
Abstract
For decades, preclinical toxicology was essentially a descriptive discipline in which treatment-related effects were carefully reported and used as a basis to calculate safety margins for drug candidates. In recent years, however, technological advances have increasingly enabled researchers to gain insights into toxicity mechanisms, supporting greater understanding of species relevance and translatability to humans, prediction of safety events, mitigation of side effects and development of safety biomarkers. Consequently, investigative (or mechanistic) toxicology has been gaining momentum and is now a key capability in the pharmaceutical industry. Here, we provide an overview of the current status of the field using case studies and discuss the potential impact of ongoing technological developments, based on a survey of investigative toxicologists from 14 European-based medium-sized to large pharmaceutical companies.
Similar content being viewed by others

Introduction
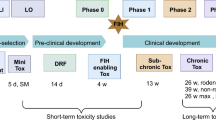
New tools and strategies in investigative toxicology are continually being implemented in pharmaceutical companies to reduce safety-related attrition in drug development1,2. A key objective of investigative toxicology is the prediction of clinical safety, which is still a high hurdle. The chemical and biological uniqueness of each drug candidate (which limits the predictive value of knowledge from previous drugs) and the limited translational relevance of many standard preclinical toxicity models to humans contributes to unforeseeable late-stage attrition. Current investigative toxicology strategies therefore adopt a tiered approach, employing progressively more complex in silico, biochemical, cellular in vitro assays and in vivo safety experiments as drug candidates progress through the pipeline to build a strong platform for identification and mitigation of hazards, and finally a risk assessment for the drug candidate. Computational approaches and prospective assays aim to predict toxicities that could lead to the termination of a drug candidate or an entire development programme, and bespoke retrospective assays are used for issue resolution and management if target-organ toxicities are identified in in vivo ‘toxicology signal-generation’ studies or later chronic studies (Fig. 1). Crucially, the goal of toxicology in the discovery phase is not to simply ‘front-load’ attrition, but to increase the likelihood of success by optimizing the safety dimension of drug design and candidate selection such that subsequent non-clinical programmes and clinical trials can successfully test clinical hypotheses.

The figure shows a roadmap of investigational toxicology activities that can be applied at each stage of the pipeline. In the earlier stages, these activities typically involve computational assessments to support target selection and batteries of routine in vitro molecular and cellular assays to support lead identification and optimization. These assays evaluate broad characteristics such as cytotoxicity, mitochondrial and genetic toxicity, as well as effects on specific cell types such as hepatocytes and cardiomyocytes to evaluate the risk of toxicity to particular organs. At later stages of the process, regular in vivo toxicology studies that support progression into clinical development may be complemented by bespoke project/target-organ-specific assays — for example, with more advanced cellular models (microphysiological systems (MPS) and multicellular assays) — to address the human relevance of preclinical in vivo findings and gain mechanistic insights (for example, on off-target effects). It is important to stress that approaches may vary significantly between companies based on internal drivers, priorities, resources and history. The focus of the figure is on approaches for small molecules, with selected activities specific for other modalities shown below. It is currently unclear whether the roadmap of toxicology activities for small molecules adequately covers the safety evaluation needs for targeted protein degraders such as proteolysis-targeting chimeras (PROTACS). There are several PROTAC-specific attributes that must be considered, including off‐target degradation, intracellular accumulation of the natural substrates for the E3 ligases used in the ubiquitin–proteasome system and proteasome saturation by ubiquitylated proteins185,186,187,188. PK, pharmacokinetic.
The key to successful investigative toxicology in drug discovery is to efficiently combine safety data with other compound-specific features, such as absorption, distribution, metabolism and excretion (ADME) and physicochemical properties, which requires timely and integrated experimental approaches to address the risks inherent in the potential target and in potential lead candidates. This involves a paradigm shift from the use of classical (and generally low-throughput) in vivo toxicology methods towards a focus on translatable mechanistic in vitro assays that can act as reliable predictive surrogates for specific aspects of in vivo studies. Such assays rely heavily on the development of human physiologically relevant model systems in combination with appropriate toxicity end points.
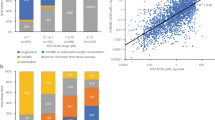
In recent years, there have been major advances in areas such as the use of induced pluripotent stem cells (iPSCs), 3D tissue models, advanced microphysiological systems (MPS) and imaging techniques that have the potential to substantially improve the predictive value of investigative toxicology assays3. There are a vast number of new platform technologies, and a comprehensive description of all of them is beyond the scope of this article. However, through a survey of 14 Europe-based medium-sized to large pharmaceutical companies (Box 1 and Fig. 2a), we identify key emerging technologies that are actually having an impact and how they are being practically and more routinely incorporated currently in drug discovery. In addition, we consider which technologies — through appropriate development — have the potential to have a ‘game-changing’ impact on investigational toxicology and how these have evolved over a 5-year period (Fig. 2b).

We assessed the perceived immediate, mid-term (<2-year) and longer-term (<5-year) benefit from the routine application of various emerging technologies and assays through surveys of pharmaceutical company experts in 2015 and 2020 (Box 1 and Supplementary information). a, Summary of current technologies or methods that offer a step change in investigational toxicology; impact according to consensus and time frame identified by experts from Europe-headquartered pharmaceutical companies (n = 14) responding to the question “Which new technologies do you foresee may become game-changers for investigational toxicology?” in 2020. Variations in perceptions across the respondents potentially reflect current practices and usage within their organizations. b, Evolution among the respondents of the impact perception for each technology or method from 2015 to 2020. The positive perception of some technologies such as organs-on-chips, genomic profiling and high-content imaging increased, while the perception of others barely changed or the time frame for impact was adjusted (systems toxicology and induced pluripotent stem cells (iPSCs)). Other technologies have clearly lost ground, with the ‘no potential game-changer’ category becoming more prominent (metabolomics, microRNAs (miRNAs) and mass spectrometry imaging). qPCR, quantitative PCR; RNA-seq, RNA sequencing.
In this article, we first summarize the key goals of investigative toxicology and highlight the current approaches used in investigative toxicology using published case studies, mainly from across the group of companies surveyed. We then discuss selected emerging technologies that have the potential to shift the current safety-testing paradigm, with a focus on those considered most likely to have the greatest near-term impact. Finally, we consider the role of collaborations and pre-competitive consortia in enabling the development, validation and effective application of these next-generation tools.
Goals and tools for investigative toxicology
Investigative toxicology can be broadly split into two approaches: the prediction of potential toxicity before a compound is tested non-clinically in vivo or clinically (prospective approach), or the provision of mechanistic understanding of non-clinical in vivo or clinical toxicity findings (retrospective approach) (Fig. 1). These approaches have two major goals: in the drug discovery phase, the aim is to guide the identification of the most promising drug candidates to pursue (that is, safe candidates that provide the best therapeutic index) and to deselect the most toxic drug candidates as early as possible. In the preclinical and clinical development phases, the purpose is to provide mechanistic safety data that enable development of a well-characterized hazard and translational risk profile to support clinical trial design (that is, risk assessment, management and mitigation) (Fig. 1). In this section, we summarize the key activities in investigative toxicology as a drug candidate progresses through the pipeline. We focus primarily on small-molecule drugs; however, a high-level comparison with the activities for other therapeutic modalities is provided in Fig. 1.
Support assessment of the target and the competitive landscape
In the traditional approach to regulatory toxicity assessment, before progressing a drug candidate to ‘first-in-human’ clinical trials, findings of the pivotal regulatory good laboratory practice (GLP) in vivo studies are presented to pharmacologists and clinicians for joint discussion, with the occasional outcome that the toxicological findings could be attributed to effects mediated by the primary target of the drug4. Over the past decade, the traditional analysis has been widened to consider the broader physiological role of the target in health and disease, based on a systematic search of the literature and databases. This search should address at least the following topics: description of the main target functions and upstream and downstream signalling pathways, description of closely related targets (orthologues and paralogues), analysis of cross-species target homology, function and tissue expression across species, and phenotypic consequences of genetic modifications to the target in animals (knockout and knock-in; conditional or not). Results of competitive landscape analyses, including identification of lead structures from competitors, if possible, should also be incorporated. Target safety assessment may also be important in the evaluation of potential candidates for in-licensing by larger companies (Box 2).
A thorough target safety assessment may contribute to the selection of the most appropriate preclinical safety species; for example, by avoiding non-relevant species that do not express the target. It could also identify potentially affected target organs and tissues that need to be integrated into screening studies, alongside biomarkers for monitoring the occurrence and extent of pharmacodynamic responses2. Ultimately, the target safety assessment may lead to a stop/go decision for the particular target based on the target patient population, as well as an experimental plan to quantify risks if the project progresses further1,5.
For example, the family of aspartic proteases from mammals (such as cathepsins, napsin A, pepsin renin and β-secretases) and non-mammals (such as plasmepsins) are an important drug target family6,7. However, inadequate or suboptimal selectivity for some aspartic proteases may hamper the development of safe and effective therapies. Evaluation of data collated from in vitro investigations, in vivo animal studies and adverse drug reactions reported in patients treated with approved aspartic protease inhibitors highlighted functional and structural toxicities associated with inhibition of mammalian aspartic proteases8. Of these, cathepsins D and E may be of most toxicological relevance, as cathepsin D is a ubiquitous lysosomal enzyme present in most cell types, and cathepsin E is found in the gut and in erythrocytes; that is, tissues likely to be exposed to high drug concentrations. Of particular toxicological relevance, human cathepsin D deficiency has been reported as a cause of congenital human neuronal ceroid-lipofuscinosis, which is a severe lysosomal storage disease characterized by neurodegeneration, developmental regression, visual loss, epilepsy and premature death owing to excessive tissue accumulation of lipopigments (lipofuscin)9,10. Moreover, several structurally distinct β-secretase (BACE1) inhibitors have been withdrawn from development after inducing ocular toxicity following chronic treatment in animal models. Quantification of cathepsin D target engagement in cells has been shown to be predictive of ocular toxicity in vivo, suggesting that off-target inhibition of cathepsin D was a principal driver of ocular toxicity for these agents7. This example illustrates the need for a thorough assessment of the risks of inhibiting closely related targets when developing drugs for targets in families such as aspartic proteases.
Use safety profiling and target-organ models to guide candidate design and selection
Prediction and/or understanding of organ-specific drug toxicities can be achieved with varying levels of confidence using in vitro cell models. Simple cell death monitoring in vitro, as a rough surrogate of toxicity potential in vivo, is widely used to eliminate compounds early in the candidate selection process2,11,12. This works adequately for phototoxicity and genotoxicity, for which there is a simple and close correlation between in vitro and in vivo outcomes. However, there are usually no such relationships between in vitro cytotoxicity and organ function alterations, and so more sophisticated models have been developed to assess compound-induced perturbations of cellular functions and thus provide information on the potential for specific tissue and organ adverse effects13.
Initially, work towards this goal focused on measuring simple end points such as cell survival through ATP content or other cytotoxicity end points in organ-derived cell lines that are accessible and amenable to high-throughput screening (HTS), which are still used heavily in industry. Primary cells with improved tissue or organ phenotypes are also used, but sourcing primary cells of suitable quality remains challenging, especially in quantities sufficient to support larger screening programmes and consistent testing over many years. In addition, the ethical and legal issues for human tissue sources require careful consideration. More recently, 2D and 3D cultures and MPS have been developed that attempt to mimic micro-environmental conditions by incorporating multicellular tissues, scaffolding and mechanical factors based on cells such as cardiomyocytes derived from iPSCs or extended pluripotent stem cells (ePSCs)14,15,16,17. Models for most major toxicologically relevant organ systems have been described18,19 (Fig. 3). These have been reviewed elsewhere and so are not covered in depth here17,18,19,20.

The figure summarizes the status of in vitro test systems that are amenable across key target organs. Key characteristics of each organ and its model options are summarized. Boxes are coloured according to the currently perceived confidence in safety translation among the survey participants. This empirical assessment is based on published data, considering the ability of the model to emulate key organ phenotypes (and stability for extended periods of culture) and toxicology validation data. More details on in vitro cell culture assays for each organ system with associated references are available in Supplementary Table 1. CNS, central nervous system; CYP, cytochrome P450; GI, gastrointestinal; iPSC, induced pluripotent stem cell; iPSC-CM, iPSC-derived cardiomyocytes; MEA, microelectrode array; MPS, microphysiological systems.
Each in vitro target-organ approach — from simple cell lines through to MPS — has intrinsic challenges and opportunities (summarized in Fig. 3), and so the use of multiple ‘fit-for-purpose’ assays best represents the current practice. Simple cell cultures with relatively straightforward end points can provide throughput and robustness for screening and specific mechanistic end point studies (for example, mitochondrial toxicity), but not the complexity often required to reflect the in vivo situation. These may be complemented with organoids or MPS, or even with organ explants and slices to provide in-depth mechanistic understanding. Indeed, connected organ models in single MPS chips are emerging and are starting to be applied for drug discovery risk assessment. A ‘heart-on-a-chip’ connected to a ‘liver-on-a-chip’ via a microfluidics system was used to derive in vitro temporal pharmacokinetic/pharmacodynamic (PK/PD) relationships for the histamine receptor antagonist terfenadine: the terfenadine-induced increase in QT interval of the ‘heart-on-a-chip’ was reduced when coupled to a metabolically competent liver compartment. Importantly, when the model was applied to a small-molecule drug discovery programme, a representative molecule was identified as having a hERG liability, due to the formation of a hERG active metabolite21. Thus, such ‘heart-on-a-chip’ coupled with a ‘liver-on-a-chip’ systems have the potential to reveal complex metabolic and toxicological relationships earlier in the drug discovery process.
Crucially, there is an opportunity for the large amounts of multi-factorial data generated to be used to feed algorithms for the mathematical modelling of drug liabilities22. However, full integration of multi-factorial data and further refinement of test systems are still required. This necessitates extensive work on qualitative and quantitative understanding of complex metabolic and toxicological relationship23,24 models to ensure that in vitro data are sufficiently predictive of in vivo non-clinical and clinical findings. This in turn will help to identify a fit-for-purpose battery of target-organ assays and test systems for use in early selection of the safest compounds through a deeper understanding of the mechanisms of drug toxicity.
The general approach and challenges for understanding the potential for target-organ toxicities can be illustrated for the liver, for which a wide range of in vitro assays has been and is being developed for toxicological assessment. Hepatic cell lines constitute an integral part of most pharmaceutical companies’ safety screening strategies and can provide an initial hazard warning system13,25,26. However, several of these transformed cells can poorly reflect native primary human hepatocytes, as revealed by transcriptomic and proteomic phenotype differences27,28,29. Consequently, their toxicological utility without a priori knowledge of the physiological and pharmacological characteristics and mechanistic end point under investigation can be relatively low30,31,32. Primary human hepatocytes in 2D cultures also have limitations to their use for toxicological studies, because cells de-differentiate27. Culturing hepatocytes in 3D ‘spheroid’ configurations appears to overcome this issue to some extent33,34. Other examples are discussed in the section on advanced cellular models below.
In addition to understanding potential target-organ toxicities through in vitro assays, another key investigative toxicology activity at this stage of the drug discovery process is pharmacological profiling to identify undesirable off-target activity that could hinder or halt the development of drug candidates. At the molecular level, secondary pharmacology studies evaluate drug effects against a broad range of molecular targets that are related to or distinct from the intended therapeutic target35,36,37. Numerous targets — including G protein-coupled receptors, transporters, ion channels, nuclear receptors and kinases — have been associated with significant side effects in non-clinical or clinical settings35,36,37, and the list is continually expanding. It has been reported that compounds with a target hit rate of ≥20% (where the target hit rate is the percentage of a panel of at least 50 targets for which more than 50% binding is noted at a compound concentration of 10 µM) are associated with a higher attrition rate37,38,39. Higher hit rates are often, but not exclusively, related to physicochemical properties of compounds, particularly lipophilicity40. These properties may also lead to artefactual potent target inhibition due to colloidal aggregation, and efforts are needed to avoid such false positives41,42. Various strategies have been used to address off-target binding43,44, as have empirical guidelines to avoid unfavourable compound profiles and/or minimize off-target effects. For example, compounds beyond the boundaries of Pfizer’s 3/75 rule (logP > 3 and tPSA < 75 Å2)40 or GSK’s 4/400 rule (logP > 4 and M > 400 Da)45 may be considered more likely to be problematic.
Various protocols for in vitro secondary pharmacology screening approaches are available, and these generally consist of binding assays, functional assays and enzymatic assays, all of which provide important information regarding the pharmacological activity of a drug (such as potency, efficacy and type of interaction), including possible undesirable side effects that may be anticipated in humans37,39,46,47. This is a cost-effective and efficient approach commonly used as a safety screen for hazard identification and elimination in the early phases of drug discovery.
During hit identification and lead series selection, the main objective is to identify toxicological hazards and understand the potential for promiscuity (that is, the incidence of off-target hits) in the initial lead chemical series. The approach consists of screening chemicals against a panel of fewer than 60 targets strongly linked to safety liabilities, thus enabling prioritization of lead molecules and chemical series for further development36,37,39,48. Data from this initial profiling can be used during the lead optimization phase to enable medicinal chemists to establish structure–activity relationships, allowing mitigation of undesirable off-target activity from the molecules by design. The profiling can also be used to select drug candidates to progress into preclinical development, as well as to trigger and influence the design of investigative in vivo toxicology studies. In later phases of drug discovery, before carrying out first-in-human trials, screening against a broader panel of targets can be performed to aid in mechanistic understanding of in vivo effects35,39.
The impact of such investigations is exemplified by our next case study: the off-target profiling of a novel antimalarial chemical series belonging to the triaminopyrimidine (TAP) class. The TAP lead series, with potent activity against a panel of clinical strains of Plasmodium falciparum that harbour resistance to known antimalarial drugs, were found to have relatively potent hERG channel activity (associated with adverse cardiovascular risk) and acetylcholine esterase activity (associated with adverse neurological risk). Extensive and focused sub-panel off-target screening was employed to develop structure–activity relationships and optimize the in vitro selectivity. The activity of the final drug candidate at the primary pharmacological target and potencies against the affected off-targets was improved (>100-fold) and translated into an acceptable in vivo toxicology and in vivo safety pharmacology profile, providing confidence in the candidate drug therapeutic index49.
In contrast, a simple routine cellular off-target assay such as in vitro phospholipidosis (PLD)50, which is commonly used either prospectively to screen out potential PLD inducers, or retrospectively to explain the nature of vacuoles seen in vivo, may be used to help understand in vivo lack of efficacy. In a recent example, a PLD assay based on high-content imaging (HCI) was used to investigate the high hit rate in coronavirus disease 2019 (COVID-19) drug-repurposing screens and why these ‘hits’ were not efficacious in vivo51. This study showed that, independently of their putative mode of antiviral activity, many of these hits were inducing PLD, which can itself inhibit severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication. So, in this case, an assay that originated from investigative toxicology activities could help early deselection of artefactual hits and prevent wastage of further resources on their study51.
Safety profiling can also be approached by combining different technologies to gain insight or to confirm findings. For example, the identification of off-targets of drug candidates with biophysical and proteomic techniques can provide the basis for developing hypotheses for observed adverse effects. Side effects associated with panobinostat, a histone deacetylase (HDAC) inhibitor, include hypothyroidism, seizures and tremors52. Thermal proteome profiling (TPP) and affinity enrichment-based chemoproteomics identified four specific off-target proteins of panobinostat, including phenylalanine hydroxylase (PAH)52. PAH has not previously been identified as a panobinostat target and was not stabilized in TPP experiments with vorinostat, a structurally distinct HDAC inhibitor. Inhibition was confirmed through enzymatic assays with rat liver PAH. Binding of panobinostat to PAH resulted in an increase in cellular phenylalanine and a decrease in tyrosine levels: a potential molecular initiating event related to panobinostat side effects52. Thus, optimizing the PAH selectivity of panobinostat analogues may improve HDAC inhibitor tolerability. More generally, earlier deployment of TPP and chemoproteomic approaches could enable a fuller assessment of off-target liability before candidates are selected for toxicology evaluation in animals.
Classical histopathology may also gain power when combined with analytical methods. Mass spectrometry imaging (MSI) approaches encompass a range of direct ionization methods that sample endogenous and exogenous compounds directly from the surface of tissue sections. Coupled mass spectrometry enables multiplexed label-free spatial analysis of compounds directly from tissue sections and is now being used in drug discovery53. Such spatial resolution when coupled to histopathological images has the potential to eliminate bulk analysis artefacts of pathological tissue sampling and focus on adverse biological perturbations at the cellular level54. The potential of MSI to complement other methods has recently been demonstrated in a study of the metabolic changes induced by cisplatin nephrotoxicity. Kidney tubule injury biomarkers and morphological effects in rats exposed to cisplatin were complemented by MSI revealing molecular alterations on a spatial level55. Early metabolic changes in the corticomedullary boundary metabolism were coincident with pathological changes. Interestingly, effects that included changes in nucleotides, antioxidants and phospholipids detected by MSI persisted longer than changes in renal biomarkers and after morphological recovery, highlighting the potential of MSI for detecting long-term liabilities.
De-risk preclinical in vivo findings and address human relevance
In vitro assays may be valuable in understanding the molecular and cellular mechanisms of pathological effects observed in animal studies in various species and clarifying their potential relevance to humans. The following two examples illustrate the impact of mechanistic understanding of toxic effects in the progress of a drug candidate either into clinical trials or onto the market.
Drug-induced neoplastic changes are a frequent phenomenon in in vivo safety studies, particularly in rodents exposed up to life time. For example, foci of altered hepatocytes are considered putative pre-neoplastic lesions that can occur spontaneously or be induced by chemicals or drugs56. Progression of these foci to hepatocellular neoplasms has been reported, but increases in foci in rodents do not necessarily lead to tumours in carcinogenicity studies57. A non-genotoxic nicotinic α7 receptor partial agonist drug candidate, RG3487, was found to induce foci of altered hepatocytes and subsequently tumours in rats58, potentially precluding human clinical studies. However, the hepatocyte alterations observed in rats were not seen in the liver of mice or dogs. To assess human relevance, primary rodent, canine and human stem cell and patient-derived 3D cell model phenotypic assays were used to explore potential mechanisms of neoplasia. Rodent phenotypic models were found to recapitulate the in vivo effect whereas human and canine models clearly showed an absence of effect, and the study revealed drug-induced effects that were not relevant in humans, such as nuclear receptor-driven liver proliferation in rodents but not in human models58. These data supported the progression of RG3487 into clinical trials.
Another example in which a high incidence of haemangiosarcoma induced by siponimod, a S1P1/5 receptor modulator, in mice but not in rat 2-year carcinogenicity studies was addressed by a mixed in vivo and in vitro approach. A 9-month investigative mouse study with multiple time-point euthanasia explored molecular mechanisms that underlie the emergence of haemangiosarcoma, in parallel with a similar rat 3-month study59. These studies showed equal activation of vascular endothelial cells (VECs) by gene expression profiling in the two species, but mitotic gene expression was only transiently induced in rats whereas it remained constantly activated in mice. Additionally, out of many cytokines assessed, placental growth factor 2 (PlGF2) was induced in blood from day 1 of treatment and sustained in mice, whereas it was weakly and transiently increased in rats. In vitro experiments using primary VECs of both species showed a similar PlGF2 induction and mitotic activation in mice but not in rats. Human primary VECs behaved like rat VECs, showing no induction of mitosis or PlGF2 over a very large concentration range59. After review by regulatory agencies, this set of studies supported the approval of siponimod for the treatment of secondary progressive multiple sclerosis60. The data from these two cases highlight the importance of humanizing toxicology research and illustrate the use of cross-species in vitro comparison at the molecular and phenotypic levels to address the human relevance of preclinical findings.
Support identification of mechanisms underlying clinical events
Investigative toxicology may also be crucial in assessing safety issues that arise during clinical development or post-approval. This support is not limited to parent drugs, but may encompass drug impurities, degradants and metabolites.
For example, the development of antibody-mediated pure red cell aplasia (PRCA) in patients with anaemia treated with aggregated forms of recombinant human erythropoietin (rhEPO) is a rare and severe example of the consequences of protein immunogenicity61,62, and so understanding the potential causes of protein aggregation is crucial for the safe use of rhEPO. Tungsten-induced rhEPO aggregates arising from the manufacturing process were hypothesized to be the root cause, and dendritic cell–T cell co-culture assays were used to differentiate non-aggregated from aggregated rhEPO and the immunogenicity of tungsten-induced rhEPO aggregates. T cell assays confirmed the immunogenicity of tungsten-induced aggregates of rhEPO in a clinical batch associated with one case of antibody-mediated PRCA, a finding that contributed to regulatory approval as part of a root-cause analysis63. Consequently, the syringe mode of production was modified to avoid traces of tungsten contamination.
Post-hoc investigations of clinical trial adverse events do not always lead to salvation of the product, but may provide solutions for follow-up drug candidates. For example, following the termination of the clinical development of an anti-CD40L antibody for the treatment of autoimmune disorders owing to unexpected thrombotic complications, studies using a blood vessel-on-a-chip model unveiled the mechanism of the prothrombotic effects64. Modifying a follow-up antibody with an Fc domain that did not bind to the FcγRIIa receptor on human platelets alleviated the prothrombotic effects in the vessel-on-a-chip model.
High-impact innovations in toxicology
On the basis of our survey of pharmaceutical companies (Box 1), which was first performed in 2015 and repeated in 2020, we were able to identify clear changes in the value of certain technologies and also prioritize those technologies that could improve the impact of investigative toxicology on drug discovery and development, according to their perceived impact at the time and over the next 2–5 years (Fig. 2). The perceived game-changing potential increased substantially over the 5 years between the two surveys for HCI, modelling and simulation, mRNA profiling, iPSCs and organs-on-chips. Decreases for game-changing potential were seen for in vivo imaging and MSI, gene editing and microRNA (miRNA). A more mixed picture developed for systems toxicology and metabolomics. In general, these changes can mainly be attributed to experience accrued by the participating partners over the 5-year period, as well as the perceived value of a technology for internal decision-making or the accessibility and deployment of some of the technologies (for example, MSI and metabolomics). Interestingly, although there has been an explosion in application of CRISPR to gain biological insights, its use is more nuanced in the investigational toxicology space because adverse pharmacology and/or toxicity will not necessarily be phenocopied by a gene deletion.
In this section, we highlight the current applicability of technologies for investigative toxicology based on experience in the companies participating in the survey, using published examples where possible. We also discuss the associated challenges and steps needed to achieve their potential, as well as the reasons for the perceptional changes described above.
Imaging technologies
Spatial resolution of drug effects on cells and tissues provides invaluable insight into drug toxicity; moreover, multi-parametric imaging biomarkers provide a basis for mechanistic understanding. There have been significant advances and application of imaging technologies, both in vitro and in vivo, including HCI, fluorescence and bioluminescence imaging, MRI, CT and high-dimensional molecular profiling of cell and tissue samples using MSI65,66.
HCI technology combining automated microscopy and quantitative image analysis can be used to investigate mechanisms of compound and organ-specific toxicity67,68,69, and in genotoxicity screening70. Consequently, HCI has emerged as an important technique for monitoring the spatial and biological changes induced by small molecules, biologics or other modalities, and integrating them to provide toxicological insights. It can discriminate subsets of cellular phenotypic features from a larger set, enabling the construction of machine-learning models for phenotypic profiling of drug candidates according to their mechanism of action in an unbiased way. For example, microtubule inhibitors and mitochondrial toxicants showed distinct clustering when 1,008 annotated reference compounds were tested in a panel of 15 reporter cell lines71. Hussain and coworkers72 predicted direct hepatocyte toxicity in humans by using a subset of 30 phenotypic features, with a test sensitivity, specificity and balanced accuracy of 73%, 92% and 83%, respectively. Data from a single high-throughput imaging assay can be repurposed to predict the biological activity of compounds in other assays, even those that target alternative pathways or biological processes73.
Cell painting is a high-throughput phenotypic profiling assay that uses fluorescent cytochemistry to visualize various organelles and HCI to derive many morphological features at the single-cell level74. Recently, this approach was used to test a set of reference chemicals in six biologically diverse human-derived cell lines. Similar biological activity profiles were obtained in the various cell lines without the requirement for cell type-specific optimization of cytochemistry protocols74,75. The availability of high-throughput systems, together with robust protocols and rapid data processing, are the main reasons why the game-changing potential of HCI significantly increased between the two surveys.
In vivo imaging using techniques such as MRI, positron emission tomography (PET), single photon emission tomography and CT offer the possibility of investigating biological events in any region of the body in real time and can provide the basis for minimally invasive biomarkers for safety profiling. Nonspecific biomarkers, such as those based on MRI relaxometry, CT density or PET with [18F]fluoro-2-deoxy-d-glucose, are better suited for preclinical general toxicology and clinical monitoring, whereas specific biomarkers, such as those based on most PET and single photon emission tomography ligands, are better suited to clarify mechanisms of toxicity or unwanted target engagement76,77.
The potential of MSI to simultaneously determine the discrete tissue distribution of the parent compound and/or its metabolites has paved the way for in situ studies on drug development, metabolism and toxicology66. MSI also has high molecular specificity and allows comprehensive, multiplexed detection and localization of hundreds of proteins, peptides and lipids directly in tissues78. In addition, single-cell imaging has advanced to the point where it is possible with nanoscale secondary ion mass spectrometry (NanoSIMS) to directly measure the absolute concentration of an organelle-associated, isotopically labelled pro-drug directly from a mass spectrometry image. A nano-electrochemistry method for single organelles has recently been developed79 that allows subcellular quantification of drugs and metabolites. Nevertheless, the game-changing potential of in vivo imaging and MSI was downgraded in the 2020 survey relative to 2015, owing to the complexity and the high costs of these technologies, which still prevent their routine use in early phases of drug development. In the future, this potential could be boosted by the development of artificial intelligence (AI) tools for digital pathology80; for example, the recently launched 6-year public–private partnership BigPicture81 aims to develop a repository of more than 3 million preclinical and clinical digitized pathology slides and AI tools82 to analyse them and recognize abnormalities within tissue sections.
‘Omics’ technologies
Toxicogenomics (TGx) — the molecular assessment of toxicological effects via transcriptomics or the cellular output of gene expression (that is, proteomics and metabolomics) — generated considerable hype in the early 2000s, but its initial impact was disappointing. In 2010, a multi-sector survey showed that despite the application of TGx data for more than 10 years, the broad implementation of TGx to improve decision-making had not been achieved83. However, it is now considered that the use of such technologies can greatly strengthen preclinical toxicology studies. Approaches such as the L1000 methodology used within the NIH Library of Integrated Network-based Cellular Signatures (LINCS) programme allow high-throughput transcriptional profiling and signature analysis84. It is hypothesized that for most biochemical networks, mRNA expression levels of key representative genes (landmark genes) can be used as surrogates for all members of that network. If correct, measurement of the expression levels of an appropriate subset of landmark genes at a relatively low cost could be used to represent the whole transcriptome information, as has been demonstrated for EPA’s ToxCast and the Tox21 data85.
Transcriptional profiling for the prioritization of compounds during early drug discovery is a common approach within the pharmaceutical industry: 42% in our survey considered this a game-changer for investigative toxicology already in 2015, and 65% in 2020, illustrating the increasing maturity of this technology (Fig. 2). Gene expression signatures of compounds can be used to capture a range of poly-pharmacological effects and can be used to monitor the impact of medicinal chemistry optimization of a lead series. In addition, it may be used to generate hypotheses as to what compound substructures are responsible for a given effect and ultimately support the medicinal chemist in designing new structures to synthesize. More recently, non-signature-based transcriptome and gene network-based approaches for the analysis of drug-induced genomic effects have been demonstrated to explain mechanisms of hepatotoxicity and predict distinct toxicity phenotypes using gene network module associations. Network responses were found to complement traditional histology-based assessment in predicting outcomes of longer-term in vivo toxicology studies and to identify a novel mechanism of hepatotoxicity involving endoplasmic reticulum stress and NRF2 activation86.
Expression of some miRNAs — small evolutionarily conserved endogenous non-coding RNAs that regulate the translation of protein-coding genes — have been associated with adverse biological effects87,88. More than 1,000 human miRNAs have been identified so far (see the miRBase database and miRNA tissue Atlas89,90), and a growing number of miRNAs have been reported to be expressed in a specific tissue or to be highly tissue-enriched90,91; for example, miR-122 in the liver92, miR-192 in the kidney cortex93 and miR-208 in the heart94. Importantly, circulating miRNAs have recently been correlated with liver and heart toxicity or injury in preclinical models of hepatotoxicity and cardiotoxicity and in patients with drug-induced liver and cardiac injury95,96. Consequently, circulating miRNAs are increasingly being investigated as tissue-specific toxicity biomarkers in investigative toxicology. However, perception of the game-changing potential of miRNAs in the survey decreased between 2015 and 2020, most probably owing to the still-limited translational potential.
Advances in NMR and mass spectroscopy workflows as well as analytical techniques now mean that metabolomic changes in cells, supernatants and also bio-fluids from in vivo animal studies or clinical samples can now be readily quantified and used either for hypothesis-free profiling (often consisting of thousands of cellular metabolites) or for biased approaches that measure more focused metabolic subsets97. Metabolic changes after drug treatment can be monitored, and together with bioinformatics processing analysis to either identify perturbed biological process or make statistical comparisons with metabolic signatures of chemical/drug libraries, can aid in the identification of toxicological mechanisms or hazards98,99. In the two surveys, views of the technology were mixed, with an increase of “already a game-changer” from 0% to 22% between 2015 and 2020, but also a concomitant increase of “no potential game-changer” from 17% to 58%. On the basis of discussions within the surveyed companies, we concluded that this heterogeneous perception is due to project- and company-specific experiences with this technology, which may provide high value in specific, targeted cases, but may also too often fail to provide interpretable data, as well as presenting a set of challenges and requiring substantial investments of money and time100.
Computational and systems-level modelling to predict drug toxicity
The integration of multiple testing platforms generates large data sets and can require the use of high-performance computing and machine-learning-based modelling to fully realize mechanistic insights or make effective predictions. Successive integration of chemical, protein target and HTS cytotoxicity data has been shown to improve predictive performance, and studies with Tox21 chemicals101 show that models based on in vitro assay data may in some specific cases perform better in predicting human toxicities than animal toxicity studies102. In addition, the combination of structural and activity data results in better models than using structure or activity data alone. For example, a Bayesian machine-learning approach, integrating in vitro assay data (such as hepatic transporter inhibition, mitochondrial toxicity and cytotoxicity in spheroid liver cell lines) with compound physicochemical properties and in vivo animal exposure, allowed binary yes/no prediction of drug-induced liver injury (DILI) with a balanced accuracy of 86%103. More recently, ‘big data compacting’ and ‘data fusion’ approaches, coupled to machine-learning techniques, have been applied to public domain transcriptomic data and have yielded impressive results in explaining dose-dependent cytotoxicity effects, predicting in vivo chemically induced pathological states in the liver and predicting human DILI from hepatocyte experimental data103,104. Thus, predictive modelling of chemical hazards by integrating numerical descriptors of chemical structures and short-term toxicity assay data benefits from chemo-informatics approaches and hybrid modelling methodologies for in vitro–in vivo extrapolation105. In one example, deep learning outperformed many other computational approaches for computational toxicity prediction, such as naive Bayes, support vector machines and random forests106.
The computational power to efficiently analyse a wealth of data using machine-learning techniques (chemogenomics) is now available, and together with enhancements in more human-relevant models (3D models, cellular co-cultures, MPS), improved transcriptomics workflows and freely available public data sources, we have the opportunity to incorporate data from early screening sources and derive more quantitative risk assessments using systems toxicology approaches based on pharmacokinetics, pharmacodynamics and physiology-based models. Indeed, investments in translational science are proving fruitful in moving from descriptive toxicology to a more quantified understanding of human translation107. The company survey revealed that physiologically based pharmacokinetic (PBPK) modelling and simulation (M&S) approaches are being adopted to predict the pharmacokinetic and pharmacodynamic behaviour of drugs in humans108. One of the first areas in which this is occurring is in vivo safety pharmacology, where it has been crucial in providing more quantified cardiovascular risk assessment108 (Box 3).
M&S approaches promise to greatly impact drug safety by providing a method for integrating in silico, in vitro and in vivo preclinical data. In particular, mechanism-based models can be used to integrate disparate data types and predict the effects of new drugs in humans109,110. Indeed, M&S approaches are now being adapted to toxicological end points that use compound-related tissue injury biomarkers. Such biomarkers can be used to bridge between toxicity in in vitro and in vivo models and enable reliable translation between animals and humans. A leading example of such approaches is the prediction of myelosuppression by oncology drugs, which has often been a dose-limiting toxicity in the past. To predict and better inform the time course of myelosuppression after drug administration, semi-physiological mathematical models have been constructed111. These models explicitly represent drug-sensitive cells that correspond to the proliferating cells in the bone marrow and drug-insensitive cells that correspond to cells that subsequently mature without proliferating, as well as the circulating white blood cells. One such model was able to translate the full time course of myelosuppression from rodent data to patients for six different drugs112. A key feature of the model was a correcting factor that accommodated species differences in protein binding and sensitivity differences determined in in vitro colony-forming unit granulocyte macrophage assays. Predictions can be made not only of the magnitude of myelosuppression caused by novel drugs, but also how they interact with myelosuppressive standard-of-care therapies. This enables oncology drug candidates with the most favourable profile to be prioritized and can also guide clinical use by informing the exploration of combination dose scheduling113.
As risk assessment begins to move from single end point measures of toxicological effects towards pathway- and system-based studies, multiple parametric data coupled with M&S will offer further insight into complex effects of small-molecule and biologic drugs. Integrating classic toxicology approaches with network models and quantitative measurements of molecular and functional changes can enable a holistic understanding of toxicological processes, permitting prediction and accurate simulation of drug toxicity86. Moreover, systems toxicology frameworks, based upon mechanistic understanding of the ways in which chemicals perturb biological systems and lead to adverse outcomes, have the potential to provide a quantitative analysis of large networks of molecular and functional changes occurring across multiple levels of biological organization114. Crucially, such frameworks provide a supportive basis for translation between in vitro and in vivo model systems and translation into improved predictions of the therapeutic index in humans. Although such approaches are at first daunting, it seems possible that links could be established mathematically and statistically by leveraging public domain toxicological and bioinformatics databases. The key to success will be in successfully integrating the toxicological network components of approved drugs and failed drug candidates and enabling the construction of more holistic or global networks115. The sum of the accumulated knowledge-based molecular events, from molecular initial events to the intermediate multiple linear and/or branched key events, to adverse outcomes can be advantageously captured as so-called adverse outcome pathways for general reuse116. The field of adverse outcome pathways, which can be considered as the systematic capture of knowledge generated by mechanistic toxicology in general, has attracted enough attention and interest to trigger an Organization for Economic Cooperation and Development (OECD) guideline117.
In our surveys, there was a slight increase in the perception of the overall game-changing perception of M&S, whereas the perception of the potential of systems toxicology declined. However, as described above, the borders of the two technologies have become blurred in recent years. Toxicologists have become familiar with M&S approaches through the use in translational in vivo exposure prediction, whereas systems toxicology was primarily seen as modelling responses in in vitro systems. Recent use of in vitro data for modelling human DILI risk together with PBPK models provides a good example of how these technologies are merging118, and indicates that the game-changing potential should be considered from the M&S perspective.
iPSCs and advanced cellular models
To generate more translationally relevant data, toxicologists are increasingly using complex human and animal MPS models (organs-on-chips or organoids) to provide insight into organ-specific and inter-organ toxicity profiles119,120,121.
iPSC-derived cardiomyocytes (iPSC-CMs) are the most advanced iPSC-derived model to be implemented across the pharmaceutical industry for the assessment of cardiotoxicity and cardiac electrophysiology122,123. Nevertheless, even the newer, more complex iPSC-CM disease models still possess a clear embryonic phenotype124, and their implementation in routine testing within the pharmaceutical industry has been generally sporadic and slow.
Progress with 3D hepatocyte culture systems (including co-culture with non-parenchymal cells and incorporation of biophysical constraints such as oxygen tension or extracellular matrix) have led to additional improvement in recapitulating physiological tissue and/or organ level functions125. In addition, liver MPS based on primary hepatocytes, as well as non-parenchymal and immune cells, have the potential to emulate the genetic, physiological and disease setting of donors and reproduce context-specific toxicology126,127,128. However, differentiation of iPSCs towards hepatocytes has been more challenging than differentiation towards cardiomyocytes, and so toxicity profiling has focused on primary cell systems129,130. Nevertheless, promising translational toxicology studies are emerging. For example, repetitive exposure of hepatocytes to bile acids sensitizes them to compounds with cholestatic liabilities131,132, and elevation of glucose levels in the culture medium resulted in features reminiscent of hepatic metabolic syndrome133. Models for insulin resistance, steatosis and altered drug metabolism have also been reported133,134,135. Advances with MPS-based liver systems now allow recapitulation of complex human liver lobule morphology and coupling to plasma flow and tissue perfusion136. Further development of these models is required to fully emulate the in vivo hepatobiliary situation and include chemically induced biliary hyperplasia in early safety assessment.
Substantial progress is also being made on other MPS tissue models, including of the gastrointestinal tract (primary tissue and stem cell-derived organoids, including ‘stretch’ models to mimic peristalsis)137,138, lung (air–liquid interface epithelia with stretch to mimic breathing dynamics coupled to endothelia)139,140 and kidney (mini-kidneys under microphysiological flow)141,142. These models still need to be developed and validated as fit for purpose for use in drug discovery and safety testing.
MPS models of individual organs could be integrated to make multi-organ configurations for investigative toxicology, as illustrated by the example connecting a heart-on-a-chip with a liver-on-a-chip discussed above21. Ideally, these systems will be integrated with robust systemic modelling of ADME pathways and toxicity circuits143. The development of MPS systems that are representative of specific human subpopulations using iPSCs derived from them could also be valuable144,145,146. With further advances, MPS have the potential to be more reliable, specific and predictive than conventional drug testing on animals and cell cultures, in line with the 3Rs framework (replacement, reduction and refinement of animals in research)147.
In our surveys, both iPSCs and organs-on-chips were perceived to have a marked increase in their game-changing potential between 2015 and 2020, which is reflected in key publications148,149. Nevertheless, substantial bio-engineering, cell sourcing and characterization (including reproducibility and scalability) challenges with these advanced systems need to be addressed before they become widely integrated in industry workflows150. In addition, the cost of MPS, either developed internally, through academic collaborations, commercially purchased or assays contracted out, is still perceived as too high for routine toxicological use, until its impact on drug development is proved151.
Genome editing
The development of genome editing platforms such as CRISPR–Cas9 has enabled researchers to gain fundamental insights into the contribution of single gene products to diseases152,153. When focusing on understanding the specific biological mechanism of toxicity, or adverse outcome pathway, having the ability to accurately and specifically delete or modify a particular gene can also provide insights into the potential safety liabilities of a drug target154,155. For example, a genome-wide CRISPR–Cas9 screen identified novel genes that are protective against, or cause susceptibility to, acetaminophen-induced liver injury156. However, although the technology is being used widely in fundamental life science research, published examples of direct application in investigative toxicology in the pharmaceutical industry are limited so far154,155. This may explain why there was a slight decline in the perception of the game-changing potential of this technology between the two surveys.
Challenges and outlook
The technologies discussed in this article are illustrative examples of some recent approaches considered most likely in our survey to address gaps in mechanistic understanding of drug safety and facilitate drug candidate selection, but the discussion is not exhaustive, as the purpose of this article is to clarify the current status of the field, rather than overview each approach in depth. There is much work remaining to advance technologies that are delivering promising results, such as organs-on-chips. Furthermore, as newer technologies emerge and are developed, there may be further potential for integration. For example, the recent integration of technologies such as single-cell genomics with in situ imaging approaches into the broader domain of spatial transcriptomics may help with mapping biological mechanisms relevant to toxicology from the cellular level to the tissue level and ultimately to the in vivo level. Single-cell genomics has been broadly adopted owing to the granularity of data it provides for heterogeneous primary cell cultures and native tissues, but unfortunately the necessary cellular dissociation from tissues leads to the loss of key information about local cellular interactions157. The potential to localize hundreds or even thousands of transcripts in situ with emerging spatial transcriptomics approaches could hugely improve understanding of biological processes, particularly for complex tissues such as the brain157,158. In addition, the growing volume of ‘big data’ generated by spatial transcriptomics is coinciding with the upsurge of AI capabilities applied to biology159,160,161. Digital pathology162, AI-assisted protein folding and function prediction163, synthetic biology164, microbiome–drug interactions165 and biosensors for instant biological parameter monitoring166 are other emerging areas with potential impact on investigative toxicology that could merit deeper evaluation.
The limited understanding of the true potential of an emerging technology and the extent of the work needed to establish it are fundamental issues that face investigational toxicologists when assessing which approaches to invest in, given that the balance between proven costs and hypothetical impacts will remain the driver for expenditure decisions within the pharmaceutical industry. Importantly, the assays and technologies used for investigative toxicology in industry are neither defined nor required by regulatory guidelines, in contrast to certain in vitro assays in genetic toxicology and phototoxicity testing. This differentiating factor carries both opportunities and challenges. Although the freedom to select, establish and verify test systems remains mainly at the discretion of individual companies, this liberty results in highly fragmented approaches to assays and systems, as evidenced by the findings of our survey. The selection of approaches is mainly driven by the history of failures in individual companies, as can be seen for off-target screens that are applied in most companies, but with a high degree of heterogeneity37. In many cases this heterogeneity, together with poor mechanistic understanding, precludes a statistical evaluation of assays or strategies to confidently predict risk. The added value of a new assay can then only be shown on a cursory basis and lacks evidence for a solid translational prediction.
Furthermore, it should be highlighted that the adoption and integration of any promising new technology into preclinical safety testing needs to be fully evaluated in the light of its translational value for human subjects and patients in clinical trials. Unlike fundamental research or ADME prediction approaches, which can afford trial and error in proving or disproving a hypothesis, technologies used to ascertain whether a candidate is sufficiently safe to enter clinical trials need proven robustness. Therefore, an important strategy for investigative toxicology is to gather assay performance data and impact metrics to justify the selection of specific assays and test systems and demonstrate the added value to drug design and compound selection. This can be illustrated by the adoption of in vitro hERG assessment (Box 3) or screening assays for genetic toxicity, where the concordance with the apical (torsade de pointes) or the corresponding regulatory end points (test battery according to ICH S2(R1)), respectively, with acceptable levels of mechanistic insight are well documented167,168,169,170,171.
Given the outlined needs for harmonization and concomitant value analysis, and that the extent of the work to address these needs is very likely to be beyond any one organization, pre-competitive consortia will continue to have a crucial role in the investigative toxicology field. It is beyond the scope of this article to provide a comprehensive list of global consortia activities, and owing to the European affiliation of the authors, we focus on those in the umbrella of the European Innovative Medicines Initiative (IMI, now the Innovative Health Initiative (IHI)), a public–private partnership between the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the European Commission.
A central aspect for harmonization and qualification of new technologies is the ability to share company internal data in a safe and trusted manner. The eTOX consortium is one example of a successful large-scale preclinical data-sharing project that has set the stage in terms of data clearance and sharing for further such consortia (Box 4), and is currently being pursued in the IMI eTRANSAFE project, which extends the scope to clinical safety data sharing for concordance analysis172. More pragmatic curated data-sharing approaches across industry partners can also be useful, as illustrated by a study from the Innovation and Quality in Pharmaceutical Development (IQ) consortium based on a blinded database composed of animal and phase I clinical data for 182 molecules that indicated that an absence of toxicity in animal studies strongly predicts a similar outcome in the clinic173.
Consortia approaches are sometimes also needed to distil technological hype into a realistic assessment of an application, as has been done with gene expression profiling in vitro. Numerous international consortia investigated gene expression correlations for specific pharmaco-toxicological end points, but essentially refuted the holistic predictive potential of the technology that was anticipated when it first emerged. As a consequence, the focus of technology has moved to the mechanistic elucidation of toxicities174,175, which was also confirmed in an EFPIA survey in which multiple respondents of the sampled companies indicated an initial intensive investment in toxicogenomics as a predictive tool174,175,176,177, with a shift over time to a more targeted approach that emphasizes mechanism178. In line with this changed view on toxicogenomics, partner organizations have joined forces in the IMI-funded TransQST initiative to share expertise, data and workload to develop novel systems toxicology models of drug-induced toxicity in the liver, kidney, gastrointestinal and cardiovascular systems24. This project is expected to have a major impact with respect to systems toxicology and computational approaches. A further example is the IMI project TransBioLine, which aims to develop qualified biomarkers for the identification of injury of the liver, kidneys, pancreas, blood vessels and central nervous system. Finally, beyond the exploration of new technologies, the advantage of consortia is also the joint training of both academic and industry staff, which was particularly fruitfully implemented in the IMI project SafeSciMet, in which specific courses were offered to train future toxicologists in the understanding of molecular toxicology and its application in the context of drug development and regulatory submission.
Although timely mechanism-based hazard identification is a key goal of investigative toxicology, additional data are needed to exert maximum influence on lead design and candidate drug selection. Risk assessments based on two crucial components are required: robust translational principles that accommodate human relevance in both healthy and disease states; and provision of quantitative translational data that place the assay in the context of human (patho)physiology and, importantly, drug exposure. Understanding how toxicity relates to drug efficacy given the required dose and exposure is crucial in determining whether a drug has an appropriate therapeutic index1,179. Unfortunately, our survey (Supplementary information) revealed that for many observed drug toxicities, there is a major gap in understanding of the molecular, cellular, physiological and histopathological processes, and how observed effects in cell and animal models used in classical toxicology studies relate to potential adverse effects in healthy volunteers and specific patient populations180,181,182. Areas in which the development of emerging technologies could deliver greatest impact in addressing these limitations are highlighted above. Importantly, these limitations mirror a similar situation with regard to efficacy testing, where the target relevance to disease progression and management is only confirmed with proof-of-concept clinical trials, normally phase IIb or even sometimes phase III. Thus, the intention of investigational toxicology in drug discovery is to identify key liabilities by building a platform of evidence early enough using in silico and in vitro models. In addition, it should be ensured that there is chemical scope to design out risks instead of only profiling the most interesting compounds at candidate selection, since at this point the scope (and generally the time) to correct deficiencies will be inherently more limited.
To efficiently exploit the available safety data for early target assessment, three hurdles have to be overcome.
-
Access to scattered information sources: tools that allow versatile and easy access to the heterogeneous data sources need to be developed. For example, for the early assessment of a target, automated processes need to be developed that allow text and data mining of the scientific literature, in parallel with searches in pharmaceutical, toxicological and clinical databases.
-
Management of divergent definitions of similarity: when querying databases for compound effect predictions, sophisticated computational tools to assess similarity beyond just chemical structure (for example, the Tanimoto similarity index) need to be available to relate chemistry to biological effect.
-
Knowledge management databases: integrating the results from complex searches requires automated processing to be able to handle the big data volumes. New approaches to data visualization, machine learning and AI will help to develop hypotheses for early candidates.
Summarizing our survey and our observations, we propose the following action points to help address these hurdles and further advance investigative toxicology.
-
Establish collaborations to evaluate existing and new assays to allow the development of standardized protocols that improve hazard identification and risk assessment (see MIP-DILI as an example31).
-
Establish baseline data sets to provide a reference for progress and enable evaluation of the impact of various investigative toxicology approaches.
-
Engage in open innovation and pre-competitive collaboration; share data within defined boundaries to fully leverage knowledge from the vast industry archive to build predictive safety models and more effectively evaluate the most promising emerging technologies.
-
Develop and share system toxicology and adverse outcome pathway frameworks for drug candidate toxicity to facilitate mechanistic insight and quantitative translational utility.
-
Advance ‘humanized’ models that reflect off-target effects, genetic background and disease.
Responding to this call to action will need broad scientific skills and experience. Investigative toxicology requires expertise in physiology, cell biology, organic chemistry, toxicology, ADME, mechanistic and molecular pharmacology and a good understanding of toxicology and safety regulations, and increasingly, computational toxicology, bioinformatics, systems pharmacology/toxicology, data analysis, genetic skills and pharmacometric skills are also needed. Translation of academically developed expertise in these latter areas into pharmaceutical toxicology will require intensive training, an exercise that the pharmaceutical industry has already successfully accomplished two decades ago with the development of the professional profile of medicinal chemists. To repeat this success, companies, professional societies and academia need to develop a common understanding of the professional profile of an investigative toxicologist and to shape the necessary training. International conferences can be instrumental on this path, as can be seen by the inclusion of specific symposia on investigative toxicology in the pharmaceutical industry during the annual conferences of the European Society of Toxicology (Eurotox)183,184.
In conclusion, we emphasize the need for continued support of existing initiatives and the establishment of programmes that foster a closer and more open collaboration between pharmaceutical investigational toxicologists, academia and technology developers and providers. These alliances and coalition networks would have the goal of increasing the impact of investigational toxicology on the discovery and development of safer medicines.
References
Weaver, R. J. & Valentin, J. P. Today’s challenges to de-risk and predict drug safety in human “Mind-the-Gap”. Toxicol. Sci. 167, 307–321 (2019).
Moggs, J. et al. Investigative safety science as a competitive advantage for Pharma. Expert Opin. Drug Metab. 8, 1071–1082 (2012).
Beilmann, M. et al. Optimizing drug discovery by investigative toxicology: current and future trends. ALTEX 36, 289–313 (2019).
ICH Topic -M3 (R2): Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (European Medicines Agency, 2009).
Roberts, R. A. Understanding drug targets: no such thing as bad news. Drug Discov. Today 23, 1925–1928 (2018).
Favuzza, P. et al. Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 27, 642–658 e612 (2020).
Zuhl, A. M. et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of beta-secretase inhibitors. Nat. Commun. 7, 13042 (2016).
Barber, J. et al. A target safety assessment of the potential toxicological risks of targeting plasmepsin IX/X for the treatment of malaria. Toxicol. Res. 10, 203–213 (2021).
Siintola, E. et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 129, 1438–1445 (2006).
Ramirez-Montealegre, D., Rothberg, P. G. & Pearce, D. A. Another disorder finds its gene. Brain 129, 1353–1356 (2006).
Hornberg, J. J. et al. Exploratory toxicology as an integrated part of drug discovery. Part II: screening strategies. Drug Discov. Today 19, 1137–1144 (2014).
Hornberg, J. J. et al. Exploratory toxicology as an integrated part of drug discovery. Part I: why and how. Drug Discov. Today 19, 1131–1136 (2014).
Atienzar, F. A. et al. Predictivity of dog co-culture model, primary human hepatocytes and HepG2 cells for the detection of hepatotoxic drugs in humans. Toxicol. Appl. Pharm. 275, 44–61 (2014).
Atienzar, F. et al. Investigative safety strategies to improve success in drug development. J. Med. Dev. Sci. 2, 2–29 (2016).
Lin, Z. W. & Will, Y. Evaluation of drugs with specific organ toxicities in organ-specific cell lines. Toxicol. Sci. 126, 114–127 (2012).
Langhans, S. A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 9, 6 (2018).
Low, L. A., Mummery, C., Berridge, B. R., Austin, C. P. & Tagle, D. A. Organs-on-chips: into the next decade. Nat. Rev. Drug Discov. 20, 345–361 (2021).
Natale, A. et al. Technological advancements for the development of stem cell-based models for hepatotoxicity testing. Arch. Toxicol. 93, 1789–1805 (2019).
Baudy, A. R. et al. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 20, 215–225 (2020).
Vulto, P. & Joore, J. Adoption of organ-on-chip platforms by the pharmaceutical industry. Nat. Rev. Drug Discov. 20, 961–962 (2021).
McAleer, C. W. et al. On the potential of in vitro organ-chip models to define temporal pharmacokinetic-pharmacodynamic relationships. Sci. Rep. 9, 9619 (2019).
Vo, A. H., Van Vleet, T. R., Gupta, R. R., Liguori, M. J. & Rao, M. S. An overview of machine learning and big data for drug toxicity evaluation. Chem. Res. Toxicol. 33, 20–37 (2020).
Hunter, F. M. I. et al. Drug safety data curation and modeling in ChEMBL: boxed warnings and withdrawn drugs. Chem. Res. Toxicol. 34, 385–395 (2021).
Ferreira, S. et al. Quantitative systems toxicology modeling to address key safety questions in drug development: a focus of the TransQST consortium. Chem. Res. Toxicol. 33, 7–9 (2020).
Weaver, R. J. et al. Test systems in drug discovery for hazard identification and risk assessment of human drug-induced liver injury. Expert. Opin. Drug Met. 13, 767–782 (2017).
Atienzar, F. A. et al. Key challenges and opportunities associated with the use of in vitro models to detect human DILI: integrated risk assessment and mitigation plans. Biomed. Res. Int. 2016, 9737820 (2016).
Bell, C. C. et al. Transcriptional, functional, and mechanistic comparisons of stem cell-derived hepatocytes, HepaRG cells, and three-dimensional human hepatocyte spheroids as predictive in vitro systems for drug-induced liver injury. Drug Metab. Dispos. 45, 419–429 (2017).
Hart, S. N. et al. A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab. Dispos. 38, 988–994 (2010).
Rogue, A., Lambert, C., Spire, C., Claude, N. & Guillouzo, A. Interindividual variability in gene expression profiles in human hepatocytes and comparison with HepaRG cells. Drug Metab. Dispos. 40, 151–158 (2012).
Castell, J. V., Jover, R., Martinez-Jimenez, C. P. & Gomez-Lechon, M. J. Hepatocyte cell lines: their use, scope and limitations in drug metabolism studies. Expert. Opin. Drug Met. 2, 183–212 (2006).
Sison-Young, R. L. et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Arch. Toxicol. 91, 1385–1400 (2017).
Kamalian, L. et al. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol. Vitr. 29, 732–740 (2015).
Jang, K. J. et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 11, eaax5516 (2019).
Proctor, W. R. et al. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch. Toxicol. 91, 2849–2863 (2017).
Whitebread, S. et al. Secondary pharmacology: screening and interpretation of off-target activities - focus on translation. Drug Discov. Today 21, 1232–1242 (2016).
Whitebread, S., Hamon, J., Bojanic, D. & Urban, L. Keynote review: in vitro safety pharmacology profiling: an essential tool for successful drug development. Drug Discov. Today 10, 1421–1433 (2005).
Bowes, J. et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 11, 909–922 (2012).
Valentin, J. P. et al. In vitro secondary pharmacological profiling: an IQ-DruSafe industry survey on current practices. J. Pharmacol. Toxicol. Methods 93, 7–14 (2018).
Jenkinson, S., Schmidt, F., Rosenbrier Ribeiro, L., Delaunois, A. & Valentin, J. P. A practical guide to secondary pharmacology in drug discovery. J. Pharmacol. Toxicol. Methods 105, 106869 (2020).
Hughes, J. D. et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 18, 4872–4875 (2008).
Irwin, J. J. et al. An aggregation advisor for ligand discovery. J. Med. Chem. 58, 7076–7087 (2015).
Ganesh, A. N., Donders, E. N., Shoichet, B. K. & Shoichet, M. S. Colloidal aggregation: from screening nuisance to formulation nuance. Nano Today 19, 188–200 (2018).
Tarcsay, A. & Keseru, G. M. Contributions of molecular properties to drug promiscuity. J. Med. Chem. 56, 1789–1795 (2013).
Van Vleet, T. R., Liguori, M. J., Lynch, J. J. 3rd, Rao, M. & Warder, S. Screening strategies and methods for better off-target liability prediction and identification of small-molecule pharmaceuticals. SLAS Discov. 24, 1–24 (2019).
Hann, M. M. & Keseru, G. M. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 11, 355–365 (2012).
Papoian, T. et al. Secondary pharmacology data to assess potential off-target activity of new drugs: a regulatory perspective. Nat. Rev. Drug Discov. 14, 294 (2015).
Roth, B. L. Drugs and valvular heart disease. N. Engl. J. Med. 356, 6–9 (2007).
Redfern, W. S. et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 58, 32–45 (2003).
Hameed, S. P. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat. Commun. 6, 6715 (2015).
Morelli, J. K. et al. Validation of an in vitro screen for phospholipidosis using a high-content biology platform. Cell Biol. Toxicol. 22, 15–27 (2006).
Tummino, T. A. et al. Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2. Science 373, 541–547 (2021).
Becher, I. et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 12, 908 (2016).
Kashimura, A., Tanaka, K., Sato, H., Kaji, H. & Tanaka, M. Imaging mass spectrometry for toxicity assessment: a useful technique to confirm drug distribution in histologically confirmed lesions. J. Toxicol. Pathol. 31, 221–227 (2018).
Goodwin, R. J. A., Takats, Z. & Bunch, J. A critical and concise review of mass spectrometry applied to imaging in drug discovery. SLAS Discov. 25, 963–976 (2020).
Sahin, M. et al. Spatial evaluation of long-term metabolic changes induced by cisplatin nephrotoxicity. Toxicol. Lett. 334, 36–43 (2020).
Bannasch, P., Moore, M. A., Klimek, F. & Zerban, H. Biological markers of preneoplastic foci and neoplastic nodules in rodent liver. Toxicol. Pathol. 10, 19–34 (1982).
Bannasch, P., Haertel, T. & Su, Q. Significance of hepatic preneoplasia in risk identification and early detection of neoplasia. Toxicol. Pathol. 31, 134–139 (2003).
Boess, F. et al. Use of early phenotypic in vivo markers to assess human relevance of an unusual rodent non-genotoxic carcinogen in vitro. Toxicology 379, 48–61 (2017).
Pognan, F. et al. Induction of hemangiosarcoma in mice after chronic treatment with S1P-modulator siponimod and its lack of relevance to rat and human. Arch. Toxicol. 92, 1877–1891 (2018).
Scott, L. J. Siponimod: a review in secondary progressive multiple sclerosis. CNS Drugs 34, 1191–1200 (2020).
Joubert, M. K. et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 287, 25266–25279 (2012).
Ratanji, K. D., Derrick, J. P., Dearman, R. J. & Kimber, I. Immunogenicity of therapeutic proteins: influence of aggregation. J. Immunotoxicol. 11, 99–109 (2014).
Rubic-Schneider, T. et al. T-cell assays confirm immunogenicity of tungsten-induced erythropoietin aggregates associated with pure red cell aplasia. Blood Adv. 1, 367–379 (2017).
Barrile, R. et al. Organ-on-Chip recapitulates thrombosis induced by an anti-CD154 monoclonal antibody: translational potential of advanced microengineered systems. Clin. Pharmacol. Ther. 104, 1240–1248 (2018).
Scarfe, L. et al. Preclinical imaging methods for assessing the safety and efficacy of regenerative medicine therapies. NPJ Regen. Med. 2, 28 (2017).
Schulz, S., Becker, M., Groseclose, M. R., Schadt, S. & Hopf, C. Advanced MALDI mass spectrometry imaging in pharmaceutical research and drug development. Curr. Opin. Biotechnol. 55, 51–59 (2019).
Uteng, M., Germano, D., Balavenkatraman, K., Pognan, F. & Wolf, A. In: In Vitro Toxicology Systems (eds Bal-Price, A. & Jennings, P.) 377–397 (Humana Press, 2014).
Chandrasekaran, S. N., Ceulemans, H., Boyd, J. D. & Carpenter, A. E. Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat. Rev. Drug Discov. 20, 145–159 (2021).
Li, S. & Xia, M. Review of high-content screening applications in toxicology. Arch. Toxicol. 93, 3387–3396 (2019).
Frieauff, W., Martus, H. J., Suter, W. & Elhajouji, A. Automatic analysis of the micronucleus test in primary human lymphocytes using image analysis. Mutagenesis 28, 15–23 (2013).
Cox, M. J. et al. Tales of 1,008 small molecules: phenomic profiling through live-cell imaging in a panel of reporter cell lines. Sci. Rep. 10, 13262 (2020).
Hussain, F., Basu, S., Heng, J. J. H., Loo, L. H. & Zink, D. Predicting direct hepatocyte toxicity in humans by combining high-throughput imaging of HepaRG cells and machine learning-based phenotypic profiling. Arch. Toxicol. 94, 2749–2767 (2020).
Simm, J. et al. Repurposing high-throughput image assays enables biological activity prediction for drug discovery. Cell Chem. Biol. 25, 611–618 e613 (2018).
Bray, M. A. et al. Cell painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 11, 1757–1774 (2016).
Willis, C., Nyffeler, J. & Harrill, J. Phenotypic profiling of reference chemicals across biologically diverse cell types using the cell painting assay. SLAS Discov. 25, 755–769 (2020).
Forootan, S. S. et al. Real-time in vivo imaging reveals localised Nrf2 stress responses associated with direct and metabolism-dependent drug toxicity. Sci. Rep. 7, 16084 (2017).
Liachenko, S. Translational imaging in toxicology. Curr. Opin. Toxicol. 23–24, 29–38 (2020).
Kampa, J. M. et al. Mass spectrometry imaging reveals lipid upregulation and bile acid changes indicating amitriptyline induced steatosis in a rat model. Toxicol. Lett. 325, 43–50 (2020).
Thomen, A. et al. Subcellular mass spectrometry imaging and absolute quantitative analysis across organelles. ACS Nano 14, 4316–4325 (2020).
Jacobsen, M. et al. Utilizing whole slide images for the primary evaluation and peer review of a GLP-compliant rodent toxicology study. Toxicol. Pathol. 49, 1164–1173 (2021).
Latonen, L. & Ruusuvuori, P. Building a central repository landmarks a new era for artificial intelligence-assisted digital pathology development in Europe. Eur. J. Cancer 150, 31–32 (2021).
Moulin, P., Grunberg, K., Barale-Thomas, E. & der Laak, J. V. IMI-bigpicture: a central repository for digital pathology. Toxicol. Pathol. 49, 711–713 (2021).
Pettit, S. et al. Current and future applications of toxicogenomics: results summary of a survey from the HESI Genomics State of Science Subcommittee. Env. Health Perspect. 118, 992–997 (2010).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452 e1417 (2017).
Franzosa, J. A. et al. High-throughput toxicogenomic screening of chemicals in the environment using metabolically competent hepatic cell cultures. NPJ Syst. Biol. Appl. 7, 7 (2021).
Sutherland, J. J. et al. Toxicogenomic module associations with pathogenesis: a network-based approach to understanding drug toxicity. Pharmacogenomics J. 18, 377–390 (2018).
Schraml, E., Hackl, M. & Grillari, J. MicroRNAs and toxicology: a love marriage. Toxicol. Rep. 4, 634–636 (2017).
Balasubramanian, S., Gunasekaran, K., Sasidharan, S., Jeyamanickavel Mathan, V. & Perumal, E. MicroRNAs and xenobiotic toxicity: an overview. Toxicol. Rep. 7, 583–595 (2020).
de Rie, D. et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 35, 872 (2017).
Isakova, A., Fehlmann, T., Keller, A. & Quake, S. R. A mouse tissue atlas of small noncoding RNA. Proc. Natl Acad. Sci. USA 117, 25634–25645 (2020).
Ludwig, N. et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 44, 3865–3877 (2016).
Jopling, C. Liver-specific microRNA-122: biogenesis and function. RNA Biol. 9, 137–142 (2012).
Mukhadi, S., Hull, R., Mbita, Z. & Dlamini, Z. The role of microRNAs in kidney disease. Noncoding RNA 1, 192–221 (2015).
Colpaert, R. M. W. & Calore, M. MicroRNAs in cardiac diseases. Cells 8, 737 (2019).
Lin, H. X., Ewing, L. E., Koturbash, I., Gurley, B. J. & Miousse, I. R. MicroRNAs as biomarkers for liver injury: current knowledge, challenges and future prospects. Food Chem. Toxicol. 110, 229–239 (2017).
Gerlach, C. V. & Vaidya, V. S. MicroRNAs in injury and repair. Arch. Toxicol. 91, 2781–2797 (2017).
Baharum, S. N. & Azizan, K. A. Metabolomics in systems biology. Adv. Exp. Med. Biol. 1102, 51–68 (2018).
Zaitsu, K., Hayashi, Y., Kusano, M., Tsuchihashi, H. & Ishii, A. Application of metabolomics to toxicology of drugs of abuse: a mini review of metabolomics approach to acute and chronic toxicity studies. Drug Metab. Pharmacokinet. 31, 21–26 (2016).
Olesti, E., Gonzalez-Ruiz, V., Wilks, M. F., Boccard, J. & Rudaz, S. Approaches in metabolomics for regulatory toxicology applications. Analyst 146, 1820–1834 (2021).
Di Minno, A. et al. Challenges in metabolomics-based tests, biomarkers revealed by metabolomic analysis, and the promise of the application of metabolomics in precision medicine. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23095213 (2022).
Tice, R. R., Austin, C. P., Kavlock, R. J. & Bucher, J. R. Improving the human hazard characterization of chemicals: a Tox21 update. Environ. Health Perspect. 121, 756–765 (2013).
Huang, R. L. et al. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat. Commun. 7, 10425 (2016).
Williams, D. P., Lazic, S. E., Foster, A. J., Semenova, E. & Morgan, P. Predicting drug-induced liver injury with Bayesian machine learning. Chem. Res. Toxicol. 33, 239–248 (2020).
Minerali, E., Foil, D. H., Zorn, K. M., Lane, T. R. & Ekins, S. Comparing machine learning algorithms for predicting drug-induced liver injury (DILI). Mol. Pharm. 17, 2628–2637 (2020).
Rusyn, I., Sedykh, A., Low, Y., Guyton, K. Z. & Tropsha, A. Predictive modeling of chemical hazard by integrating numerical descriptors of chemical structures and short-term toxicity assay data. Toxicol. Sci. 127, 1–9 (2012).
Huang, R. L. & Xia, M. H. Editorial: Tox21 challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental toxicants and drugs. Front. Environ. Sci. 5, 3 (2017).
Clark, M. & Steger-Hartmann, T. A big data approach to the concordance of the toxicity of pharmaceuticals in animals and humans. Regul. Toxicol. Pharm. 96, 94–105 (2018).
Tylutki, Z., Mendyk, A. & Polak, S. Physiologically based pharmacokinetic-quantitative systems toxicology and safety (PBPK-QSTS) modeling approach applied to predict the variability of amitriptyline pharmacokinetics and cardiac safety in populations and in individuals. J. Pharmacokinet. Pharmacodyn. 45, 663–677 (2018).
Andersen, M. E., Clewell, H. J. III & Frederick, C. B. Applying simulation modeling to problems in toxicology and risk assessment–a short perspective. Toxicol. Appl. Pharmacol. 133, 181–187 (1995).
Diaz Ochoa, J. G. et al. A multi-scale modeling framework for individualized, spatiotemporal prediction of drug effects and toxicological risk. Front. Pharmacol. 3, 204 (2012).
Friberg, L. E., Henningsson, A., Maas, H., Nguyen, L. & Karlsson, M. O. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J. Clin. Oncol. 20, 4713–4721 (2002).
Friberg, L. E., Sandstrom, M. & Karlsson, M. O. Scaling the time-course of myelosuppression from rats to patients with a semi-physiological model. Invest N. Drugs 28, 744–753 (2010).
Collins, T. A. et al. Translational modeling of drug-induced myelosuppression and effect of pretreatment myelosuppression for AZD5153, a selective BRD4 inhibitor. CPT Pharmacomet. Syst. Pharmacol. 6, 357–364 (2017).
Sturla, S. J. et al. Systems toxicology: from basic research to risk assessment. Chem. Res. Toxicol. 27, 314–329 (2014).
Lysenko, A., Sharma, A., Boroevich, K. A. & Tsunoda, T. An integrative machine learning approach for prediction of toxicity-related drug safety. Life Sci. Alliance 1, e2002800098 (2018).
Vinken, M. et al. Adverse outcome pathways: a concise introduction for toxicologists. Arch. Toxicol. 91, 3697–3707 (2017).
Draft Guidance Document for the Scientific Review of Adverse Outcome Pathways (OECD, 2020); https://www.oecd.org/chemicalsafety/testing/Draft_GD_AOP_scientific_review_27_July.pdf
Watkins, P. B. DILIsym: quantitative systems toxicology impacting drug development. Curr. Opin. Toxicol. 23–24, 67–73 (2020).
van Duinen, V., Trietsch, S. J., Joore, J., Vulto, P. & Hankemeier, T. Microfluidic 3D cell culture: from tools to tissue models. Curr. Opin. Biotechnol. 35, 118–126 (2015).
Esch, E. W., Bahinski, A. & Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 14, 248–260 (2015).
Godoy, P. et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 87, 1315–1530 (2013).
Archer, C. R. et al. Characterization and validation of a human 3D cardiac microtissue for the assessment of changes in cardiac pathology. Sci. Rep. 8, 10160 (2018).
Ribas, J. et al. Cardiovascular organ-on-a-chip platforms for drug discovery and development. Appl. Vitr. Toxicol. 2, 82–96 (2016).
Reilly, L., Munawar, S., Zhang, J., Crone, W. C. & Eckhardt, L. L. Challenges and innovation: disease modeling using human-induced pluripotent stem cell-derived cardiomyocytes. Front. Cardiovasc. Med. 9, 966094 (2022).
Serras, A. S. et al. A critical perspective on 3D liver models for drug metabolism and toxicology studies. Front. Cell Dev. Biol. 9, 626805 (2021).
Gupta, R. et al. Comparing in vitro human liver models to in vivo human liver using RNA-Seq. Arch. Toxicol. 95, 573–589 (2021).
Jiang, J., Pieterman, C. D., Ertaylan, G., Peeters, R. L. M. & de Kok, T. M. C. M. The application of omics-based human liver platforms for investigating the mechanism of drug-induced hepatotoxicity in vitro. Arch. Toxicol. 93, 3067–3098 (2019).
Ribeiro, A. J. S., Yang, X. N., Patel, V., Madabushi, R. & Strauss, D. G. Liver microphysiological systems for predicting and evaluating drug effects. Clin. Pharmacol. Ther. 106, 139–147 (2019).
Meier, F. et al. Hepatic differentiation of human iPSCs in different 3D models: a comparative study. Int. J. Mol. Med. 40, 1759–1771 (2017).
Goldring, C. et al. Stem cell-derived models to improve mechanistic understanding and prediction of human drug-induced liver injury. Hepatology 65, 710–721 (2017).
Hendriks, D. F., Fredriksson Puigvert, L., Messner, S., Mortiz, W. & Ingelman-Sundberg, M. Hepatic 3D spheroid models for the detection and study of compounds with cholestatic liability. Sci. Rep. 6, 35434 (2016).
Ni, X. et al. Functional human induced hepatocytes (hiHeps) with bile acid synthesis and transport capacities: a novel in vitro cholestatic model. Sci. Rep. 6, 38694 (2016).
Davidson, M. D., Ballinger, K. R. & Khetani, S. R. Long-term exposure to abnormal glucose levels alters drug metabolism pathways and insulin sensitivity in primary human hepatocytes. Sci. Rep. 6, 28178 (2016).
Kozyra, M. et al. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 8, 14297 (2018).
Lauschke, V. M., Shafagh, R. Z., Hendriks, D. F. G. & Ingelman-Sundberg, M. 3D primary hepatocyte culture systems for analyses of liver diseases, drug metabolism, and toxicity: emerging culture paradigms and applications. Biotechnol. J. 14, e1800347 (2019).
Kang, Y. B., Eo, J., Mert, S., Yarmush, M. L. & Usta, O. B. Metabolic patterning on a chip: towards in vitro liver zonation of primary rat and human hepatocytes. Sci. Rep. 8, 8951 (2018).
Peters, M. F. et al. Developing in vitro assays to transform gastrointestinal safety assessment: potential for microphysiological systems. Lab Chip 20, 1177–1190 (2020).
Kim, H. J., Huh, D., Hamilton, G. & Ingber, D. E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12, 2165–2174 (2012).
Huh, D. et al. Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 (2010).
Bluhmki, T. et al. Development of a miniaturized 96-Transwell air-liquid interface human small airway epithelial model. Sci. Rep. 10, 13022 (2020).
Specioso, G. et al. Apical medium flow influences the morphology and physiology of human proximal tubular cells in a microphysiological system. Bioengineering 9, 516 (2022).
Fransen, M. F. J. et al. Bioprinting of kidney in vitro models: cells, biomaterials, and manufacturing techniques. Essays Biochem. 65, 587–602 (2021).
Ishida, S. Organs-on-a-chip: current applications and consideration points for in vitro ADME-Tox studies. Drug Metab. Pharmacokinet. 33, 49–54 (2018).
Matsa, E. et al. Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 19, 311–325 (2016).
McSweeney, K. M., Bozza, W. P., Alterovitz, W. L. & Zhang, B. L. Transcriptomic profiling reveals p53 as a key regulator of doxorubicin-induced cardiotoxicity. Cell Death Discov. 5, 102 (2019).
Burridge, P. W. et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556 (2016).
Knight, D. J., Deluyker, H., Chaudhry, Q., Vidal, J. M. & de Boer, A. A call for action on the development and implementation of new methodologies for safety assessment of chemical-based products in the EU–a short communication. Regul. Toxicol. Pharmacol. 119, 104837 (2021).
Steger-Hartmann, T. & Raschke, M. Translating in vitro to in vivo and animal to human. Curr. Opin. Toxicol. 23–24, 6–10 (2020).
Marx, U. et al. Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. ALTEX 37, 365–394 (2020).
Ewart, L. & Roth, A. Opportunities and challenges with microphysiological systems: a pharma end-user perspective. Nat. Rev. Drug Discov. 20, 327–328 (2021).
Hargrove-Grimes, P., Low, L. A. & Tagle, D. A. Microphysiological systems: stakeholder challenges to adoption in drug development. Cell Tissues Organs 211, 269–281 (2022).
Li, H. et al. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target. Ther. 5, 1 (2020).
Xu, Y. & Li, Z. CRISPR-Cas systems: overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 18, 2401–2415 (2020).
Lujan, H., Romer, E., Salisbury, R., Hussain, S. & Sayes, C. Determining the biological mechanisms of action for environmental exposures: applying CRISPR/Cas9 to toxicological assessments. Toxicol. Sci. 175, 5–18 (2020).
Sobh, A., Russo, M. & Vulpe, C. D. CRISPR Screens in toxicology research: an overview. Curr. Protoc. 1, e136 (2021).
Shortt, K. et al. Identification of novel regulatory genes in APAP induced hepatocyte toxicity by a genome-wide CRISPR–Cas9 screen. Sci. Rep. 9, 1396 (2019).
Williams, C. G., Lee, H. J., Asatsuma, T., Vento-Tormo, R. & Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 14, 68 (2022).
Rao, A., Barkley, D., Franca, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Tandon, A. et al. Deep learning image analysis of high-throughput toxicology assay images. SLAS Discov. 27, 29–38 (2022).
Zhang, J., Norinder, U. & Svensson, F. Deep learning-based conformal prediction of toxicity. J. Chem. Inf. Model. 61, 2648–2657 (2021).
Zhu, H. Big data and artificial intelligence modeling for drug discovery. Annu. Rev. Pharmacol. Toxicol. 60, 573–589 (2020).
Baxi, V., Edwards, R., Montalto, M. & Saha, S. Digital pathology and artificial intelligence in translational medicine and clinical practice. Mod. Pathol. 35, 23–32 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Cubillos-Ruiz, A. et al. Engineering living therapeutics with synthetic biology. Nat. Rev. Drug Discov. 20, 941–960 (2021).
Zimmermann, M., Patil, K. R., Typas, A. & Maier, L. Towards a mechanistic understanding of reciprocal drug-microbiome interactions. Mol. Syst. Biol. 17, e10116 (2021).
Tao, Y., Chen, L., Pan, M., Zhu, F. & Zhu, D. Tailored biosensors for drug screening, efficacy assessment, and toxicity evaluation. ACS Sens. 6, 3146–3162 (2021).
Kamber, M., Fluckiger-Isler, S., Engelhardt, G., Jaeckh, R. & Zeiger, E. Comparison of the Ames II and traditional Ames test responses with respect to mutagenicity, strain specificities, need for metabolism and correlation with rodent carcinogenicity. Mutagenesis 24, 359–366 (2009).
Wallis, R. M. Integrated risk assessment and predictive value to humans of non-clinical repolarization assays. Br. J. Pharmacol. 159, 115–121 (2010).
Ewart, L. et al. The concordance between nonclinical and phase I clinical cardiovascular assessment from a cross-company data sharing initiative. Toxicol. Sci. 142, 427–435 (2014).
Gintant, G. An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol. Ther. 129, 109–119 (2011).
Park, E. et al. Abstr. P301. Can nonclinical repolarization assays predict the results of clinical thorough QT studies? A HESI-FDA retrospective analysis. Int. J. Toxicol. 36, 70 (2017).
Pognan, F. et al. The eTRANSAFE project on translational safety assessment through integrative knowledge management: achievements and perspectives. Pharmaceuticals 14, 237 (2021).
Monticello, T. M. et al. Current nonclinical testing paradigm enables safe entry to first-in-human clinical trials: the IQ consortium nonclinical to clinical translational database. Toxicol. Appl. Pharmacol. 334, 100–109 (2017).
Suter, L., Babiss, L. E. & Wheeldon, E. B. Toxicogenomics in predictive toxicology in drug development. Chem. Biol. 11, 161–171 (2004).
Uehara, T. et al. Prediction model of potential hepatocarcinogenicity of rat hepatocarcinogens using a large-scale toxicogenomics database. Toxicol. Appl. Pharmacol. 255, 297–306 (2011).
Nie, A. Y. et al. Predictive toxicogenomics approaches reveal underlying molecular mechanisms of nongenotoxic carcinogenicity. Mol. Carcinog. 45, 914–933 (2006).
Vahle, J. L., Anderson, U., Blomme, E. A. G., Hoflack, J. C. & Stiehl, D. P. Use of toxicogenomics in drug safety evaluation: current status and an industry perspective’. Regul. Toxicol. Pharm. 96, 18–29 (2018).
Harrill, A. H. & Rusyn, I. Systems biology and functional genomics approaches for the identification of cellular responses to drug toxicity. Expert Opin. Drug Metab. Toxicol. 4, 1379–1389 (2008).
Muller, P. Y. & Milton, M. N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 11, 751–761 (2012).
Madorran, E., Stozer, A., Bevc, S. & Maver, U. In vitro toxicity model: upgrades to bridge the gap between preclinical and clinical research. Bosn. J. Basic. Med. 20, 157–168 (2020).
Van Norman, G. A. Limitations of animal studies for predicting toxicity in clinical trials: part 2: potential alternatives to the use of animals in preclinical trials. JACC Basic Transl. Sci. 5, 387–397 (2020).
Van Norman, G. A. Limitations of animal studies for predicting toxicity in clinical trials: is it time to rethink our current approach? JACC Basic Transl. Sci. 4, 845–854 (2019).
Lison, D. & Wallace, H. ABSTRACTS of the 54th congress of the european societies of toxicology (EUROTOX 2018) toxicology out of the box Brussels, Belgium, 2nd–5th of September, 2018 Preface. Toxicol. Lett. 295, S1 (2018).
Abstracts of the 55th Congress of the European Societies of Toxicology (EUROTOX 2019). Toxicology–Science providing solutions. Toxicol. Lett. 314, 1–310 (2019).
Bekes, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Moreau, K. et al. Proteolysis-targeting chimeras in drug development: a safety perspective. Br. J. Pharmacol. 177, 1709–1718 (2020).
Garber, K. The PROTAC gold rush. Nat. Biotechnol. 40, 12–16 (2022).
Jones, L. H. et al. Targeted protein degraders: a call for collective action to advance safety assessment. Nat. Rev. Drug Discov. 21, 401–402 (2022).
Wang, T. et al. Investigation of correlation among safety biomarkers in serum, histopathological examination, and toxicogenomics. Int. J. Toxicol. 30, 300–312 (2011).
Wang, L., Plump, A. & Ringel, M. Racing to define pharmaceutical R&D external innovation models. Drug Discov. Today 20, 361–370 (2015).
Khanna, I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov. Today 17, 1088–1102 (2012).
Hofmann, C. Pharma partnering: other people’s science. Mol. Biol. Cell 28, 9–11 (2017).
Stockbridge, N., Morganroth, J., Shah, R. R. & Garnett, C. Dealing with global safety issues was the response to QT-liability of non-cardiac drugs well coordinated? Drug Saf. 36, 167–182 (2013).
Gintant, G., Sager, P. T. & Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 15, 457–471 (2016).
Woosley, R. L., Chen, Y., Freiman, J. P. & Gillis, R. A. Mechanism of the cardiotoxic actions of terfenadine. JAMA 269, 1532–1536 (1993).
Shah, R. R. The significance of QT interval in drug development. Br. J. Clin. Pharmacol. 54, 188–202 (2002).
ICH S7B: The Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals CPMP/ICH/423/02 (European Medicines Agency, 2005).
Lindgren, S. et al. Benchmarking safety pharmacology regulatory packages and best practice. J. Pharmacol. Toxicol. Methods 58, 99–109 (2008).
Sager, P. T., Gintant, G., Turner, J. R., Pettit, S. & Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 167, 292–300 (2014).
Vargas, H. M. et al. Time for a fully integrated nonclinical-clinical risk assessment to streamline QT prolongation liability determinations: a pharma industry perspective. Clin. Pharmacol. Ther. 109, 310–318 (2021).
De Ponti, F. in Antitargets: Prediction and Prevention of Drug Side Effects Methods and Principles in Medicinal Chemistry (eds Vaz, R. J. & Klabunde, T.) Ch. 3, 53–88 (Wiley‐VCH, 2008).
Strauss, D. G. et al. Comprehensive in vitro proarrhythmia assay (CiPA) update from a cardiac safety research consortium/health and environmental sciences institute/FDA meeting. Ther. Innov. Regul. Sci. 53, 519–525 (2019).
Asakura, K. et al. Improvement of acquisition and analysis methods in multi-electrode array experiments with iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 75, 17–26 (2015).
Blinova, K. et al. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol. Sci. 155, 234–247 (2017).
Fermini, B. et al. A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J. Biomol. Screen. 21, 1–11 (2016).
Li, Z. et al. Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether-a-go-go-related gene) channel-drug binding kinetics and multichannel pharmacology. Circ. Arrhythm. Electrophysiol. 10, e004628 (2017).
O’Hara, T., Virag, L., Varro, A. & Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7, e1002061 (2011).
Peng, S., Lacerda, A. E., Kirsch, G. E., Brown, A. M. & Bruening-Wright, A. The action potential and comparative pharmacology of stem cell-derived human cardiomyocytes. J. Pharmacol. Toxicol. Methods 61, 277–286 (2010).
Pugsley, M. K., Curtis, M. J. & Hayes, E. S. Biophysics and molecular biology of cardiac ion channels for the safety pharmacologist. Handb. Exp. Pharmacol. 229, 149–203 (2015).
Vicente, J. et al. Assessment of multi-ion channel block in a phase I randomized study design: results of the CiPA phase I ECG biomarker validation study. Clin. Pharmacol. Ther. 105, 943–953 (2019).
Colatsky, T. et al. The comprehensive in vitro proarrhythmia assay (CiPA) initiative – update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20 (2016).
Authier, S. et al. Proarrhythmia liability assessment and the comprehensive in vitro Proarrhythmia Assay (CiPA): an industry survey on current practice. J. Pharmacol. Toxicol. Methods 86, 34–43 (2017).
ICH Guideline E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-antiarrhythmic Drugs (R3) – Questions and Answers EMA/CHMP/ICH/310133/2008 (European Medicines Agency, 2016).
Darpo, B., Garnett, C., Keirns, J. & Stockbridge, N. Implications of the IQ-CSRC prospective study: time to revise ICH E14. Drug Saf. 38, 773–780 (2015).
Darpo, B. & Ferber, G. The new S7B/E14 question and answer draft guidance for industry: contents and commentary. J. Clin. Pharmacol. 61, 1261–1273 (2021).
Ando, H. et al. A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 84, 111–127 (2017).
Kanda, Y., Yamazaki, D., Kurokawa, J., Inutsuka, T. & Sekino, Y. Points to consider for a validation study of iPS cell-derived cardiomyocytes using a multi-electrode array system. J. Pharmacol. Toxicol. Methods 81, 196–200 (2016).
Final Concept Paper: ICH S7B and E14 Q&A (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), 2018).
ICH E14/S7B Implementation Working Group: Clinical and Nonclinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential. Questions and Answers (European Medicines Agency, 2022).
Strauss, D. G., Wu, W. W., Li, Z., Koerner, J. & Garnett, C. Translational models and tools to reduce clinical trials and improve regulatory decision making for QTc and proarrhythmia risk (ICH E14/S7B Updates). Clin. Pharmacol. Ther. 109, 319–333 (2021).
Bajusz, D., Racz, A. & Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. J. Cheminformatics 7, 20 (2015).
Montanari, F. et al. Flagging drugs that inhibit the bile salt export pump. Mol. Pharm. 13, 163–171 (2016).
Ozer, J. S. et al. A panel of urinary biomarkers to monitor reversibility of renal injury and a serum marker with improved potential to assess renal function. Nat. Biotechnol. 28, 486–494 (2010).
Harpur, E. et al. Biological qualification of biomarkers of chemical-induced renal toxicity in two strains of male rat. Toxicol. Sci. 122, 235–252 (2011).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
All the authors are employed by pharmaceutical companies, as listed in their addresses. In addition, some or all may hold shares or stock options in their respective companies.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks Bob van de Water, Wendy Hu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
BigPicture: https://bigpicture.eu/
CiPA project: https://cipaproject.org/about-cipa
eTRANSAFE: https://etransafe.eu
Innovative Medicines Initiative: https://www.imi.europa.eu/
LINCS: https://lincsproject.org
mIRbase database: https://www.mirbase.org/cgi-bin/mirna_summary.pl?org=hsa
Predictive Safety Testing Consortium: https://c-path.org/programs/pstc/
SafeSciMet: https://www.imi.europa.eu/projects-results/project-factsheets/safescimet
TransBioLine: https://transbioline.com/project/
TransQST: https://transqst.org
Supplementary information
Glossary
- Hazards
-
In drug discovery and development, hazards relate to the potential for a drug candidate to cause toxicity to humans. Hazards are often qualified according to the type of toxicity, such as genetic toxicity, irritation or hepatic toxicity.
- hERG channel
-
The human ether-à-go-go-related gene (hERG), also known as KCNH2, codes for a protein known as Kv11.1, the α-subunit of a potassium ion channel. This ion channel (sometimes simply denoted as ‘hERG’) is best known for its contribution to the electrical activity of the heart: the hERG channel mediates the repolarizing IKr current in the cardiac action potential, which helps to coordinate the heart’s beating.
- Metabolomics
-
The comprehensive analysis of hundreds of metabolites in a biological sample, whereas metabonomics is the quantitative measurement of the dynamic multi-parametric metabolic response of living systems to pathophysiological stimuli or genetic modification.
- Risk assessment
-
This involves consideration of the likelihood, the severity and the reversibility of the safety hazard at a given drug exposure (often relative to the anticipated efficacious exposure) required for a given toxicity to occur.
- Therapeutic index
-
(TI). The margin between the dose or the concentration of a compound that produces the desired effect and the dose that produces unwanted side effects. TI is frequently used as a synonym for safety margin (SM); however, in industry, TI is commonly used to describe strictly calculated margins from in vitro assays, whereas SM is more often used to describe in vivo margins based on animal exposures and predicted or determined efficacious doses in humans.
- Tox21
-
An association of four US government agencies (National Institute of Environmental Health Sciences (NIEHS), National Center for Advancing Translational Sciences (NCATS), FDA and National Center for Competency Testing (NCCT)) to research, develop, evaluate and translate innovative test methods in toxicology, including robotic high-throughput screening to better predict how substances may affect humans and the environment.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pognan, F., Beilmann, M., Boonen, H.C.M. et al. The evolving role of investigative toxicology in the pharmaceutical industry. Nat Rev Drug Discov 22, 317–335 (2023). https://doi.org/10.1038/s41573-022-00633-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-022-00633-x
This article is cited by
-
Non-toxicity of Plant Candicidal Peptides for Mammalian Cell Lines and Galleria mellonella Model to Improving Selectivity for Clinical Use
International Journal of Peptide Research and Therapeutics (2024)
-
Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy
Molecular Cancer (2023)
-
CSL-Tox: an open-source analytical framework for the comparison of short-term and long-term toxicity end points and assessing the need of chronic studies in drug development
Scientific Reports (2023)
-
A semiconductor 96-microplate platform for electrical-imaging based high-throughput phenotypic screening
Nature Communications (2023)
