
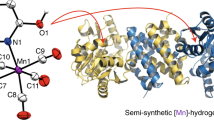
Abstract
Metal cofactors considerably widen the catalytic space of naturally occurring enzymes whose specific and enantioselective catalytic activity constitutes a blueprint for economically relevant chemical syntheses. To optimize natural enzymes and uncover novel reactivity, we need a detailed understanding of cofactor–protein interactions, which can be challenging to obtain in the case of enzymes with sophisticated cofactors. As a case study, we summarize recent research on the [FeFe]-hydrogenases, which interconvert protons, electrons and dihydrogen at a unique iron-based active site. We can now chemically synthesize the complex cofactor and incorporate it into an apo-protein to afford functional enzymes. By varying both the cofactor and the polypeptide components, we have obtained detailed knowledge on what is required for a metal cluster to process H2. In parallel, the design of artificial proteins and catalytically active nucleic acids are advancing rapidly. In this Perspective, we introduce these fields and outline how chemists and biologists can use this knowledge to develop novel tailored semisynthetic catalysts.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others
References
Yannone, S. M., Hartung, S., Menon, A. L., Adams, M. W. & Tainer, J. A. Metals in biology: defining metalloproteomes. Curr. Opin. Biotechnol. 23, 89–95 (2012).
Waldron, K. J., Rutherford, J. C., Ford, D. & Robinson, N. J. Metalloproteins and metal sensing. Nature 460, 823–830 (2009).
Andreini, C., Bertini, I., Cavallaro, G., Holliday, G. L. & Thornton, J. M. Metal ions in biological catalysis: from enzyme databases to general principles. J. Biol. Inorg. Chem. 13, 1205–1218 (2008).
Österberg, R. Origins of metal ions in biology. Nature 249, 382–383 (1974).
Anbar, A. D. Elements and evolution. Science 322, 1481–1483 (2008).
Liu, J. et al. Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469 (2014).
Hosseinzadeh, P. & Lu, Y. Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim. Biophys. Acta 1857, 557–581 (2016).
Valdez, C. E., Smith, Q. A., Nechay, M. R. & Alexandrova, A. N. Mysteries of metals in metalloenzymes. Acc. Chem. Res. 47, 3110–3117 (2014).
Kepp, K. P. Heme: from quantum spin crossover to oxygen manager of life. Coord. Chem. Rev. 344, 363–374 (2017).
Lubitz, W., Ogata, H., Rüdiger, O. & Reijerse, E. Hydrogenases. Chem. Rev. 114, 4081–4148 (2014).
Hoffman, B. M., Lukoyanov, D., Yang, Z.-Y., Dean, D. R. & Seefeldt, L. C. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev. 114, 4041–4062 (2014).
Berggren, G. et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 499, 66–69 (2013).
Esselborn, J. et al. Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat. Chem. Biol. 9, 607–609 (2013).
Esselborn, J. et al. A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem. Sci. 7, 959–968 (2016).
Siebel, J. F. et al. Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity. Biochemistry 54, 1474–1483 (2015).
Holm, R. H., Kennepohl, P. & Solomon, E. I. Structural and functional aspects of metal sites in biology. Chem. Rev. 96, 2239–2314 (1996).
Sheng, Y. et al. Superoxide dismutases and superoxide reductases. Chem. Rev. 114, 3854–3918 (2014).
Vallee, B. L. & Williams, R. J. Metalloenzymes: the entatic nature of their active sites. Proc. Natl Acad. Sci. USA 59, 498–505 (1968).
Kounosu, A. et al. Engineering a three-cysteine, one-histidine ligand environment into a new hyperthermophilic archaeal Rieske-type [2Fe-2S] ferredoxin from Sulfolobus solfataricus. J. Biol. Chem. 279, 12519–12528 (2004).
Bak, D. W. & Elliott, S. J. Alternative FeS cluster ligands: tuning redox potentials and chemistry. Curr. Opin. Chem. Biol. 19, 50–58 (2014).
Smith, L. J., Kahraman, A. & Thornton, J. M. Heme proteins — diversity in structural characteristics, function, and folding. Proteins 78, 2349–2368 (2010).
Colquhoun, H. M., Stoddart, J. F. & Williams, D. J. Second-sphere coordination — a novel role for molecular receptors. Angew. Chem. Int. Ed. 25, 487–507 (1986).
Meyer, J. Iron–sulfur protein folds, iron–sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 13, 157–170 (2008).
Paddock, M. L. et al. MitoNEET is a uniquely folded 2Fe−2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl Acad. Sci. USA 104, 14342–14347 (2007).
Zuris, J. A. et al. Engineering the redox potential over a wide range within a new class of FeS proteins. J. Am. Chem. Soc. 132, 13120–13122 (2010).
Bak, D. W., Zuris, J. A., Paddock, M. L., Jennings, P. A. & Elliott, S. J. Redox characterization of the FeS protein MitoNEET and impact of thiazolidinedione drug binding. Biochemistry 48, 10193–10195 (2009).
Liu, X., Kim, C. N., Yang, J., Jemmerson, R. & Wang, X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 (1996).
Kagan, V. E. et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232 (2005).
Miller, G. T., Zhang, B., Hardman, J. K. & Timkovich, R. Converting a c-type to a b-type cytochrome: Met61 to His61 mutant of Pseudomonas cytochrome c-551. Biochemistry 39, 9010–9017 (2000).
Raphael, A. L. & Gray, H. B. Axial ligand replacement in horse heart cytochrome c by semisynthesis. Proteins 6, 338–340 (1989).
Kroll, T. et al. Resonant inelastic X-ray scattering on ferrous and ferric bis-imidazole porphyrin and cytochrome c: nature and role of the axial methionine–Fe bond. J. Am. Chem. Soc. 136, 18087–18099 (2014).
Mara, M. W. et al. Metalloprotein entatic control of ligand–metal bonds quantified by ultrafast x-ray spectroscopy. Science 356, 1276–1280 (2017).
Kagan, V. E. et al. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic. Biol. Med. 46, 1439–1453 (2009).
Kapralov, A. A. et al. The hierarchy of structural transitions induced in cytochrome c by anionic phospholipids determines its peroxidase activation and selective peroxidation during apoptosis in cells. Biochemistry 46, 14232–14244 (2007).
Vance, C. K. & Miller, A.-F. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high Em. Biochemistry 40, 13079–13087 (2001).
Vance, C. K. & Miller, A.-F. A simple proposal that can explain the inactivity of metal-substituted superoxide dismutases. J. Am. Chem. Soc. 120, 461–467 (1998).
Miller, A. F. Redox tuning over almost 1 V in a structurally conserved active site: lessons from Fe-containing superoxide dismutase. Acc. Chem. Res. 41, 501–510 (2008).
Fernández-Gacio, A., Codina, A., Fastrez, J., Riant, O. & Soumillion, P. Transforming carbonic anhydrase into epoxide synthase by metal exchange. ChemBioChem 7, 1013–1016 (2006).
Okrasa, K. & Kazlauskas, R. J. Manganese-substituted carbonic anhydrase as a new peroxidase. Chem. Eur. J. 12, 1587–1596 (2006).
Key, H. M., Dydio, P., Clark, D. S. & Hartwig, J. F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 534, 534–537 (2016).
Oohora, K., Kihira, Y., Mizohata, E., Inoue, T. & Hayashi, T. C(sp3)–H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J. Am. Chem. Soc. 135, 17282–17285 (2013).
Ogata, H., Nishikawa, K. & Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 520, 571–574 (2015).
Vignais, P. M. & Billoud, B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272 (2007).
Hiromoto, T. et al. The crystal structure of C176A mutated [Fe]-hydrogenase suggests an acyl–iron ligation in the active site iron complex. FEBS Lett. 583, 585–590 (2009).
Shima, S. et al. The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 321, 572–575 (2008).
Ogata, H. et al. Activation process of [NiFe] hydrogenase elucidated by high-resolution X-ray analyses: conversion of the ready to the unready state. Structure 13, 1635–1642 (2005).
Volbeda, A. et al. Crystal structure of the nickel–iron hydrogenase from Desulfovibrio gigas. Nature 373, 580 (1995).
Peters, J. W., Lanzilotta, W. N., Lemon, B. J. & Seefeldt, L. C. X-Ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282, 1853–1858 (1998).
Nicolet, Y., Piras, C., Legrand, P., Hatchikian, C. E. & Fontecilla-Camps, J. C. Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure 7, 13–23 (1999).
Nicolet, Y. et al. Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans. J. Am. Chem. Soc. 123, 1596–1601 (2001).
Shima, S. & Thauer, R. K. A third type of hydrogenase catalyzing H2 activation. Chem. Rec. 7, 37–46 (2007).
Peters, J. W. et al. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 1853, 1350–1369 (2015).
Schilter, D., Camara, J. M., Huynh, M. T., Hammes-Schiffer, S. & Rauchfuss, T. B. Hydrogenase enzymes and their synthetic models: the role of metal hydrides. Chem. Rev. 116, 8693–8749 (2016).
Winkler, M., Esselborn, J. & Happe, T. Molecular basis of [FeFe]-hydrogenase function: an insight into the complex interplay between protein and catalytic cofactor. Biochim. Biophys. Acta 1827, 974–985 (2013).
Meyer, J. [FeFe] hydrogenases and their evolution: a genomic perspective. Cell. Mol. Life Sci. 64, 1063–1084 (2007).
Silakov, A., Wenk, B., Reijerse, E. & Lubitz, W. 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge. Phys. Chem. Chem. Phys. 11, 6592–6599 (2009).
Tard, C. & Pickett, C. J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 109, 2245–2274 (2009).
Mulder, D. W. et al. Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydAΔEFG. Nature 465, 248–251 (2010).
Mulder, D. W. et al. Activation of HydAΔEFG requires a preformed [4Fe-4S] cluster. Biochemistry 48, 6240–6248 (2009).
Li, H. & Rauchfuss, T. B. Iron carbonyl sulfides, formaldehyde, and amines condense to give the proposed azadithiolate cofactor of the Fe-only hydrogenases. J. Am. Chem. Soc. 124, 726–727 (2002).
Roy, S. & Jones, A. K. Metalloenzymes: cutting out the middleman. Nat. Chem. Biol. 9, 603–605 (2013).
Kertess, L. et al. Chalcogenide substitution in the [2Fe] cluster of [FeFe]-hydrogenases conserves high enzymatic activity. Dalton Trans. 46, 16947–16958 (2017).
Winkler, M. & Happe, T. in Biohydrogen (ed. Rögner, M.) 41–60 (de Gruyter, 2015).
Greco, C. et al. Structural insights into the active-ready form of [FeFe]-hydrogenase and mechanistic details of its inhibition by carbon monoxide. Inorg. Chem. 46, 7256–7258 (2007).
Knörzer, P. et al. Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. J. Biol. Chem. 287, 1489–1499 (2012).
Lampret, O. et al. Interplay between CN− ligands and the secondary coordination sphere of the H-cluster in [FeFe]-hydrogenases. J. Am. Chem. Soc. 139, 18222–18230 (2017).
Morra, S. et al. Site saturation mutagenesis demonstrates a central role for cysteine 298 as proton donor to the catalytic site in CaHydA [FeFe]-hydrogenase. PLoS ONE 7, e48400 (2012).
Cornish, A. J., Gärtner, K., Yang, H., Peters, J. W. & Hegg, E. L. Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum. J. Biol. Chem. 286, 38341–38347 (2011).
Hong, G., Cornish, A. J., Hegg, E. L. & Pachter, R. On understanding proton transfer to the biocatalytic [Fe-Fe]H sub-cluster in [Fe-Fe]H2ases: QM/MM MD simulations. Biochim. Biophys. Acta 1807, 510–517 (2011).
Sode, O. & Voth, G. A. Electron transfer activation of a second water channel for proton transport in [FeFe]-hydrogenase. J. Chem. Phys. 141, 22D527 (2014).
Megarity, C. F. et al. Electrochemical investigations of the mechanism of assembly of the active-site H-cluster of [FeFe]-hydrogenases. J. Am. Chem. Soc. 138, 15227–15233 (2016).
Kertess, L. et al. Influence of the [4Fe−4S] cluster coordinating cysteines on active site maturation and catalytic properties of C. reinhardtii [FeFe]-hydrogenase. Chem. Sci. 8, 8127–8137 (2017).
Noth, J. et al. [FeFe]-Hydrogenase with chalcogenide substitutions at the H-cluster maintains full H2 evolution activity. Angew. Chem. Int. Ed. 55, 8396–8400 (2016).
Reich, H. J. & Hondal, R. J. Why nature chose selenium. ACS Chem. Biol. 11, 821–841 (2016).
Reijerse, E. J. et al. Direct observation of an iron-bound terminal hydride in [FeFe]-hydrogenase by nuclear resonance vibrational spectroscopy. J. Am. Chem. Soc. 139, 4306–4309 (2017).
Winkler, M. et al. Accumulating the hydride state in the catalytic cycle of [FeFe]-hydrogenases. Nat. Commun. 8, 16115 (2017).
Adamska-Venkatesh, A. et al. Artificially maturated [FeFe] hydrogenase from Chlamydomonas reinhardtii: a HYSCORE and ENDOR study of a non-natural H-cluster. Phys. Chem. Chem. Phys. 17, 5421–5430 (2015).
Adamska-Venkatesh, A. et al. New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster. J. Am. Chem. Soc. 136, 11339–11346 (2014).
Mulder, D. W., Guo, Y., Ratzloff, M. W. & King, P. W. Identification of a catalytic iron-hydride at the H-Cluster of [FeFe]-Hydrogenase. J. Am. Chem. Soc. 139, 83–86 (2017).
Shafaat, H. S., Rüdiger, O., Ogata, H. & Lubitz, W. [NiFe] hydrogenases: a common active site for hydrogen metabolism under diverse conditions. Biochim. Biophys. Acta 1827, 986–1002 (2013).
Perotto, C. U. et al. Heterobimetallic [NiFe] complexes containing mixed CO/CN− ligands: analogs of the active site of the [NiFe] hydrogenases. Inorg. Chem. 57, 2558–2569 (2018).
Kaur-Ghumaan, S. & Stein, M. [NiFe] hydrogenases: how close do structural and functional mimics approach the active site? Dalton Trans. 43, 9392–9405 (2014).
Senger, M., Stripp, S. T. & Soboh, B. Proteolytic cleavage orchestrates cofactor insertion and protein assembly in [NiFe]-hydrogenase biosynthesis. J. Biol. Chem. 292, 11670–11681 (2017).
Soboh, B. et al. [NiFe]-hydrogenase maturation in vitro: analysis of the roles of the HybG and HypD accessory proteins. Biochem. J. 464, 169–177 (2014).
Buurman, G., Shima, S. & Thauer, R. K. The metal-free hydrogenase from methanogenic archaea: evidence for a bound cofactor. FEBS Lett. 485, 200–204 (2000).
Bai, L. et al. Towards artificial methanogenesis: biosynthesis of the [Fe]-hydrogenase cofactor and characterization of the semi-synthetic hydrogenase. Faraday Discuss. 198, 37–58 (2017).
Shima, S. et al. Reconstitution of [Fe]-hydrogenase using model complexes. Nat. Chem. 7, 995–1002 (2015).
Bornscheuer, U. T. et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012).
Turner, N. J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 5, 567–573 (2009).
Renata, H., Wang, Z. J. & Arnold, F. H. Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed. 54, 3351–3367 (2015).
Koder, R. L. & Dutton, P. L. Intelligent design: the de novo engineering of proteins with specified functions. Dalton Trans. 0, 3045–3051 (2006).
Fehl, C. & Davis, B. G. Proteins as templates for complex synthetic metalloclusters: towards biologically programmed heterogeneous catalysis. Proc. Math. Phys. Eng. Sci. 472, 20160078 (2016).
Jeschek, M., Panke, S. & Ward, T. R. Artificial metalloenzymes on the verge of new-to-nature metabolism. Trends Biotechnol. 36, 60–72 (2018).
Schwizer, F. et al. Artificial metalloenzymes: reaction scope and optimization strategies. Chem. Rev. 118, 142–231 (2018).
Lichtenstein, B. R. et al. Engineering oxidoreductases: maquette proteins designed from scratch. Biochem. Soc. Trans. 40, 561–566 (2012).
Sommer, D. J. et al. Reengineering cyt b 562 for hydrogen production: a facile route to artificial hydrogenases. Biochim. Biophys. Acta 1857, 598–603 (2016).
Sommer, D. J., Vaughn, M. D. & Ghirlanda, G. Protein secondary-shell interactions enhance the photoinduced hydrogen production of cobalt protoporphyrin IX. Chem. Commun. 50, 15852–15855 (2014).
Sano, Y., Onoda, A. & Hayashi, T. A hydrogenase model system based on the sequence of cytochrome c: photochemical hydrogen evolution in aqueous media. Chem. Commun. 47, 8229–8231 (2011).
Song, W. J. & Tezcan, F. A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014).
Salgado, E. N., Faraone-Mennella, J. & Tezcan, F. A. Controlling protein−protein interactions through metal coordination: assembly of a 16-helix bundle protein. J. Am. Chem. Soc. 129, 13374–13375 (2007).
Brodin, J. D. et al. Evolution of metal selectivity in templated protein interfaces. J. Am. Chem. Soc. 132, 8610–8617 (2010).
Sigman, J. A., Kwok, B. C. & Lu, Y. From myoglobin to heme–copper oxidase: design and engineering of a CuB center into sperm whale myoglobin. J. Am. Chem. Soc. 122, 8192–8196 (2000).
Miner, K. D. et al. A designed functional metalloenzyme that reduces O2 to H2O with over one thousand turnovers. Angew. Chem. Int. Ed. 51, 5589–5592 (2012).
Yu, Y. et al. A designed metalloenzyme achieving the catalytic rate of a native enzyme. J. Am. Chem. Soc. 137, 11570–11573 (2015).
Onoda, A., Kihara, Y., Fukumoto, K., Sano, Y. & Hayashi, T. Photoinduced hydrogen evolution catalyzed by a synthetic diiron dithiolate complex embedded within a protein matrix. ACS Catal. 4, 2645–2648 (2014).
Huang, P.-S., Boyken, S. E. & Baker, D. The coming of age of de novo protein design. Nature 537, 320–327 (2016).
Ljubetič, A., Gradišar, H. & Jerala, R. Advances in design of protein folds and assemblies. Curr. Opin. Chem. Biol. 40, 65–71 (2017).
Ljubetič, A. et al. Design of coiled-coil protein-origami cages that self-assemble in vitro and in vivo. Nat. Biotechnol 35, 1094–1101 (2017).
Dou, J. et al. Sampling and energy evaluation challenges in ligand binding protein design. Protein Sci. 26, 2426–2437 (2017).
Chevalier, A. et al. Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79 (2017).
Tinberg, C. E. et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature 501, 212–216 (2013).
Zanghellini, A. et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 15, 2785–2794 (2006).
Butlerow, A. Formation synthétique d’une substance sucrée. CR Acad. Sci. 53, 145–147 (1861).
Siegel, J. B. et al. Computational protein design enables a novel one-carbon assimilation pathway. Proc. Natl Acad. Sci. USA 112, 3704–3709 (2015).
Siegel, J. B. et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels–Alder reaction. Science 329, 309–313 (2010).
Grzyb, J. et al. De novo design of a non-natural fold for an iron–sulfur protein: alpha-helical coiled-coil with a four-iron four-sulfur cluster binding site in its central core. Biochim. Biophys. Acta 1797, 406–413 (2010).
Grzyb, J. et al. Empirical and computational design of iron–sulfur cluster proteins. Biochim. Biophys. Acta 1817, 1256–1262 (2012).
Antonkine, M. L. et al. Synthesis and characterization of de novo designed peptides modelling the binding sites of [4Fe−4S] clusters in photosystem I. Biochim. Biophys. Acta 1787, 995–1008 (2009).
Scott, M. P. & Biggins, J. Introduction of a [4Fe-4S (S-cys)4]+1,+2 iron-sulfur center into a four-α helix protein using design parameters from the domain of the Fx cluster in the Photosystem I reaction center. Protein Sci. 6, 340–346 (1997).
Roy, A., Sarrou, I., Vaughn, M. D., Astashkin, A. V. & Ghirlanda, G. De novo design of an artificial bis[4Fe-4S] binding protein. Biochemistry 52, 7586–7594 (2013).
Roy, A. et al. A de novo designed 2[4Fe-4S] ferredoxin mimic mediates electron transfer. J. Am. Chem. Soc. 136, 17343–17349 (2014).
Jones, A. K., Lichtenstein, B. R., Dutta, A., Gordon, G. & Dutton, P. L. Synthetic hydrogenases: incorporation of an iron carbonyl thiolate into a designed peptide. J. Am. Chem. Soc. 129, 14844–14845 (2007).
Lombardi, A. et al. Retrostructural analysis of metalloproteins: application to the design of a minimal model for diiron proteins. Proc. Natl Acad. Sci. USA 97, 6298–6305 (2000).
Calhoun, J. R. et al. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J. Mol. Biol. 334, 1101–1115 (2003).
Reig, A. J. et al. Alteration of the oxygen-dependent reactivity of de novo Due Ferri proteins. Nat. Chem. 4, 900–906 (2012).
Chin, J. W. Expanding and reprogramming the genetic code. Nature 550, 53–60 (2017).
Hu, C., Chan, S. I., Sawyer, E. B., Yu, Y. & Wang, J. Metalloprotein design using genetic code expansion. Chem. Soc. Rev. 43, 6498–6510 (2014).
Mills, J. H. et al. Computational design of an unnatural amino acid dependent metalloprotein with atomic level accuracy. J. Am. Chem. Soc. 135, 13393–13399 (2013).
Liu, X. et al. Significant increase of oxidase activity through the genetic incorporation of a tyrosine–histidine cross-link in a myoglobin model of heme–copper oxidase. Angew. Chem. Int. Ed. 51, 4312–4316 (2012).
Kaes, C., Katz, A. & Hosseini, M. W. Bipyridine: the most widely used ligand. A review of molecules comprising at least two 2,2′-bipyridine units. Chem. Rev. 100, 3553–3590 (2000).
Drienovská, I. et al. Design of an enantioselective artificial metallo-hydratase enzyme containing an unnatural metal-binding amino acid. Chem. Sci. 8, 7228–7235 (2017).
Roy, S., Shinde, S., Hamilton, G. A., Hartnett, H. E. & Jones, A. K. Artificial [FeFe]-hydrogenase: on resin modification of an amino acid to anchor a hexacarbonyldiiron cluster in a peptide framework. Eur. J. Inorg. Chem. 2011, 1050–1055 (2011).
Roy, A., Madden, C. & Ghirlanda, G. Photo-induced hydrogen production in a helical peptide incorporating a [FeFe] hydrogenase active site mimic. Chem. Commun. 48, 9816–9818 (2012).
Roy, S., Nguyen, T.-A. D., Gan, L. & Jones, A. K. Biomimetic peptide-based models of [FeFe]-hydrogenases: utilization of phosphine-containing peptides. Dalton Trans. 44, 14865–14876 (2015).
Hyster, T. K. & Ward, T. R. Genetic optimization of metalloenzymes: enhancing enzymes for non-natural reactions. Angew. Chem. Int. Ed. 55, 7344–7357 (2016).
Jeschek, M. et al. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 537, 661–665 (2016).
Khanna, N., Esmieu, C., Mészáros, L. S., Lindblad, P. & Berggren, G. In vivo activation of an [FeFe] hydrogenase using synthetic cofactors. Energy Environ. Sci. 10, 1563–1567 (2017).
Mészáros, L. S., Németh, B., Esmieu, C., Ceccaldi, P. & Berggren, G. In vivo EPR characterization of semi-synthetic [FeFe] hydrogenases. Angew. Chem. Int. Ed. 130, 2626–2629 (2018).
Dunn, M. R., Jimenez, R. M. & Chaput, J. C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 1, 0076 (2017).
Darmostuk, M., Rimpelova, S., Gbelcova, H. & Ruml, T. Current approaches in SELEX: an update to aptamer selection technology. Biotechnol. Adv. 33, 1141–1161 (2015).
Wang, F., Liu, X. & Willner, I. DNA switches: from principles to applications. Angew. Chem. Int. Ed. 54, 1098–1129 (2015).
Lu, C.-H., Cecconello, A. & Willner, I. Recent advances in the synthesis and functions of reconfigurable interlocked DNA nanostructures. J. Am. Chem. Soc. 138, 5172–5185 (2016).
Hu, Y., Cecconello, A., Idili, A., Ricci, F. & Willner, I. Triplex DNA nanostructures: from basic properties to applications. Angew. Chem. Int. Ed. 56, 15210–15233 (2017).
Hong, F., Zhang, F., Liu, Y. & Yan, H. DNA origami: scaffolds for creating higher order structures. Chem. Rev. 117, 12584–12640 (2017).
Jones, M. R., Seeman, N. C. & Mirkin, C. A. Programmable materials and the nature of the DNA bond. Science 347, 1260901 (2015).
Kruger, K. et al. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 31, 147–157 (1982).
Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N. & Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35, 849–857 (1983).
Gilbert, W. Origin of life: the RNA world. Nature 319, 618 (1986).
Cech, T. R. The ribosome is a ribozyme. Science 289, 878–879 (2000).
Ward, W. L., Plakos, K. & DeRose, V. J. Nucleic acid catalysis: metals, nucleobases, and other cofactors. Chem. Rev. 114, 4318–4342 (2014).
Breaker, R. R. & Joyce, G. F. A DNA enzyme that cleaves RNA. Chem. Biol. 1, 223–229 (1994).
Wilson, T. J., Liu, Y. & Lilley, D. M. J. Ribozymes and the mechanisms that underlie RNA catalysis. Front. Chem. Sci. Eng. 10, 178–185 (2016).
Felletti, M. & Hartig, J. S. Ligand-dependent ribozymes. Wiley Interdiscip. Rev. RNA 8, e1395 (2017).
Hollenstein, M. DNA catalysis: the chemical repertoire of DNAzymes. Molecules 20, 20777–20804 (2015).
Seelig, B. & Jäschke, A. A small catalytic RNA motif with Diels–Alderase activity. Chem. Biol. 6, 167–176 (1999).
Seelig, B., Keiper, S., Stuhlmann, F. & Jäschke, A. Enantioselective ribozyme catalysis of a bimolecular cycloaddition reaction. Angew. Chem. Int. Ed. 39, 4576–4579 (2000).
Murray, J. M. & Doudna, J. A. Creative catalysis: pieces of the RNA world jigsaw. Trends Biochem. Sci. 26, 699–701 (2001).
Talini, G., Gallori, E. & Maurel, M.-C. Natural and unnatural ribozymes: back to the primordial RNA world. Res. Microbiol. 160, 457–465 (2009).
Boersma, A. J., Megens, R. P., Feringa, B. L. & Roelfes, G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 39, 2083–2092 (2010).
Serganov, A. et al. Structural basis for Diels–Alder ribozyme-catalyzed carbon–carbon bond formation. Nat. Struct. Mol. Biol. 12, 218–224 (2005).
Emahi, I., Gruenke, P. R. & Baum, D. A. Effect of aptamer binding on the electron-transfer properties of redox cofactors. J. Mol. Evol. 81, 186–193 (2015).
Emahi, I., Mulvihill, I. M. & Baum, D. A. Pyrroloquinoline quinone maintains redox activity when bound to a DNA aptamer. RSC Adv. 5, 7450–7453 (2015).
Thoa, T. T. T., Minagawa, N., Aigaki, T., Ito, Y. & Uzawa, T. Regulation of photosensitisation processes by an RNA aptamer. Sci. Rep. 7, 43272 (2017).
Winkler, W. C., Nahvi, A., Roth, A., Collins, J. A. & Breaker, R. R. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428, 281–286 (2004).
Ferré-D’Amaré, A. R. The glmS ribozyme: use of a small molecule coenzyme by a gene-regulatory RNA. Q. Rev. Biophys. 43, 423–447 (2010).
Bingaman, J. L. et al. The GlcN6P cofactor plays multiple catalytic roles in the glmS ribozyme. Nat. Chem. Biol. 13, 439–445 (2017).
Carmi, N., Shultz, L. A. & Breaker, R. R. In vitro selection of self-cleaving DNAs. Chem. Biol. 3, 1039–1046 (1996).
Hsiao, C. et al. RNA with iron(II) as a cofactor catalyses electron transfer. Nat. Chem. 5, 525–528 (2013).
Tsukiji, S., Pattnaik, S. B. & Suga, H. An alcohol dehydrogenase ribozyme. Nat. Struct. Biol. 10, 713–717 (2003).
Sen, D. & Poon, L. C. RNA and DNA complexes with hemin [Fe(III) heme] are efficient peroxidases and peroxygenases: how do they do it and what does it mean? Crit. Rev. Biochem. Mol. Biol. 46, 478–492 (2011).
Golub, E., Albada, H. B., Liao, W.-C., Biniuri, Y. & Willner, I. Nucleoapzymes: hemin/G-quadruplex DNAzyme–aptamer binding site conjugates with superior enzyme-like catalytic functions. J. Am. Chem. Soc. 138, 164–172 (2016).
Saito, K., Tai, H., Hemmi, H., Kobayashi, N. & Yamamoto, Y. Interaction between the heme and a G-quartet in a heme–DNA complex. Inorg. Chem. 51, 8168–8176 (2012).
Poon, L. C.-H. et al. Guanine-rich RNAs and DNAs that bind heme robustly catalyze oxygen transfer reactions. J. Am. Chem. Soc. 133, 1877–1884 (2011).
Kong, D., Lei, Y., Yeung, W. & Hili, R. Enzymatic synthesis of sequence-defined synthetic nucleic acid polymers with diverse functional groups. Angew. Chem. Int. Ed. 55, 13164–13168 (2016).
Sefah, K. et al. In vitro selection with artificial expanded genetic information systems. Proc. Natl Acad. Sci. USA 111, 1449–1454 (2014).
Kimoto, M., Yamashige, R., Matsunaga, K.-i., Yokoyama, S. & Hirao, I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 31, 453–457 (2013).
Zhou, C. et al. DNA-catalyzed amide hydrolysis. J. Am. Chem. Soc. 138, 2106–2109 (2016).
Santoro, S. W., Joyce, G. F., Sakthivel, K., Gramatikova, S. & Barbas, C. F. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 122, 2433–2439 (2000).
Andersen, E. S. Prediction and design of DNA and RNA structures. New Biotechnol. 27, 184–193 (2010).
Gong, S., Wang, Y., Wang, Z. & Zhang, W. Computational methods for modeling aptamers and designing riboswitches. Int. J. Mol. Sci. 18, e2442 (2017).
Lah, M. S. et al. Structure–function in Escherichia coli iron superoxide dismutase: comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry 34, 1646–1660 (1995).
Borgstahl, G. E. O., Pokross, M., Chehab, R., Sekher, A. & Snell, E. H. Cryo-trapping the six-coordinate, distorted-octahedral active site of manganese superoxide dismutase. J. Mol. Biol. 296, 951–959 (2000).
Carrell, C. J., Zhang, H., Cramer, W. A. & Smith, J. L. Biological identity and diversity in photosynthesis and respiration: structure of the lumen-side domain of the chloroplast Rieske protein. Structure 5, 1613–1625 (1997).
van den Heuvel, R. H. H. et al. The active conformation of glutamate synthase and its binding to ferredoxin. J. Mol. Biol. 330, 113–128 (2003).
Zhang, H. et al. Characterization and crystallization of the lumen side domain of the chloroplast Rieske iron–sulfur protein. J. Biol. Chem. 271, 31360–31366 (1996).
Bottin, H. & Lagoutte, B. Ferredoxin and flavodoxin from the cyanobacterium Synechocystis sp PCC 6803. Biochim. Biophys. Acta 1101, 48–56 (1992).
Lederer, F., Glatigny, A., Bethge, P. H., Bellamy, H. D. & Mathews, F. S. Improvement of the 2.5 Å resolution model of cytochrome b 562 by redetermining the primary structure and using molecular graphics. J. Mol. Biol. 148, 427–448 (1981).
Durley, R. C. E. & Mathews, F. S. Refinement and structural analysis of bovine cytochrome b 5 at 1.5 Å resolution. Acta Cryst. 52, D65–D76 (1996).
Poulos, T. L., Finzel, B. C. & Howard, A. J. Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry 25, 5314–5322 (1986).
Barker, P. D., Butler, J. L., de Oliveira, P., Hill, H. A. O. & Hunt, N. I. Direct electrochemical studies of cytochromes b 562. Inorg. Chim. Acta 252, 71–77 (1996).
Reid, L. S., Taniguchi, V. T., Gray, H. B. & Mauk, A. G. Oxidation-reduction equilibrium of cytochrome b 5. J. Am. Chem. Soc. 104, 7516–7519 (1982).
Sligar, S. G. & Gunsalus, I. C. A thermodynamic model of regulation: modulation of redox equilibria in camphor monoxygenase. Proc. Natl Acad. Sci. USA 73, 1078–1082 (1976).
Conlan, A. R. et al. Mutation of the His ligand in mitoNEET stabilizes the 2Fe−2S cluster despite conformational heterogeneity in the ligand environment. Acta Cryst. 67, D516–D523 (2011).
Tsukihara, T. et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl Acad. Sci. USA 100, 15304–15309 (2003).
Vojtěchovský, J., Chu, K., Berendzen, J., Sweet, R. M. & Schlichting, I. Crystal structures of myoglobin–ligand complexes at near-atomic resolution. Biophys. J. 77, 2153–2174 (1999).
Acknowledgements
The authors appreciate funding from the VolkswagenStiftung (Design of [FeS]-cluster containing Metallo-DNAzymes (Az 93412)) and the Deutsche Forschungsgemeinschaft (DFG) (the Cluster of Excellence RESOLV EXC1069 and GRK 2341: Microbial Substrate Conversion).
Author information
Authors and Affiliations
Contributions
All authors contributed to researching the article, discussing the content and writing and editing of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- β-lactamase
-
An enzyme that hydrolyses β-lactam-containing antibiotics, such as penicillins or cephalosporins. Most β-lactam antibiotics inhibit biosynthesis of the bacterial cell wall. Bacteria expressing genes that encode for β-lactamase are resistant to such antibiotics.
- Chlorophycean microalgae
-
The Chlorophyta (green algae) diverged from land plants over a billion years ago. Some unicellular green algae, such as the model organism Chlamydomonas reinhardtii, synthesize [FeFe]-hydrogenase enzymes that are located within their chloroplasts — the organelles in which photosynthesis takes place. In C. reinhardtii, [FeFe]-hydrogenases accept electrons from ferredoxin, which, in turn, can be reduced by photosynthetic electron transport. The alga can thus dispose of excess light energy through H+ reduction.
- Directed evolution
-
A molecular biology technique that mimics the process of natural evolution — mutagenesis and selection. A sequence that encodes a protein whose characteristics are to be altered is randomly mutagenized (for example, by error-prone PCR), and the resulting proteins are screened for optimized features, such as optimum temperature or solvent stability. Several rounds of mutagenesis and selection are usually performed.
- Entatic state
-
Translated literally, entatic means under tension or stretched. In the case of metalloenzymes, the ‘entatic state’ refers to an unusual geometric or electronic configuration of a metal ion that would not occur in the presence of unrestrained ligands. This poised state is enforced by the protein and enables the protein-specific reactivity of a given metal centre.
- Error-prone PCR
-
The DNA sequence that codes for a protein can be randomly mutated by modulating the PCR conditions such that DNA polymerase introduces mistakes into the newly polymerized DNA strand. Thereby, random changes in the amino acid composition of the encoded protein are generated. The technique is usually coupled with (high-throughput) screening techniques to identify proteins with desired variations in their properties.
- Genetic code expansion
-
One can enable organisms to introduce unnatural amino acid residues into proteins using their natural translation machinery. For this purpose, a novel (orthogonal) tRNA is evolved together with its aminoacyl-tRNA synthetase, which couples the respective (unnatural) amino acid to the tRNA. The tRNA is designed to recognize a stop codon, which can then be introduced into the sequence that encodes the protein whose amino acid content is to be changed. Most often, the rare amber stop codon is utilized.
- G-quadruplex
-
A specific structure of guanine-rich nucleic acids in which so-called G-tetrads are stacked. G-tetrads are built from four guanine bases that form a square planar structure.
- Hydrogenases
-
These enzymes can oxidize H2 or produce H2 from protons. Organisms utilize hydrogenases to make use of the energy content of H2 (for example, through the Knallgas reaction) or to dispose of excess electrons (for example, during fermentation).
- Pleiotropic effects
-
This term was originally used to describe genes that had more than one effect. However, its general meaning in biology refers to something that has multiple, often unrelated impacts. The exchange of an amino acid in a protein by site-directed mutagenesis can have a structural effect, for example, when the two residues differ in size, or a functional effect, when the chemical properties of the residues differ. If both effects are observed, the mutation is said to have pleiotropic effects.
Rights and permissions
About this article

Cite this article
Hemschemeier, A., Happe, T. The plasticity of redox cofactors: from metalloenzymes to redox-active DNA. Nat Rev Chem 2, 231–243 (2018). https://doi.org/10.1038/s41570-018-0029-3
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41570-018-0029-3
This article is cited by
-
Design of a recombinant asparaginyl ligase for site-specific modification using efficient recognition and nucleophile motifs
Communications Chemistry (2024)
-
A trade-off for covalent and intercalation binding modes: a case study for Copper (II) ions and singly modified DNA nucleoside
Scientific Reports (2019)
