically modified guides that allow for the first demonstration of CRISPR–Cas9 gene editing in human HSCs.
Zeng, J. et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 26, 535–541 (2020).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). This paper describes the first demonstration of prime editing, a flexible platform for making gene edits without the use of a double-strand break.
Liao, W., Du, C. & Wang, J. The cGAS–STING pathway in hematopoiesis and its physiopathological significance. Front. Immunol. 11, 573915 (2020).
Dever, D. P. et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016).
Genovese, P. et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510, 235–240 (2014). This work is the first demonstration of gene editing mediated by homology directed repair in HSCs; innate immune signalling of HSCs was overcome using integration-deficient lentiviral vectors.
Lombardo, A. et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 25, 1298–1306 (2007).
Schiroli, G. et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell 24, 551–565.e8 (2019).
Hosel, M. et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 55, 287–297 (2012).
Zhu, J., Huang, X. & Yang, Y. The TLR9–MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest. 119, 2388–2398 (2009).
DeWitt, M. A. et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 8, 360ra134 (2016).
Pien, G. C. et al. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J. Clin. Invest. 119, 1688–1695 (2009).
Li, C. et al. Cytotoxic-T-lymphocyte-mediated elimination of target cells transduced with engineered adeno-associated virus type 2 vector in vivo. J. Virol. 83, 6817–6824 (2009).
Manno, C. S. et al. Successful transduction of liver in hemophilia by AAV–factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347 (2006). This paper reports adaptive immune responses to gene-therapy reagents as a major barrier to efficacy in clinical trials.
Meadows, A. S., Pineda, R. J., Goodchild, L., Bobo, T. A. & Fu, H. Threshold for pre-existing antibody levels limiting transduction efficiency of systemic rAAV9 gene delivery: relevance for translation. Mol. Ther. Methods Clin. Dev. 13, 453–462 (2019).
Li, A. et al. AAV–CRISPR gene editing is negated by pre-existing immunity to Cas9. Mol. Ther. 28, 1432–1441 (2020).
Long, B. R. et al. The impact of pre-existing immunity on the non-clinical pharmacodynamics of AAV5-based gene therapy. Mol. Ther. Methods Clin. Dev. 13, 440–452 (2019).
Mingozzi, F. & High, K. A. Overcoming the host immune response to adeno-associated virus gene delivery vectors: the race between clearance, tolerance, neutralization, and escape. Annu. Rev. Virol. 4, 511–534 (2017).
Mingozzi, F. et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422 (2007).
Wagner, D. L. et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 25, 242–248 (2019).
Charlesworth, C. T. et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 25, 249–254 (2019).
Boutin, S. et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 21, 704–712 (2010).
Shirley, J. L., de Jong, Y. P., Terhorst, C. & Herzog, R. W. Immune responses to viral gene therapy vectors. Mol. Ther. 28, 709–722 (2020).
George, L. A. et al. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol. Ther. 28, 2073–2082 (2020).
Chew, W. L. et al. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 13, 868–874 (2016).
Rust, B. J. et al. Envelope-specific adaptive immunity following transplantation of hematopoietic stem cells modified with VSV-G lentivirus. Mol. Ther. Methods Clin. Dev. 19, 438–446 (2020).
Milani, M. et al. Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol. Med. 9, 1558–1573 (2017).
Calcedo, R. et al. Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for α-1-antitrypsin deficiency. Proc. Natl Acad. Sci. USA 114, 1655–1659 (2017).
Mendell, J. R. et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 363, 1429–1437 (2010).
Riddell, S. R. et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat. Med. 2, 216–223 (1996). This paper reports the first clinical trial in which gene-modified cells are cleared from the body owing to an adaptive immune response against the introduced transgene.
Uchida, N. et al. Busulfan combined with immunosuppression allows efficient engraftment of gene-modified cells in a rhesus macaque model. Mol. Ther. 27, 1586–1596 (2019).
Uchida, N. et al. Evaluation of engraftment and immunological tolerance after reduced intensity conditioning in a rhesus hematopoietic stem cell gene therapy model. Gene Ther. 21, 148–157 (2014).
Bubnic, S. J., Nagy, A. & Keating, A. Donor hematopoietic cells from transgenic mice that express GFP are immunogenic in immunocompetent recipients. Hematology 10, 289–295 (2005).
Rosenzweig, M. et al. Induction of cytotoxic T lymphocyte and antibody responses to enhanced green fluorescent protein following transplantation of transduced CD34+ hematopoietic cells. Blood 97, 1951–1959 (2001).
Drysdale, C. M., Tisdale, J. F. & Uchida, N. Immunoresponse to gene-modified hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 16, 42–49 (2020).
Uchida, N. et al. Total body irradiation must be delivered at high dose for efficient engraftment and tolerance in a rhesus stem cell gene therapy model. Mol. Ther. Methods Clin. Dev. 3, 16059 (2016).
Kung, S. K. et al. Induction of transgene-specific immunological tolerance in myeloablated nonhuman primates using lentivirally transduced CD34+ progenitor cells. Mol. Ther. 8, 981–991 (2003).
Manilay, J. O., Pearson, D. A., Sergio, J. J., Swenson, K. G. & Sykes, M. Intrathymic deletion of alloreactive T cells in mixed bone marrow chimeras prepared with a nonmyeloablative conditioning regimen. Transplantation 66, 96–102 (1998).
Styczynski, J. et al. Death after hematopoietic stem cell transplantation: changes over calendar year time, infections and associated factors. Bone Marrow Transpl. 55, 126–136 (2020).
Ogonek, J. et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 7, 507 (2016).
Mikhael, N. L. & Elsorady, M. Clinical significance of T cell receptor excision circle (TREC) quantitation after allogenic HSCT. Blood Res. 54, 274–281 (2019).
Hakim, F. T. et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J. Clin. Invest. 115, 930–939 (2005).
Bhatt, K. H. et al. Short-course rapamycin treatment enables engraftment of immunogenic gene-engineered bone marrow under low-dose irradiation to permit long-term immunological tolerance. Stem Cell. Res. Ther. 8, 57 (2017).
Beagles, K. E., Peterson, L., Zhang, X., Morris, J. & Kiem, H. P. Cyclosporine inhibits the development of green fluorescent protein (GFP)-specific immune responses after transplantation of GFP-expressing hematopoietic repopulating cells in dogs. Hum. Gene Ther. 16, 725–733 (2005).
Tarantal, A. F. et al. Nonmyeloablative conditioning regimen to increase engraftment of gene-modified hematopoietic stem cells in young rhesus monkeys. Mol. Ther. 20, 1033–1045 (2012).
Swart, J. F. et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat. Rev. Rheumatol. 13, 244–256 (2017).
Sack, B. K., Herzog, R. W., Terhorst, C. & Markusic, D. M. Development of gene transfer for induction of antigen-specific tolerance. Mol. Ther. Methods Clin. Dev. 1, 14013 (2014).
Coleman, M. A. et al. Tolerance induction with gene-modified stem cells and immune-preserving conditioning in primed mice: restricting antigen to differentiated antigen-presenting cells permits efficacy. Blood 121, 1049–1058 (2013).
Furlan, R. A tolerizing mRNA vaccine against autoimmunity? Mol. Ther. 29, 896–897 (2021).
Weischendorff, S. et al. Reduced plasma amino acid levels during allogeneic hematopoietic stem cell transplantation are associated with systemic inflammation and treatment-related complications. Biol. Blood Marrow Transpl. 25, 1432–1440 (2019).
De La Serna, J. et al. Toxicity and efficacy of busulfan and fludarabine myeloablative conditioning for HLA-identical sibling allogeneic hematopoietic cell transplantation in AML and MDS. Bone Marrow Transplant. 51, 961–966 (2016).
Xun, C. Q., Thompson, J. S., Jennings, C. D., Brown, S. A. & Widmer, M. B. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood 83, 2360–2367 (1994).
Squeri, G. et al. Targeting a pre-existing anti-transgene T cell response for effective gene therapy of MPS-I in the mouse model of the disease. Mol. Ther. 27, 1215–1227 (2019).
Cavazzana-Calvo, M. et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467, 318–322 (2010).
Gouw, S. C. et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood 119, 2922–2934 (2012).
Ponder, K. P. Immune response hinders therapy for lysosomal storage diseases. J. Clin. Invest. 118, 2686–2689 (2008).
Lutzko, C. et al. Genetically corrected autologous stem cells engraft, but host immune responses limit their utility in canine α-l-iduronidase deficiency. Blood 93, 1895–1905 (1999).
Gentner, B. et al. Hematopoietic stem- and progenitor-cell gene therapy for Hurler syndrome. N. Engl. J. Med. 385, 1929–1940 (2021).
Yin, L. et al. CRISPR–Cas13a inhibits HIV-1 infection. Mol. Ther. Nucleic Acids 21, 147–155 (2020).
Xu, L. et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N. Engl. J. Med. 381, 1240–1247 (2019).
Burke, B. P. et al. Engineering cellular resistance to HIV-1 infection in vivo using a dual therapeutic lentiviral vector. Mol. Ther. Nucleic Acids 4, e236 (2015).
Kim, M. Y. et al. Genetic inactivation of CD33 in hematopoietic stem cells to tnable CAR T cell immunotherapy for acute myeloid leukemia. Cell 173, 1439–1453.e19 (2018).
Voss, J. E. et al. Reprogramming the antigen specificity of B cells using genome-editing technologies. eLife 8, e42995 (2019).
Greiner, V. et al. CRISPR-mediated editing of the B cell receptor in primary human B cells. iScience 12, 369–378 (2019).
De Oliveira, S. N. et al. Modification of hematopoietic stem/progenitor cells with CD19-specific chimeric antigen receptors as a novel approach for cancer immunotherapy. Hum. Gene Ther. 24, 824–839 (2013).
Vatakis, D. N. et al. Antitumor activity from antigen-specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc. Natl Acad. Sci. USA 108, E1408–E1416 (2011).
Irving, A. T., Ahn, M., Goh, G., Anderson, D. E. & Wang, L. F. Lessons from the host defences of bats, a unique viral reservoir. Nature 589, 363–370 (2021).
Bai, T. et al. Expansion of primitive human hematopoietic stem cells by culture in a zwitterionic hydrogel. Nat. Med. 25, 1566–1575 (2019).
Fares, I. et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 345, 1509–1512 (2014).
Boitano, A. E. et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329, 1345–1348 (2010).
Demirci, S., Leonard, A. & Tisdale, J. F. Hematopoietic stem cells from pluripotent stem cells: clinical potential, challenges, and future perspectives. Stem Cell Transl. Med. 9, 1549–1557 (2020).
Sandler, V. M. et al. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature 511, 312–318 (2014).
Riddell, J. et al. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell 157, 549–564 (2014). This paper is the first to demonstrate trans-differentiation of cells to HSCs.
Dendrou, C. A., Petersen, J., Rossjohn, J. & Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 18, 325–339 (2018).
Robinson, J., Soormally, A. R., Hayhurst, J. D. & Marsh, S. G. E. The IPD-IMGT/HLA Database — new developments in reporting HLA variation. Hum. Immunol. 77, 233–237 (2016).
Xu, H. et al. Targeted disruption of HLA genes via CRISPR–Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 24, 566–578.e7 (2019).
Deuse, T. et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 37, 252–258 (2019).
Gornalusse, G. G. et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 35, 765–772 (2017). This paper reports that hypoimmune cells allow for allogeneic transplantation without rejection or HLA matching.
Deuse, T. et al. Hypoimmune induced pluripotent stem cell-derived cell therapeutics treat cardiovascular and pulmonary diseases in immunocompetent allogeneic mice. Proc. Natl Acad. Sci. USA 118, e2022091118 (2021).
Liu, X. et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat. Immunol. 12, 416–424 (2011).
Madsen, L. et al. Mice lacking all conventional MHC class II genes. Proc. Natl Acad. Sci. USA 96, 10338–10343 (1999).
Grusby, M. J. et al. Mice lacking major histocompatibility complex class I and class II molecules. Proc. Natl Acad. Sci. USA 90, 3913–3917 (1993).
Burger, P., Hilarius-Stokman, P., de Korte, D., van den Berg, T. K. & van Bruggen, R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 119, 5512–5521 (2012).
Noyan, F. et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am. J. Transplant. 17, 917–930 (2017).
Todo, S. et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology 64, 632–643 (2016).
MacDonald, K. G. et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 126, 1413–1424 (2016).
Akbarpour, M. et al. Insulin B chain 9-23 gene transfer to hepatocytes protects from type 1 diabetes by inducing Ag-specific FoxP3+ Tregs. Sci. Transl. Med. 7, 289ra281 (2015).
Cunningham, E. C. et al. Gene therapy for tolerance: high-level expression of donor major histocompatibility complex in the liver overcomes naive and memory alloresponses to skin grafts. Transplantation 95, 70–77 (2013).
Calne, R. Y. et al. Induction of immunological tolerance by porcine liver allografts. Nature 223, 472–476 (1969).
Bernardo, M. E. & Aiuti, A. The role of conditioning in hematopoietic stem-cell gene therapy. Hum. Gene Ther. 27, 741–748 (2016).
Chanut, F. J. A. et al. Conditioning regimens in long-term pre-clinical studies to support development of ex vivo gene therapy: review of nonproliferative and proliferative changes. Hum. Gene Ther. 32, 66–76 (2021).
Agarwal, R. et al. First report of non-genotoxic conditioning with JSP191 (anti-CD117) and hematopoietic stem cell transplantation in a newly diagnosed patient with severe combined immune deficiency. Blood 136, 10–10 (2020).
Myburgh, R. et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia 34, 2688–2703 (2020).
Chhabra, A. et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci. Transl. Med. 8, 351ra105 (2016).
George, B. M. et al. Antibody conditioning enables MHC-mismatched hematopoietic stem cell transplants and organ graft tolerance. Cell Stem Cell 25, 185–192.e3 (2019). This paper reports that antibody-mediated conditioning allows for HSCT between completely MHC-mismatched donor and recipient mice.
Taya, Y. et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 354, 1152–1155 (2016).
Ochi, K., Morita, M., Wilkinson, A. C., Iwama, A. & Yamazaki, S. Non-conditioned bone marrow chimeric mouse generation using culture-based enrichment of hematopoietic stem and progenitor cells. Nat. Commun. 12, 3568 (2021).
Shimoto, M., Sugiyama, T. & Nagasawa, T. Numerous niches for hematopoietic stem cells remain empty during homeostasis. Blood 129, 2124–2131 (2017). This work is the first demonstration of robust HSC engraftment without conditioning in immunocompetent mice.
Rio, P. et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 25, 1396–1401 (2019).
Wilkinson, A. C. et al. Cas9-AAV6 gene correction of β-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat. Commun. 12, 686 (2021).
Ginn, S. L. et al. Limiting thymic precursor supply increases the risk of lymphoid malignancy in murine X-linked severe combined immunodeficiency. Mol. Ther. Nucleic Acids 6, 1–14 (2017).
Pasi, K. J. et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 382, 29–40 (2020).
Jacobson, S. G. et al. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 372, 1920–1926 (2015).
Goldstein, J. M. et al. In situ modification of tissue stem and progenitor cell genomes. Cell Rep. 27, 1254–1264.e7 (2019).
Humbert, O. et al. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Adv. 2, 987–999 (2018).
Gillmore, J. D. et al. CRISPR–Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021). This work is the first clinical trial to use the CRISPR–Cas9 system in vivo.
Krishnamurthy, S. et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 10, 4906 (2019).
Staahl, B. T. et al. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 35, 431–434 (2017).
[No authors listed.] High-dose AAV gene therapy deaths. Nat. Biotechnol. 38, 910 (2020).
Sibbald, B. Death but one unintended consequence of gene-therapy trial. CMAJ 164, 1612 (2001).
Rothgangl, T. et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 39, 949–957 (2021).
Brown, B. D., Venneri, M. A., Zingale, A., Sergi Sergi, L. & Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 12, 585–591 (2006).
Dunleavy, K. With the pricing situation ‘untenable’ in Europe, Bluebird will wind down its operations in the ‘broken’ market. Fierce Pharma https://www.fiercepharma.com/pharma/situation-untenable-bluebird-will-wind-down-its-operations-broken-europe (2021).
Ferrari, G., Thrasher, A. J. & Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 22, 216–234 (2021).
Cavazzana, M., Bushman, F. D., Miccio, A., Andre-Schmutz, I. & Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat. Rev. Drug Discov. 18, 447–462 (2019).
Aiuti, A., Roncarolo, M. G. & Naldini, L. Gene therapy for ADA–SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 9, 737–740 (2017).
Tebas, P. et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910 (2014).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Yla-Herttuala, S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European Union. Mol. Ther. 20, 1831–1832 (2012).
Porteus, M. H. & Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 300, 763 (2003).
Naldini, L. et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267 (1996).
Blaese, R. M. et al. T lymphocyte-directed gene therapy for ADA–SCID: initial trial results after 4 years. Science 270, 475–480 (1995).
Civin, C. I. et al. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-1a cells. J. Immunol. 133, 157–165 (1984).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Gyurkocza, B. & Sandmaier, B. M. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood 124, 344–353 (2014).
Acknowledgements
C.T.C. is supported by the National Science Foundation (NSF). A.C.W. acknowledges support from the Medical Research Council (MRC) and the Kay Kendall Leukaemia Fund. H.N. is supported by the National Institutes of Health (NIH) (grants R01DK116944; R01HL147124) and the Virginia and D.K. Ludwig Fund for Cancer Research (D.K. Ludwig Fund).
Author information
Authors and Affiliations
Contributions
All authors contributed to conceiving, writing and editing this Review.
Corresponding authors
Ethics declarations
Competing interests
H.N. is a co-founder and shareholder in Megakaryon, Century Therapeutic and Celaid Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks P. Genovese and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov/
Glossary
- Sickle cell disease
-
A disease caused by a specific point mutation in the haemoglobin-β (HBB) gene, which leads to the formation of haemoglobin tetramers that can polymerize with each other and that cause red blood cells to become highly fragile and adopt a characteristic sickle shape.
- Conditioning regimens
-
Treatments given prior to haematopoietic stem cell transplantation (HSCT) to ablate components of the haematopoietic system in the recipient in order to ensure engraftment of transplanted cells.
- Autoimmune haemolytic anaemia
-
An autoimmune disease whereby patients produce autoantibodies that target red blood cells, causing them to lyse prematurely and thus leading to anaemia.
- cGAS–STING pathway
-
A cytosolic DNA-sensing signalling pathway, in which binding of cGAS to double-stranded DNA (dsDNA) in the cytoplasm leads to the downstream activation of STING and, subsequently, the activation of an inflammatory transcriptional programme in cells.
- Adenosine deaminase deficiency–severe combined immunodeficiency
-
(ADA–SCID). A disease caused by mutations in the adenosine deaminase (ADA) gene. ADA is an essential enzyme in the purine salvage pathway, deficiency of which prevents the maturation of B cells, T cells and natural killer cells.
- β-Thalassaemia
-
A disease caused by mutations in the haemoglobin-β (HBB) gene that prevent functional HBB expression, leading to an inability of red blood cells to form haemoglobin tetramers.
- CRISPR–Cas9
-
A CRISPR–Cas gene-editing platform adapted from bacteria that can be directed to make double-strand breaks at specific sequences of DNA.
- Base editors
-
Gene-editing platforms that allow for the alteration of single nucleotides in the genome without requiring a double-strand break in DNA, through fusion of a catalytically dead DNA endonuclease Cas9 to a deaminase enzyme.
- Prime editing
-
A gene-editing platform that allows for the modification of small sequences (up to ~40 bp) in the genome through fusion of a catalytically dead DNA endonuclease Cas9 to a reverse transcriptase.
- Homology directed repair
-
A DNA repair pathway that corrects double-strand breaks using a homologous DNA sequence. This pathway may be used to change specific sequences in the genome or to introduce transgenes in specific locations in the genome.
- Mucopolysaccharidosis type 1
-
(MPS1). A disease caused by mutations in α-l-iduronidase (IDUA), which lead to a build-up of glycosaminoglycan in lysosomes.
- Small interfering RNAs
-
Small, 20–27 bp, double-stranded RNA molecules that bind endogenous mRNAs, leading to their downregulation through the RNA-induced silencing complex (RISC) pathway.
- CRISPR–Cas13
-
A CRISPR–Cas gene-editing platform adapted from bacteria that can be used to target mRNAs for cleavage, preventing their translation within cells.
- Chimeric antigen receptor T cell
-
(CAR T cell). A T cell that has been genetically modified to express a chimeric receptor that consists of the intracellular portion of the T cell receptor (TCR) fused to an extracellular domain that can bind an antigen of interest, causing the T cell to become activated.
Rights and permissions
About this article

Cite this article
Charlesworth, C.T., Hsu, I., Wilkinson, A.C. et al. Immunological barriers to haematopoietic stem cell gene therapy. Nat Rev Immunol 22, 719–733 (2022). https://doi.org/10.1038/s41577-022-00698-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-022-00698-0
This article is cited by
-
Base Editors-Mediated Gene Therapy in Hematopoietic Stem Cells for Hematologic Diseases
Stem Cell Reviews and Reports (2024)
-
AAV-based in vivo gene therapy for neurological disorders
Nature Reviews Drug Discovery (2023)
-
APEX1 Nuclease and Redox Functions are Both Essential for Adult Mouse Hematopoietic Stem and Progenitor Cells
Stem Cell Reviews and Reports (2023)
-
Stimuli-responsive nanoformulations for CRISPR-Cas9 genome editing
Journal of Nanobiotechnology (2022)
