
Abstract
The core pathology of coronavirus disease 2019 (COVID-19) is infection of airway cells by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that results in excessive inflammation and respiratory disease, with cytokine storm and acute respiratory distress syndrome implicated in the most severe cases. Thrombotic complications are a major cause of morbidity and mortality in patients with COVID-19. Patients with pre-existing cardiovascular disease and/or traditional cardiovascular risk factors, including obesity, diabetes mellitus, hypertension and advanced age, are at the highest risk of death from COVID-19. In this Review, we summarize new lines of evidence that point to both platelet and endothelial dysfunction as essential components of COVID-19 pathology and describe the mechanisms that might account for the contribution of cardiovascular risk factors to the most severe outcomes in COVID-19. We highlight the distinct contributions of coagulopathy, thrombocytopathy and endotheliopathy to the pathogenesis of COVID-19 and discuss potential therapeutic strategies in the management of patients with COVD-19. Harnessing the expertise of the biomedical and clinical communities is imperative to expand the available therapeutics beyond anticoagulants and to target both thrombocytopathy and endotheliopathy. Only with such collaborative efforts can we better prepare for further waves and for future coronavirus-related pandemics.
Key points
-
Venous thromboembolism, arterial thrombosis and thrombotic microangiopathy substantially contribute to increased morbidity and mortality in patients with COVID-19.
-
A complex interaction between coagulopathy, thrombocytopathy and endotheliopathy contributes to COVID-19-associated thromboinflammation.
-
Coagulopathy, thrombocytopathy and endotheliopathy are characteristic features associated with cardiovascular risk factors such as diabetes mellitus, obesity and ageing.
-
The combination of cardiovascular risk factors and infection with SARS-CoV-2 leads to exacerbated thrombosis and increased mortality.
-
Age has an important role in COVID-19 pathogenesis; young patients without a predisposition to coagulopathy, thrombocytopathy and endotheliopathy can have a distinct multisystem inflammatory syndrome that includes a Kawasaki disease-like syndrome in very young individuals.
-
Combination therapies targeting inflammation, coagulopathy, thrombocytopathy and endotheliopathy are likely to be more successful than a single agent in tackling the COVID-19-associated thrombotic complications.
Similar content being viewed by others
Introduction
The unprecedented outbreak of coronavirus disease 2019 (COVID-19) was declared a pandemic by the WHO, with >34 million people infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19, and with>1 million COVID-19-related deaths worldwide1. COVID-19 can lead to a disease spectrum ranging from mild respiratory symptoms to acute respiratory distress syndrome (ARDS) and death2,3,4. SARS-CoV-2 is now the third highly pathogenic and transmissible coronavirus identified in humans. Human coronaviruses were first discovered in the 1960s5, but it was not until the 21st century that coronaviruses were recognized as major threats to public health. SARS-CoV6,7,8,9, Middle East respiratory syndrome coronavirus (MERS-CoV)10 and SARS-CoV-2 all cause severe respiratory tract infections and have been associated with global pandemics. SARS-CoV was first reported in China in 2003 and infected >8,000 individuals, causing 774 deaths worldwide11. A decade later, MERS was first reported in Saudi Arabia and infected >2,494 individuals and caused 858 deaths, with an extremely high death rate of 34% in part owing to the lack of effective therapies12,13.
SARS-CoV, MERS-CoV and SARS-CoV-2 belong to the Betacoronavirus genus, which is one of four genera of coronavirus14. Phylogenetic analysis revealed that SARS-CoV-2 is closely related to two bat-derived SARS-like coronaviruses, bat-SL-CoVZC45 and bat-SL-CoVZXC21 (with around 88% sequence identity), SARS-CoV (approximately 79% sequence identity) and MERS-CoV (approximately 50% sequence identity)15. Homology modelling revealed that the receptor-binding domain structures in SARS-CoV and SARS-CoV-2 are similar, despite some amino acid variations15. MERS-CoV infects human cells by binding to the dipeptidyl peptidase 4 receptor16, whereas both SARS-CoV17 and SARS-CoV-2 (refs18,19) use angiotensin-converting enzyme 2 (ACE2) as a receptor to infect cells. For SARS-CoV-2 infection, in addition to ACE2, one or more proteases including transmembrane protease serine 2 (TMPRSS2), basigin (also known as CD147) and potentially cathepsin B or cathepsin L are required18,19. Coronaviruses also have a propensity to bind acetylated sialic acid residues, which are abundant on cell membrane proteins including on megakaryocytes and endothelial cells20,21.
Although research into the underlying mechanisms of coronavirus-mediated diseases is still ongoing and a strong consensus has not yet been reached regarding the optimal treatment and prevention of these diseases (especially of COVID-19), reports of important commonalities in the haematological effects of the three diseases are emerging. In this Review, we identify these shared features, with a focus on the effects on platelets and endothelial cells and how these mechanisms intersect with cardiovascular risk factors. We highlight how cardiovascular risk factors, including diabetes mellitus, obesity and ageing, in combination with SARS-CoV-2 infection can lead to catastrophic thrombosis (Fig. 1). Reviewing the literature and the lessons from previous coronavirus-related pandemics as well as the initial studies and observations from the current COVID-19 pandemic can provide important insights into disease pathogenesis, prognostic indicators and novel therapeutic strategies for COVID-19 and for future coronavirus-related pandemics.

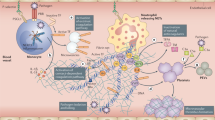
a | In the absence of disease or stressors, platelets and endothelial cells do not directly interact. b | Cardiovascular risk factors such as diabetes mellitus and obesity can induce thrombocytopathy, endotheliopathy, coagulopathy and inflammation, all of which promote cardiovascular disease. c | Coronavirus disease 2019 (COVID-19) through either direct actions on platelets and the endothelium or indirect effects through inflammation and coagulopathy can lead to increased thrombus formation and to emboli and haemorrhage. All these phenotypes have been characterized in blood and autopsy reports from patients with COVID-19. Combination therapies targeting thrombocytopathy, endotheliopathy, inflammation and coagulopathy are needed to reduce morbidity and mortality in patients with COVID-19. A list of drugs or drug classes that might be beneficial for targeting thrombocytopathy, endotheliopathy or both in patients with COVID-19 is provided. All are currently under investigation in this clinical setting. At the top are drugs that predominantly affect platelets (such as aspirin and P2Y purinoceptor 12 (P2Y12) inhibitors), the agents in the middle can affect both, and those at the bottom (complement inhibitors and statins) predominantly target the endothelium. Given the intricate relationship between platelets and the endothelium, the beneficial effects of these drugs on one might improve the other. PDE3, phosphodiesterase 3.
COVID-19-associated thromboinflammation
Thromboinflammation (the coordinated activation of the inflammatory and thrombotic responses) is a major cause of morbidity and mortality in patients with COVID-19 (Fig. 2). The initial reports from hospitals in Wuhan, China, showed that patients with severe COVID-19 can develop acute lung injury and hypoxaemic respiratory failure, which were the major causes of death in these patients22,23,24,25,26. Notably, laboratory studies revealed evidence of coagulopathy with significantly elevated levels of d-dimer in the plasma, a mild prolongation of prothrombin time and borderline thrombocytopenia in a large proportion of patients hospitalized with COVID-19 (refs25,27). Subsequent reports that were based largely on case series and cross-sectional studies from China28,29,30, Netherlands31,32,33, France34,35,36 and Italy37 indicated that the incidence of thromboembolic events, including deep vein thrombosis and pulmonary embolism, is high in critically ill patients with COVID-19 in the intensive care unit (ICU). Post-mortem examination of a small cohort of patients with COVID-19 in Germany found evidence of venous thromboembolism in 7 out of 12 COVD-19-related deaths, with massive pulmonary embolism arising from lower extremity deep vein thrombi as the direct cause of death in 4 out of the 12 patients38. Other studies have reported a high frequency of platelet–fibrin thrombi in the small arteries and capillaries of the pulmonary vasculature in post-mortem lung tissue from patients with COVID-19, suggesting a hypercoagulable state that can predispose to both venous and arterial thrombosis in critically ill patients with COVID-19 (refs39,40,41,42,43). Alveolar capillary microthrombi were nine times more prevalent in patients with COVID-19 than in patients with influenza in a small series of patient autopsy evaluations43. These occluding microthrombi have been found not only in the lungs, but also in the heart, kidneys and liver in patients with COVID-19 (ref.44), supporting the presence of widespread thrombotic microangiopathy in these patients.

Imaging examples of thrombotic complications in patients with coronavirus disease 2019 (COVID-19) and with multiple cardiovascular risk factors, including diabetes mellitus, obesity, old age and hypertension. a | CT scan showing pulmonary emboli (arrow) involving the anterobasilar arteries in the left lower lobe of the lung. b | Doppler ultrasound image showing short-segment non-occlusive deep vein thrombus in the right lateral subclavian vein (arrow). c | CT scan showing a 10-mm diameter thrombus in the abdominal aorta (arrow).
In addition to the pulmonary pathologies, thrombotic complications associated with COVID-19 also affect the cardiovascular and cerebrovascular systems. Substantial evidence indicates that COVID-19 morbidity is increased in patients with underlying cardiovascular conditions (such as hypertension, diabetes mellitus and pre-existing cardiovascular disease) and can provoke myocardial injury leading to complications such as heart failure, myocarditis and cardiac arrhythmias45,46,47. Several case series have found that ST-segment elevation myocardial infarction (STEMI) can be the first clinical presentation of COVID-19 (refs48,49). Moreover, a high burden of coronary thrombus has been observed in patients with COVID-19 who develop STEMI50.
An increased frequency of peripheral artery thrombosis manifesting as acute limb ischaemia with high rates of revascularization failure has been reported in patients with COVID-19 compared with patients presenting with acute limb ischaemia during a similar period in 2019 (ref.51). One study described four cases of acute limb ischaemia, two of which occurred in young healthy individuals without risk factors for ischaemia52. Consistent with these findings, a case series from a single institution showed that COVID-19 was associated with the incidence of large-artery ischaemic stroke, particularly in young individuals53. These observations are concerning given the relative absence of cardiovascular disease risk factors and other comorbidities in these young patient populations compared with older patients. Additionally, autopsy reports of patients with COVID-19 indicate that thrombosis in small vessels (platelet-rich thrombotic microangiopathy), with or without haemorrhage, is associated with ARDS43,54.
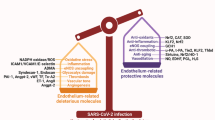
The combined presence of venous thrombosis, arterial thrombosis and small-vessel thrombosis is rare in a single disease entity and can point towards a unique mechanism of disease. Interestingly, a similar constellation of vascular complications is found in antiphospholipid syndrome55. Indeed, data are now emerging indicating that some patients with COVID-19 can develop antiphospholipid syndrome56. The exact mechanisms of thrombosis in antiphospholipid syndrome are still a matter of intense investigation but it has become clear that the origin of thrombosis is at the interface between the blood and the endothelium. Similarly, the cause of thrombosis in patients with COVID-19 is the focus of intense investigation, and coagulation abnormalities (coagulopathy), complement activation and cytokine release (inflammation), platelet hyperactivity and apoptosis (thrombocytopathy), and endothelial dysfunction (endotheliopathy) are emerging as potential major contributors to the pathogenesis of thrombosis in these patients (discussed in detail below). The process is likely to be very complex because inflammation, coagulopathy, thrombocytopathy and endotheliopathy are distinct and yet intricately related processes.
Given the high risk of thrombosis in patients with COVID-19, interim guidance from the International Society on Thrombosis and Haemostasis (ISTH) recommends thrombosis prophylaxis with heparin therapy in all patients hospitalized with COVID-19 who do not have contraindications for antithrombotic therapy57,58. Therapeutic doses of anticoagulants are recommended if evidence of thrombosis such as venous thromboembolism is found. Anticoagulant therapy with low molecular weight heparin (enoxaparin 40–60 mg per day) seems to be associated with a better prognosis than no anticoagulant therapy in patients with severe COVID-19 who have a sepsis-induced coagulopathy score of ≥4 or have markedly elevated d-dimer levels in the plasma (greater than sixfold of the upper limit of normal)59. Although effective to some extent, anticoagulant therapy alone seems to be inadequate because patients with COVID-19 receiving anticoagulants still have major thrombotic events. Detailed understanding of the complex mechanisms leading to COVID-19-associated thromboinflammation is urgently required.
COVID-19-associated coagulopathy
Coagulation and fibrinolysis
COVID-19-associated coagulopathy is characterized by a prothrombotic state with high rates of thrombosis and microvascular complications28,29,30,31,32,33. The aetiology of COVID-19-associated coagulopathy remains controversial and is likely to be heterogeneous, with contributions from many cell types. Early reports from China provided initial evidence for the presence of coagulopathy in patients with COVID-19 (refs28,29,30). Elevated plasma levels of d-dimers (which are fibrin degradation fragments indicative of excessive clotting) and mild thrombocytopenia are often observed in hospitalized patients with COVID-19, and high d-dimer levels with severe thrombocytopenia correlate with a more severe disease course, including higher rates of admission to the ICU and increased mortality25,27,60,61,62. Interestingly, platelet counts in patients with COVID-19 have been found to be on average higher than in patients with comparable, non-COVID-19 respiratory illnesses63, probably related to the megakaryocyte response, as described below. In a case series of 183 patients with COVID-19, a pattern of coagulopathy that fulfilled the ISTH criteria for disseminated intravascular coagulation (DIC) — elevation of d-dimer levels, moderate-to-severe thrombocytopenia, prolongation of prothrombin time and decreased fibrinogen levels — was seen in 71% of patients who were admitted to the ICU and did not survive, whereas only 0.6% of survivors met the ISTH criteria64,65. However, subsequent reports showed that although d-dimer levels were consistently elevated across all studies, other signs of DIC, such as prolongation of the prothrombin time, severe thrombocytopenia (platelet count <50 × 109/l) and decreased fibrinogen, were not observed in most patients with COVID-19 (refs35,66,67,68). Indeed, platelet counts were typically normal or mildly elevated and fibrinogen levels were markedly increased in critically ill patients with COVID-19, findings that are contradictory to a consumptive coagulopathy such as DIC. In addition, depletion of endogenous anticoagulants such as antithrombin or of the fibrinolytic regulator α2 antiplasmin, which is a hallmark of DIC, is not commonly observed in patients with COVID-19 (ref.69). Studies of thrombin generation in patients with COVID-19 have yielded mixed results, because both high levels of thrombin–antithrombin III complexes (which suggests increased thrombin generation) and normal levels of prothrombin fragment 1.2 (which suggests no increase in thrombin generation) have been found in different single-institution studies70,71. Similarly, several studies have found elevations in the plasma levels of tissue-type plasminogen activator, urokinase-type plasminogen activator, plasminogen activator inhibitor 1 (PAI1) and carboxypeptidase B2 (also known as thrombin-activatable fibrinolysis inhibitor) in hospitalized patients with COVID-19, indicating variable effects of COVID-19 on fibrinolytic activity71,72,73. Studies using thrombin-generation assays (such as endogenous thrombin potential) and viscoelastic haemostatic assays (such as thromboelastography or rotational thromboelastometry) have found that patients with COVID-19 have a hypercoagulable state with increased thrombin generation and hypofibrinolysis66,67,73,74. The overall interpretation from these diverse reports is that although COVID-19-associated coagulopathy shares some pathophysiological features with DIC, the coagulopathy observed in patients with COVID-19 is a unique entity that leads to a variable hypercoagulable state dependent on cellular involvement (such as endothelial cells, leukocytes and platelets) as well as on the timing of sample collection during the disease process.
Endothelial effects on coagulopathy
COVID-19-associated coagulopathy has two central features in common with DIC, endotheliopathy and a marked inflammatory response, although the degree of each in COVID-19 can be higher than in other disease states. Studies from our institution have highlighted many features of endotheliopathy that correlate with severity of disease in patients with COVID-19. An analysis of a cohort of 68 patients hospitalized with COVID-19 identified elevations in the levels of numerous circulating markers of endothelial injury, such as von Willebrand factor (vWF), PAI1, soluble thrombomodulin, angiopoietin 2 and follistatin69,75. Notably, particularly high levels of vWF, PAI1 and angiopoietin 2 were observed in patients who were admitted to the ICU69,75, and elevated levels of soluble thrombomodulin, PAI1, angiopoietin 2 and follistatin in hospitalized patients with COVID-19, all correlated with increased mortality69,75. At present, whether the endotheliopathy associated with COVID-19 is a result of direct viral infection of endothelial cells, which has been reported in some autopsy studies43,76, or is a consequence of the virus-induced inflammatory response is uncertain. Further details on this aspect are provided in the endotheliopathy section.
Inflammation and coagulopathy
The inflammatory response in patients with severe COVID-19 is particularly striking, with persistent fever, elevated levels of inflammatory markers (such as C-reactive protein, erythrocyte sedimentation rate, ferritin and various cytokines, including IL-1β, IL-6 and TNF) leading to a hyperinflammatory response known as cytokine storm, which is associated with poor outcomes77,78,79,80,81. Early reports postulated that this inflammatory response might resemble the response seen in macrophage activation syndrome or haemophagocytic lymphohistiocytosis81,82,83,84,85. However, later studies have identified unique profiles of neutrophil, macrophage, lymphocyte and other immune responses in patients with COVID-19, together with evidence of NETosis and complement activation81,82,83,84,85. An intriguing finding is the correlation between elevated circulating levels of inflammatory cytokines and abnormal coagulation parameters77,86. Notably, plasma IL-6 levels have been shown to correlate directly with fibrinogen levels in patients with COVID-19 (ref.66). As detailed above, the process of thromboinflammation with hypercoagulability and a severe inflammatory state is thought to be an important factor in COVID-19 pathogenesis86,87. Indeed, levels of prothrombotic acute phase reactants, such as fibrinogen, vWF and factor VIII, that are commonly elevated during inflammatory conditions, are increased in patients with COVID-19 compared with healthy individuals35,67,69,88, implicating the endothelium, platelets and the coagulation system as potential mediators of coagulopathy and thrombosis in COVID-19. Collectively, the available clinical and laboratory data suggest that COVID-19 can be associated with a massive inflammatory response combined with a hypercoagulable state that predisposes patients to thrombotic events. Therefore, novel anti-inflammatory strategies, such as antagonists for IL-6 or IL-1β signalling89,90,91, in addition to antithrombotic therapies might have a greater effect in preventing thrombosis and death in patients with COVID-19 than either therapy alone.
COVID-19-associated thrombocytopathy
Platelets are considered to be first responders to vascular injury92, capable of rapidly responding to blood perturbations such as inflammation or hyperglycaemia through activation of stress-induced pathways93,94,95. Platelets are short-lived (7–10 days), small (2–4 μm diameter) and anucleate cells and, therefore, were previously thought to be capable of only very limited functions, such as mediating thrombosis96. However, we now know that platelets are more complex and diverse than previously thought, containing fundamental machinery for many critical cellular processes, including autophagy97,98, programmed cell death99 and rapid de novo protein synthesis100,101. Studies from our group and others have identified many roles of platelets in vascular homeostasis and immune responses95,100,102 in conditions including inflammatory, cardiovascular, neurodegenerative and oncological diseases103. Platelets interact with many other cell types, including circulating blood cells, endothelial cells and other cells of the vessel wall, either directly or through the release of signalling mediators. Therefore, platelets can function as a blood component that bridges the immune system (through interactions with various leukocytes) and thrombosis (via platelet activation and release of haemostatic and inflammatory mediators)104. Thrombocytopathy (platelet dysfunction) is also a prominent feature of COVID-19, as described in the section below, and can encompass thrombocytopenia and platelet hyperactivation, contributing to excessive thrombosis and a dysfunctional immune response.
Thrombocytopenia and platelet hyperactivation
A reduction in platelet count (thrombocytopenia) was commonly observed in patients with SARS or MERS (Table 1). In an early epidemiological report on SARS from 2003, three of nine patients with SARS (33%) in Canada were documented to have thrombocytopenia (defined as a platelet count of <130 × 109 cells per litre)105. Subsequent studies from Asia, particularly Hong Kong106,107,108, Taiwan109,110 and China111, also indicated the presence of thrombocytopenia in patients with SARS. Thrombocytopenia in these patients was generally mild, with moderate-to-severe thrombocytopenia being associated with severe end-stage disease106,107,108,109,110,111. Studies in children with SARS also demonstrated the presence of thrombocytopenia in five out of ten patients (50%) in one study112 and 12 out of 44 patients (27%) in another study70. Thrombocytopenia in patients with SARS was often associated with clinically significant coagulopathy113,114. Similar findings were observed in patients with MERS-CoV infection, for which many of the studies were from Saudi Arabia115,116,117,118,119,120.
SARS-CoV-2 infection was also associated with thrombocytopenia in early reports from China18,22,23,25,27,64,121,122,123,124. The incidence of thrombocytopenia in patients with SARS, MERS or COVID-19 is approximately 47%, 37% and 25%, respectively (Table 1). Of note, the threshold to define thrombocytopenia was lower in most COVID-19 studies (most used <100 × 109 platelets per litre) than in SARS and MERS studies (<100–150 × 109 platelets per litre) (Table 1), largely explaining why the incidence of thrombocytopenia in patients with COVID-19 is lower than in patients with SARS or MERS. The incidence of thrombocytopenia in patients with severe COVID-19 (requiring mechanical ventilation or ICU admission) has been reported to be as high as 35% (122 of 263 patients in a French study125). In patients with COVID-19, thrombocytopenia can be associated with low fibrinogen level and increased risk of bleeding, particularly in patients receiving anticoagulant therapy126. However, in patients with severe COVID-19, low fibrinogen levels and bleeding events are rare whereas thrombotic complications are more common126. One explanation is that platelets are hyperactivated in patients with COVID-19 (refs69,75,127). Previous studies by our group and others have also demonstrated high rates of platelet apoptosis in patients with cardiometabolic disease and comorbidities such as diabetes mellitus94. Furthermore, the presence of apoptotic platelets promotes hyperactivation of surviving platelets128. Given that patients with severe COVID-19 often have cardiovascular comorbidities, platelet apoptosis is likely to be an important contributor to COVID-19 pathogenesis. Moreover, other mechanisms implicated in COVID-19 pathogenesis, including hypoxia, inflammation, immune system activation and endothelial activation and dysfunction76, can further induce platelet activation and apoptosis, leading to increased thrombosis72,94,128,129 (Fig. 3).

Schematic diagram summarizing the normal physiological platelet response to vessel wall damage, that is, haemostasis (panel a) and the thrombocytopathy associated with coronavirus disease 2019 (COVID-19; panel b). Possible additional inducers of pathological platelet hyperactivation include cardiovascular risk factors such as old age, diabetes mellitus, obesity and hypoxia from lung disease, and immune factors leading to increased intracellular levels of reactive oxygen species (ROS). The consequences of pathological platelet hyperactivation include increased platelet microvesicle and granule release that contribute to increased thrombosis and cytokine storm (local and circulating). Increased platelet activation can result in platelet–leukocyte conjugates (primarily neutrophils) and platelet apoptosis and/or aggregation, which in turn can further lead to increased thrombosis and inflammation. The presence of impaired fibrinolysis and endotheliopathy can contribute to increased thrombosis and can lead to life-threatening thrombosis and thromboembolism. The lungs are the most susceptible organ to thrombosis but these events can occur in many organs, including the heart, kidneys and liver. With platelet hyperactivation, apoptosis and increased thrombus formation, platelet clearance and consumption are higher, leading to thrombocytopenia. Thrombocytopenia occurs only if megakaryocytes are unable to produce sufficient platelets to compensate for the platelet loss. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Several autopsy studies in patients with COVID-19 have reported thrombosis in arteries and in veins, particularly within small vessels (platelet-rich thrombotic microangiopathy), with or without haemorrhage43,54,130. In these patients, increased small-vessel thrombosis was associated with the typical ARDS pathology of diffusely oedematous lungs43,54,130. The presence of thrombosis throughout the lungs suggests that hyperactivated platelets might additionally influence disease in the lungs by exacerbating inflammation. Activated platelets are increasingly recognized to regulate leukocyte activity during infections and allergic reactions102, further contributing to the leukocyte cytokine release. Activated platelets express P-selectin and CD40L on the cell surface, can interact with neutrophils and can release α-granules and complement C3 as well as various cytokines, including CC-chemokine ligand 2 (CCL2), CCL3, CCL7, IL-1β, IL-7, IL-8 and hepatocyte growth factor131,132. Levels of all these cytokines have been reported to be significantly increased in patients with COVID-19 compared with healthy individuals133. In vitro and in vivo studies have shown that, in response to dengue virus infection, platelets release IL-1β on microparticles, which contributes to an increase in endothelial permeability134. This platelet response involving IL-1β and other cytokines might possibly contribute to COVID-19-associated ARDS.
In addition to cytokine release, another important event in ARDS, as seen in COVID-19 pathology, is neutrophil recruitment to the pulmonary vasculature135. The phenomenon of activated platelets binding to neutrophils and rolling of platelet-bound neutrophils on the endothelium, known as ‘secondary capture’, has a crucial role in initiating immunothrombosis136. The binding of activated platelets to neutrophils facilitates the transmigration of platelets to the alveolar lumen and contributes to the formation of oedematous lungs, which in turn can cause further platelet activation135. Neutrophil extracellular traps (NETs) have been shown to trigger thromboinflammation in patients with COVID-19 (ref.135), leading to vascular thrombosis and a high risk of death137. Taken together, platelet activation and apoptosis can contribute to the severe pathology of COVID-19, including thrombosis and the cytokine storm.
Mechanisms of thrombocytopathy and thromboinflammation
On the basis of the current knowledge of COVID-19 pathology, several mechanisms of COVID-19-related thrombocytopathy are possible. Hypoxia, oxidative stress, nutrient stress and other stressors affect platelet mitochondria metabolism and function, leading to platelet hyperactivation and apoptosis138. Studies by our group and others have shown that many of the comorbid conditions observed in patients with COVID-19 (such as diabetes mellitus, ageing and obesity), which involve oxidative stress, can lead to platelet hyperactivity and apoptosis128. We have reported the regulation of platelet reactivity by ageing-associated changes in redox homeostasis138. Thrombocytopenia can arise from reduced production or increased loss of platelets by consumption, clearance or death. The three major mechanisms of platelet clearance are platelet senescence (loss of sialic acid), apoptosis (scavenger receptor-mediated loss) and Fc receptor-mediated platelet clearance by macrophages139. Consumption of platelets into a growing thrombus or platelet apoptosis might explain the thrombocytopenia observed in some patients with COVID-19, which has been associated with increased risk of death compared with patients with normal platelet counts61. Alternatively, SARS-CoV-2-induced production of autoantibodies that target platelet surface antigens can cause increased platelet destruction140. Antiphospholipid antibodies have also been detected in critically ill patients with COVID-19 (ref.56). Hypoxia, caused by environmental conditions141 or disease states such as chronic obstructive pulmonary disease142, has been associated with platelet hyperactivation and a prothrombotic state. COVID-19 pathology involves mild-to-severe hypoxia with peripheral blood oxygen saturation falling below 94% even with oxygen therapy, and hypoxia is a crucial parameter to monitor in all patients with COVID-19 (ref.143). The hypoxia in patients with COVID-19 might contribute to thrombocytopathy directly or indirectly. A French study found that 58% of patients with COVID-19 who had thrombocytopenia at the time of hospital admission required oxygen supplementation compared with 41% of patients with platelet counts in the normal range at the time of hospital admission125. A thrombotic consumptive microangiopathy mediated by complement (C3a and C5a) might also contribute to thrombocytopathy in COVID-19 (ref.144). All these processes can contribute to dysregulated platelet function and can ultimately predispose patients with COVID-19 to thromboinflammation.
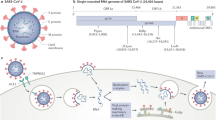
In addition to environmental stressors, viral infections also directly affect platelets, leading to increased apoptosis and removal. A study in patients with SARS-CoV-2 infection showed that most patients had platelet hyperreactivity and alterations in the platelet transcriptome compared with healthy individuals127. Of note, SARS-CoV-2 RNA traces were detected in platelet samples from two of the 25 patients included in the study, but ACE2 mRNA or protein was not detected in any of the platelet samples127. Whether platelets have sufficient ACE2 expression required for SARS-CoV-2 entry into the cell145,146 has yet to be definitively demonstrated but other potential methods of SARS-CoV-2 entry into platelets independent of ACE2 are emerging19. Studies of influenza virus infection indicate that platelets have many of the required receptors for influenza virus entry, and influenza virus infection of platelets potentially leads to platelet apoptosis147,148,149,150,151. In addition, given that platelets have features of innate immune cells, viral entry might occur as a part of a platelet-mediated antiviral response to SARS-CoV-2. Platelet uptake and degradation of influenza virus and HIV, which are single-stranded RNA viruses like SARS-CoV-2, occur through an endosomal, Toll-like receptor (TLR)-mediated pathway104,152,153,154. SARS-CoV-2 entry into platelets is likely to involve a similar mechanism leading to platelet activation104.
Various aetiologies have been suggested to underlie SARS-associated thrombocytopathy, including viral infection of platelets, endothelial damage caused by mechanical ventilation and potential viral infection of endothelial cells that in turn triggers platelet activation, apoptosis and aggregation, and thrombosis in the lung (DIC) that causes vast platelet consumption155. A similar picture is likely to occur in COVID-19-associated thrombocytopathy, with direct infection of platelets by SARS-CoV-2, endothelial damage caused by mechanical ventilation and viral infection of endothelial cells, and thrombosis.
Increased production of inflammatory chemokines and cytokines (many from platelets) and an excessive or uncontrolled innate immune response are typical characteristics of ARDS77,78,79,80,81, including in the ARDS seen in COVID-19. A cytokine storm has been suggested as a major cause of the lung damage occurring in patients with COVID-19 (refs77,78,79,80,81). Complement system activation is a natural immune response to bacterial and viral infections. Complement activation can induce platelet activation and aggregation, leading to fibrin deposition to promote thrombosis156,157. Furthermore, platelet apoptosis leads to the release of a wide variety of pro-inflammatory and procoagulant factors158 and highly thrombogenic apoptotic bodies72,94,128. The formation of immune complexes is another potential mechanism for platelet hyperactivation and thrombocytopenia in COVID-19 and might be similar to that seen in heparin-induced thrombocytopenia (HIT). HIT involves the formation of heparin–platelet factor 4 (PF4)–antibody complexes, which recognize the FcγRIIa receptor on the platelet surface, causing platelet activation and clearance159. The activated platelets can trigger NETosis leading to thrombosis in the inflamed region (immunothrombosis), which also involves endothelial cell dysfunction135,159. SARS-CoV-2 is likely to form immune complexes with reactive antibodies in the host, as observed in severely ill patients with influenza (H1N1) infection160. Influenza virus–IgG complexes can bind platelet FcγRIIa and cause platelet activation161. Virus-induced platelet activation can lead to increased platelet–leukocyte conjugates162, potentially triggering NETosis. Interestingly, platelet–PF4 deposits in the lungs have been found in autopsy studies in patients with COVID-19 and correlate with increased NETosis and microthrombus formation135.
Megakaryocytopathy
Megakaryocyte function is crucial to compensate for rapid platelet consumption or loss. Furthermore, megakaryocytes can contribute to the severe pathology (megakaryocytopathy) seen in some patients with SARS-CoV-2 infection. Autopsy studies have revealed abnormal megakaryocyte distribution and proplatelet formation in the tissues of patients with COVID-19. One study found CD61+ megakaryocytes (possibly representing resident pulmonary megakaryocytes) with nuclear hyperchromasia and atypia in lung tissues from patients with COVID-19 (ref.130). Another study identified megakaryocytes in the cardiac microvasculature in all patients with COVID-19 who were studied (n = 7) as well as in the kidneys and lungs, and the numbers of megakaryocytes in the heart and lungs were higher than in patients with ARDS from other causes44. Megakaryocyte numbers were also higher in the bone marrow and showed focal clustering and morphology consistent with proplatelet formation. These findings suggest that COVID-19 has a unique pathology, with systemic increases in megakaryocyte numbers. In both autopsy studies, the megakaryocytes in the lung had morphological characteristics suggestive of active platelet production. This observation supports the finding that the lungs are a site of platelet biogenesis with both intravascular and extravascular reservoirs of megakaryocytes163,164, and suggests that these pulmonary megakaryocytes are increased in severely ill patients with COVID-19. The capacity of this potentially compensatory increase in platelet production to maintain normal peripheral platelet counts in patients with COVID-19 is inconsistent, with some patients developing thrombocytopenia and others not62. Thrombocytopenia in some patients with COVID-19 might be due to decreased platelet production in the lungs caused by ARDS and hypoxia during coronavirus infections164. Whether the platelet hyperactivation seen in patients with COVID-19 (ref.127) results from activation of the megakaryocytes that are producing the platelets is not yet known.
Megakaryocytes express receptors that can interact with some types of virus147,148,149,150,151, have pattern recognition receptors, such as C‐type lectin receptors and TLRs, that recognize viruses and have a role in fighting infections165. However, neither megakaryocytes nor platelets seem to express ACE2, the SARS-CoV-2 entry receptor127,166. Despite the lack of ACE2 expression, electron microscopy has revealed the presence of coronavirus virions in megakaryocytes in the lungs in autopsy samples from patients with COVID-19 (ref.44). In addition, SARS-CoV-2 RNA has been detected in the platelets of some patients with COVID-19 (ref.127). The researchers in this study suggested that the platelets might take up SARS-CoV-2 or SARS-CoV-2 RNA independently of ACE2, but another possibility is that the detected SARS-CoV-2 RNA comes from the infected megakaryocytes from which the platelets were derived. Megakaryocytes and/or platelets might be able to take up intact virus via ACE2-independent mechanisms such as endocytosis through TLRs104. Moreover, through a mechanism termed emperipolesis, megakaryocytes can engulf other cells such as neutrophils, which results in the formation of platelets that contain components of the non-megakaryocyte donor cell167.
COVID-19-associated endotheliopathy
The functions of endothelial cells include maintaining vascular integrity and barrier function, as well as preventing inflammation by limiting their interactions with immune cells and platelets168. Endotheliopathy, or endothelial dysfunction, is emerging as an important pathological feature in COVID-19. Transmission electron microscopy of blood vessels from autopsy specimens from patients with COVID-19 has revealed the presence of endothelial cell damage and apoptosis43,169. Damage to the endothelium and blood vessels has crucial roles in promoting angiogenesis. Post-mortem analysis of lung samples has shown that both intussusceptive (non-sprouting) and sprouting angiogenesis are more frequent in patients with COVID-19 than in patients with influenza43. Whether endothelial dysfunction is primarily a result of direct infection of endothelial cells by SARS-CoV-2 remains to be determined, but biomarkers of endothelial dysfunction, such as thrombomodulin, vWF, angiopoietin 2 and PAI1, are frequently elevated in patients with COVID-19 compared with healthy controls, and seem to have prognostic relevance, being associated with severe disease69,75. Endothelial dysfunction leading to arteriopathy and thrombosis is a major contributor to the pathophysiology of thrombotic complications associated with COVID-19, including myocardial infarction and strokes69,75.
Mechanisms of endotheliopathy and thromboinflammation
ACE2, the cell-entry receptor for SARS-CoV-2, is expressed in the respiratory epithelium and also in endothelial cells, as shown by several studies43. Moreover, ACE2 has been shown to be upregulated in response to interferon in human epithelial cells (but not so far in endothelial cells), which might be a mechanism that contributes to potentiating the cellular uptake of SARS-CoV-2 (ref.170). Interestingly, several single-cell analyses of human lungs have not reliably demonstrated detectable levels of ACE2 expression in lung endothelial cells170,171, which requires future investigation. Nevertheless, endothelial injury, whether through direct viral infection or immune-mediated damage, is a crucial feature of severe COVID-19 across multiple studies, and the underlying mechanisms will need to be elucidated as we seek novel treatment strategies.
Resting endothelial cells maintain vascular homeostasis through antiplatelet, anticoagulant and anti-inflammatory effects172 (Fig. 4). A combination of the direct viral infection of endothelial cells by SARS-CoV-2 and the endothelial cell response to the inflammatory process associated with COVID-19 that upregulates immune cell responses, production of cytokines78 and activation of complement144,157,173 probably mediates endothelial damage and microvascular thrombosis. Studies by our group have demonstrated the contribution of endotheliopathy to COVID-19 severity, showing that increased circulating levels of markers of endothelial cell damage, including thrombomodulin, angiopoietin 2 and vWF, are significantly correlated with increased mortality in patients with COVID-19 (refs69,75). In the section on therapies, we discuss a number of potential therapeutic strategies that directly target endotheliopathy with the goal of stabilizing the endothelium, which might be an important therapeutic target in preventing thrombosis in patients with COVID-19.

a | The normal resting endothelium is crucial for maintaining vascular homeostasis through production of several anti-inflammatory and antithrombotic factors, including nitric oxide (NO), prostaglandin I2 (PGI2; also known as prostacyclin), thrombomodulin, activated protein C, tissue factor pathway inhibitor (TFPI) and antithrombin III (ATIII). b | In disease states, such as obesity and diabetes mellitus, increased oxidative stress with elevated intracellular levels of reactive oxygen species (ROS) can promote the production of pro-inflammatory cytokines (such as IL-1β, IL-6 and TNF), decrease the bioavailability of NO and PGI2 and induce endothelial cell apoptosis, leading to endothelial damage and dysfunction. Furthermore, the release of pro-inflammatory and prothrombotic factors (such as von Willebrand factor (vWF) and thrombin) can lead to vascular inflammation, platelet aggregation and thrombosis. Simultaneous invasion of the endothelium by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) — the virus that causes coronavirus disease 2019 (COVID-19) — via the angiotensin-converting enzyme 2 (ACE2) receptor can exacerbate endothelial dysfunction and damage, further promoting vascular inflammation and thrombosis. The dashed arrows represent complex molecular signalling mechanisms that remain to be defined in the context of COVID-19. TF, tissue factor.
Several studies have also found the presence of acquired antiphospholipid antibodies in patients with COVID-19 (ref.174). Antiphospholipid antibodies, which include lupus anticoagulant, anti-cardiolipin antibody and anti-β2-glycoprotein I, predispose individuals to arterial and venous thrombosis174. A small case series including three patients with COVID-19 reported multiple cerebral infarctions in the setting of positive antiphospholipid antibodies56. Several larger studies found a high frequency of antiphospholipid antibodies in patients with COVID-19 who had prolonged activated partial thromboplastin time35,175. In another study, the percentage of patients testing positive for a lupus anticoagulant was significantly higher among patients with COVID-19 than in individuals without COVID-19 (ref.176). Several lines of evidence indicate that antiphospholipid antibodies can interact with a variety of vascular and haematopoietic components, including endothelial cells, platelets and complement factors, to promote thrombosis177,178,179,180,181. However, although antiphospholipid antibodies are a common occurrence in viral infections, they are often transient and do not always imply an increased risk of thrombosis182. Furthermore, false-positive results in lupus anticoagulant tests can occur in patients receiving anticoagulation therapy183,184. Nevertheless, whether antiphospholipid antibodies have a major role in the pathophysiology of thrombosis associated with COVID-19 requires further investigation. Additionally, dermatological evidence of a vascular inflammatory phenomenon manifesting as chilblains has been reported in patients with COVID-19 (refs90,185).
Endotheliopathy associated with ageing
Age is the major risk factor for COVID-19-related death, with 304.9 deaths per 1,000 cases in patients aged ≥85 years compared with 0.3 deaths per 1,000 cases among patients aged 5–17 years186. The endotheliopathy associated with old age and the healthy endothelium associated with young age might be reasons for the severe consequences of COVID-19 in very old patients and the protection in young patients. Ageing is associated with endothelial dysfunction187,188, and endothelial dysfunction is a major contributor to vascular pathologies and cardiovascular diseases in old individuals189,190. Oxidative and nitrosative stress, which are increased in old individuals, can induce endothelial dysfunction191. NADPH oxidases and mitochondria can generate reactive oxygen species (ROS), and dysregulation of these pathways can cause accumulation of ROS187,192,193. In aged endothelial cells, increased ROS levels can decrease the availability of nitric oxide (NO), which is a potent vasodilator and antiplatelet agent with cardioprotective effects189,190,191,194. Decreased NO levels can lead to vasoconstriction and platelet activation and promote cardiovascular diseases195. Another function of the vascular endothelium is to maintain a balance between pro-inflammatory and anti-inflammatory factors196. Chronic inflammation has been associated with age-related endothelial dysfunction, characterized by elevated levels of C-reactive protein, pro-inflammatory cytokines and adhesion molecules that recruit immune cells, impair mitochondrial function and disrupt cellular energy metabolism191. Furthermore, the number of apoptotic endothelial cells increases with advancing age196,197. The age-related decline in NO bioavailability together with the upregulation of mitochondrial oxidative stress and chronic inflammation have all been suggested to induce endothelial cell apoptosis196.
An example of a potential area for further exploration in COVID-19 pathology is cerebromicrovascular dysfunction and damage as a result of age-related endothelial dysfunction. Overactivation of poly(ADP-ribose) polymerase 1, as can be observed in viral infections, can lead to NAD+ depletion and subsequent endothelial damage and dysfunction198,199. Additionally, age-related impairment in oxidative stress resilience caused by dysfunction of the nuclear factor erythroid 2-related factor 2 (NRF2) antioxidant defence pathway in vascular endothelial cells might also have a role in COVID-19-associated endotheliopathy200. Pharmacological activators of NRF2 have been proposed as potential treatment options for COVID-19 (ref.201). NRF2 has powerful anti-inflammatory and anti-apoptotic effects in endothelial cells201. Of note, NRF2 dysfunction exacerbates the endothelial damaging effect of hypertension and diabetes, conditions also known to increase the risk of COVID-19-related death201. Therefore, the underlying mechanisms of age-related endothelial dysfunction are complex and probably involve multiple molecular and cellular pathways.
Multisystem inflammatory syndrome and Kawasaki-like disease in children and adolescents
The most important risk factor for COVID-19 is age. Endotheliopathy and thrombocytopathy occur with ageing, and healthy children and adolescents have little to none of the endothelial damage and platelet dysfunction seen in adults189,190. Children were initially thought to be at very low risk of developing COVID-19. Children with SARS-CoV-2 infection typically had mild or no COVID-19 symptoms, and hospitalization rates were low202,203,204. However, as the COVID-19 pandemic progressed, reports of children hospitalized with an unusual multiorgan inflammatory syndrome similar to Kawasaki disease were published202,203,204. The condition is characterized by high levels of inflammation in multiple organ systems, including the cardiovascular, respiratory, haematological, gastrointestinal and mucocutaneous systems and was later defined as multisystem inflammatory syndrome in children (MIS-C)205,206,207,208. The delayed onset of MIS-C, several weeks after the initial COVID-19 symptoms205,206,207, suggests that the syndrome is the result of an excessive immune response rather than the viral infection itself209. Notably, whereas adults with severe COVID-19 frequently have cardiovascular comorbidities, the majority of children with COVID-19 are healthy and without the cardiovascular risk factors that predispose to severe disease205,206,207,208. Mortality among paediatric patients is low despite ICU admission being common205,206, probably owing to the lack of accompanying endotheliopathy and thrombocytopathy in paediatric patients compared with older patients189,190.
Around 33% of all patients with MIS-C show features similar to those in patients with Kawasaki disease, particularly patients aged ≤5 years205,206,207. Kawasaki disease is a vasculitis in which damage to the vascular endothelium can lead to arterial vasculopathy and aneurysm formation in the coronary vasculature and other arteries205,206,210. Neutrophils and other immune cells enter the vessel wall, promoting endothelial damage and subsequent myofibroblast hyperproliferation210. Thrombocytopathy and endotheliopathy are shared factors in the pathogenesis of both COVID-19 and Kawasaki disease211. Endothelial function can remain impaired for years after Kawasaki disease resolves212. Intravenous immunoglobulins and aspirin are the main therapeutic approaches for Kawasaki disease, to suppress the immune response and platelet function213,214,215,216,217. Given that MIS-C has similar symptoms and causes to those of Kawasaki disease, these drugs could potentially target the effects of MIS-C and might be considered in adult patients with COVID-19.
Cardiovascular risk factors and COVID-19
In healthy individuals, platelets do not directly interact with the endothelium, with both being in a quiescent state (Fig. 1). Quiescent endothelial cells secrete a number of crucial mediators that act synergistically to prevent local platelet aggregation, including NO (through the cGMP pathway)218 and prostacyclin (also known as prostaglandin I2, which acts through the cAMP pathway)219,220. Endothelial cells can also synthesize and release tissue plasminogen activator that converts plasminogen to plasmin, thereby activating fibrinolysis and clot dissolution221,222,223. Thrombomodulin on the endothelial cell surface binds thrombin with high affinity to prevent platelet activation and fibrinogen cleavage. These factors are essential to maintaining blood fluidity, particularly in the microvasculature because the endothelium in these small vessels is exposed to larger fractions of blood (owing to the smaller diameter) than the endothelium in larger vessels221. After vascular stress or injury, cross-activation of platelets and the endothelium is essential to the response and repair processes: platelets release several endothelial cell-targeted vasoactive substances such as thromboxane (which acts through thromboxane receptors), ADP (via P2Y purinoceptors), serotonin (via 5-hydroxytryptamine and serotonergic receptors) and PAI1 (which effectively inhibits the action of endothelial-associated plasminogen activator)221. vWF is released from both endothelial cells and platelet storage granules and mediates platelet adhesion to the endothelium. Evaluation of circulating vWF multimers in patients with COVID-19 might be informative because COVID-19 pathology has signatures of thrombotic thrombocytopenia purpura, which is characterized by thrombocytopenia, microthrombi, endothelial cell dysregulation and increased circulating levels of vWF multimers224,225. Elevated circulating levels of vWF seem to have prognostic relevance in COVID-19, being associated with severe disease69. Similarly, increased release of thrombomodulin is also associated with high mortality in patients with COVID-19 (ref.69). All these factors probably contribute to the thrombotic microangiopathy found in multiple autopsy studies of patients with COVID-19 (ref.44).
In inflammatory conditions, the crosstalk between platelets, the coagulation system and the endothelium is perturbed and can exacerbate the inflammatory process226. Oxidative stress and inflammatory stress associated with hypertension227,228, diabetes mellitus72,94,128, old age138,229,230, lung disease142,231 and many other systemic diseases can lead to platelet and endothelial dysfunction221. We and others have demonstrated that diabetes mellitus, obesity, old age and lung disease are associated with platelet activation and induce platelet apoptosis via increased oxidative stress and mitochondrial damage72,128,159,232. Moreover, these cardiovascular risk factors can also lead to endothelial cell activation, damage and dysfunction233,234,235,236. Thrombocytopathy and endotheliopathy, in conjunction with inflammation and coagulopathy, are exacerbated in patients with SARS-CoV-2 infection, leading to increased thrombosis and risk of embolization (Fig. 1). This fatal combination of thrombocytopathy and endotheliopathy is highlighted by observations that increased levels of markers of platelet and endothelial cell activation and damage are associated with an increased risk of ICU admission and death in patients with COVID-19 (refs69,75). Thrombocytopathy and endotheliopathy lead to the formation of occluding microthrombi not only in the lungs but also in many other organs including the heart, kidneys and liver, supporting the observation of widespread thrombotic microangiopathy in patients with COVID-19 (ref.44). This platelet–endothelial interaction in health and disease might hold the key to developing effective new targeted therapies.
Potential therapeutic approaches
Evidence-based guidelines for the treatment of COVID-19 have been established by many institutions, professional societies and countries, including the NIH237 and the Centers for Disease Control and Prevention in the USA238. Supportive care and respiratory support remain the cornerstone of therapy. Evidence of improvement (symptoms, disease duration and/or survival) has been observed with some antiviral therapies (remdesivir239), antibodies (convalescent serum240), anti-inflammatory agents (dexamethasone241), immunomodulatory therapies (tocilizumab89) and anticoagulants (heparin242), although placebo-controlled, randomized clinical trials are needed to prove efficacy definitively. Despite these therapies, the case fatality rate of COVID-19 remains high, especially in elderly patients186. Therefore, new targets and strategies are urgently needed. As described in this Review, thrombosis is a major cause of death in patients with COVID-19. Targeting the coagulation pathway with agents such as heparin seems to be effective, particularly for venous thromboembolism, and has been highlighted in several previous reviews59,243. Targeting thrombocytopathy and endotheliopathy might provide additional new therapeutic strategies of particular benefit to those patients with cardiovascular risk factors. Given the close interactions between platelets and vascular endothelial cells, a number of FDA-approved drugs targeting these cell types in vascular disease, which are currently under investigation for COVID-19, might help to reduce COVID-19 morbidity and mortality.
On the basis of our current understanding of the mechanisms of thrombocytopathy and endotheliopathy, therapies directed against platelet activation and apoptosis and/or improving endothelial cell health might be effective for treating patients with COVID-19. Importantly, therapies aimed at preventing platelet and endothelial cell dysfunction should also be considered. Although anticoagulants are currently in use for the treatment of patients with COVID-19, specific antiplatelet agents might also be worth investigating. The two main naturally occurring agents that benefit both the endothelium and platelets are endothelial-derived NO (through the cGMP pathway) and prostacyclin (through the cAMP pathway) (Figs 1,4). Prostacyclin is a naturally occurring metabolic product of the fatty acid arachidonic acid, predominantly synthesized in endothelial cells by cyclooxygenase 2 and prostacyclin synthase. Prostacyclin is an approved therapy for pulmonary hypertension and is available in several forms, including inhaled epoprostenol, iloprost and treprostinil, as well as in intravenous and oral forms244. Prostacyclin is antithrombotic, being a potent inhibitor of platelet activation, and reduces arterial tone through antiproliferative and vasodilatory actions on vascular smooth muscle cells245,246. In addition, growing evidence indicates that prostacyclin also has anti-inflammatory effects through reducing the production of the pro-inflammatory cytokines IL-1β and IL-6 and increasing vWF proteolysis246,247. NO has pleiotropic actions that protect against vascular disease by promoting vascular smooth muscle cell relaxation (vasodilatation), inhibiting platelet activation and reducing leukocyte adhesion and inflammation, while also being an antioxidant248,249. These collective effects suggest that inhaled NO might be a promising strategy for the treatment of patients with COVID-19, and multiple trials of inhaled NO are in progress250.
Given that prostacyclin has beneficial effects in platelets and the endothelium by increasing cAMP levels, other oral agents that raise cAMP levels in these cell types might also be potential therapeutic strategies for COVID-19. These include the phosphodiesterase 3 inhibitors, such as dipyridamole and cilostazil, which are currently used for stroke prevention and peripheral vascular disease251,252,253. A small study has suggested a benefit of dipyridamole in patients with COVID-19, showing a reduction in biomarkers of disease severity (such as d-dimer levels) and improved clinical outcomes252. Antiplatelet drugs alone, such as aspirin and P2Y purinoceptor 12 inhibitors, might be another option to provide antithrombotic protection in these patients. Although reviews on COVID-19 antithrombosis mention aspirin only for the acute treatment of arterial thrombotic complications of COVID-19, prophylactic antiplatelet agents are now being included in therapeutic algorithms for the management of patients with COVID-19, and many clinical trials on the prophylactic use of antiplatelet agents are proposed or are underway254,255,256. Low-dose aspirin has long been used for the treatment of Kawasaki disease, features of which have been associated with COVID-19. In addition, platelets are increasingly recognized to have a role in Kawasaki disease211. However, aspirin and other antiplatelet agents do not address endotheliopathy. Other agents such as antioxidants (vitamin C and vitamin E) and fish oils have been explored as potentially being beneficial to both platelets and the endothelium221. However, the efficacy of these agents remains uncertain, particularly in a rapid, acute disease process. Additional therapeutic strategies that might afford protection against endothelial injury or death are also being actively investigated in the context of COVID-19. Complement inhibition, which can protect endothelial cells against complement-mediated injury, is being tested in a large, randomized, placebo-controlled trial257,258,259. Statins, which have pleiotropic effects on endothelial cells in addition to their lipid-lowering effects260, are also being actively investigated for the treatment of patients with COVID-19 (refs261,262,263). Other, broader immunomodulatory agents might afford endothelial protection by suppressing the innate immune response that can injure endothelial cells. Current therapies for COVID-19 remain inadequate as they do not substantially reduce morbidity and mortality, but as new mechanistic insights emerge from concerted biomedical research efforts, opportunities will continue to arise to identify potentially safe and effective therapeutic strategies.
Conclusions
Platelets and endothelial cells are two highly symbiotic cell types whose dysfunction contributes to COVID-19-associated inflammation, coagulopathy, thrombosis and death. The mechanisms of thrombocytopathy and endotheliopathy remain poorly understood in the context of COVID-19. However, the mechanisms of thrombocytopathy and endotheliopathy are well studied in the context of the cardiovascular risk factors (such as diabetes mellitus, obesity and ageing) that have been associated with increased COVID-19 morbidity and mortality. Patients with COVID-19 and cardiovascular risk factors such as diabetes mellitus, obesity and old age have a high incidence of venous thromboembolism, arterial thrombosis and thrombotic microangiopathy, which contribute to the high mortality seen in these patients28,29,30,31,32,33,34,35,36,37. Correlations between the increased mortality and the presence of elevated levels of biomarkers of endothelial and platelet damage and dysfunction in patients with COVID-19, and autopsy reports demonstrating changes in endothelial morphology and extensive thrombosis, have confirmed the crucial importance of endotheliopathy and thrombocytopathy in COVID-19 (refs43,54,69,75,130). Despite improved understanding of the management of patients with COVID-19, the overall mortality in these patients remains high, particularly among patients at high cardiovascular risk28,29,30,31,32,33,34,35,36,37. As we approach 1.5 million COVID-19-related deaths worldwide, new therapeutic strategies are urgently needed. Now is the time to harness our understanding of thrombocytopathy and endotheliopathy and directly target and prevent this platelet and endothelial dysfunction. The necessary collaboration between the scientific and medical communities with diverse and complementary expertise will help us to navigate the current and future coronavirus pandemics.
References
Johns Hopkins Coronavirus Resource Center. COVID-19 Dashboard. https://coronavirus.jhu.edu/map.html (2020).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Wu, Y. et al. SARS-CoV-2 is an appropriate name for the new coronavirus. Lancet 395, 949–950 (2020).
Kahn, J. S. & McIntosh, K. History and recent advances in coronavirus discovery. Pediatric Infect. Dis. J. 24, S223–S227 (2005).
Drosten, C. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976 (2003).
Ksiazek, T. G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003).
Peiris, J. S. M. et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361, 1319–1325 (2003).
Rota, P. A. et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394–1399 (2003).
Zaki, A. M., Boheemen, S. V., Bestebroer, T. M., Osterhaus, A. D. M. E. & Fouchier, R. A. M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012).
Peiris, J. S. M., Yuen, K. Y., Osterhaus, A. D. M. E. & Stöhr, K. The severe acute respiratory syndrome. N. Engl. J. Med. 349, 2431–2441 (2003).
World Health Organization. MERS monthly summary Nov 2019. https://www.who.int/emergencies/mers-cov/en/ (2020).
Zumla, A., Hui, D. S. & Perlman, S. Middle East respiratory syndrome. Lancet 386, 995–1007 (2015).
Cui, J., Li, F. & Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2018).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013).
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11, 875–879 (2005).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Tortorici, M. A. et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 26, 481–489 (2019).
Kim, C. H. SARS-CoV-2 evolutionary adaptation toward host entry and recognition of receptor O-acetyl sialylation in virus-host interaction. Int. J. Mol. Sci. 21, 4549 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Chen, N. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513 (2020).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Guan, W. J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Wu, C. et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 180, 934–943 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Cui, S., Chen, S., Li, X., Liu, S. & Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 18, 1421–1424 (2020).
Ren, B. et al. Extremely high incidence of lower extremity deep venous thrombosis in 48 patients with severe COVID-19 in Wuhan. Circulation 142, 181–183 (2020).
Zhang, L. et al. Deep vein thrombosis in hospitalized patients with coronavirus disease 2019 (COVID-19) in Wuhan, China: prevalence, risk factors, and outcome. Circulation 142, 114–128 (2020).
Klok, F. A. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 191, 145–147 (2020).
Klok, F. A. et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb. Res. 191, 148–150 (2020).
Middeldorp, S. et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 18, 1995–2002 (2020).
Poissy, J. et al. Pulmonary embolism in COVID-19 patients: awareness of an increased prevalence. Circulation 142, 184–186 (2020).
Helms, J. et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 46, 1089–1098 (2020).
Nahum, J. et al. Venous thrombosis among critically ill patients with coronavirus disease 2019 (COVID-19). JAMA Netw. Open 3, e2010478 (2020).
Lodigiani, C. et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 191, 9–14 (2020).
Wichmann, D. et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann. Intern. Med. 173, 268–277 (2020).
Dolhnikoff, M. et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 18, 1517–1519 (2020).
Carsana, L. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect. Dis. 20, 1135–1140 (2020).
Menter, T. et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology 77, 198–209 (2020).
Fox, S. E. et al. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir. Med. 8, 681–686 (2020).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128 (2020).
Amy, V. et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine 24, 100434 (2020).
Mehra, M. R., Desai, S. S., Kuy, S., Henry, T. D. & Patel, A. N. Cardiovascular disease, drug therapy, and mortality in Covid-19. N. Engl. J. Med. 382, e102 (2020).
Guo, T. et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 811–818 (2020).
Inciardi, R. M. et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 819–824 (2020).
Stefanini, G. G. et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation 141, 2113–2116 (2020).
Bangalore, S. et al. ST-segment elevation in patients with Covid-19 – a case series. N. Engl. J. Med. 382, 2478–2480 (2020).
Roffi, M., Guagliumi, G. & Ibanez, B. The obstacle course of reperfusion for STEMI in the COVID-19 pandemics. Circulation 141, 1951–1953 (2020).
Bellosta, R. et al. Acute limb ischemia in patients with COVID-19 pneumonia. J. Vasc. Surg. S0741-5214, 31080–31086, https://doi.org/10.1016/j.jvs.2020.04.483 (2020).
DeBaun, M. R. Initiating adjunct low dose-hydroxyurea therapy for stroke prevention in children with SCA during the COVID-19 pandemic. Blood 135, 1997–1999 (2020).
Oxley, T. J. et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N. Engl. J. Med. 382, e60 (2020).
Barton, L. M., Duval, E. J., Stroberg, E., Ghosh, S. & Mukhopadhyay, S. COVID-19 autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 153, 725–733 (2020).
Corban, M. T. et al. Antiphospholipid syndrome: role of vascular endothelial cells and implications for risk stratification and targeted therapeutics. J. Am. Coll. Cardiol. 69, 2317–2330 (2017).
Zhang, Y. et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N. Engl. J. Med. 382, e38 (2020).
Thachil, J. et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 18, 1023–1026 (2020).
Barrett, C. D., Moore, H. B., Yaffe, M. B. & Moore, E. E. ISTH interim guidance on recognition and management of coagulopathy in COVID-19: a comment. J. Thromb. Haemost. 18, 2060–2063 (2020).
Tang, N. et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 18, 1094–1099 (2020).
Lippi, G. & Favaloro, E. J. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb. Haemost. 120, 876–878 (2020).
Yang, X. et al. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemost. 18, 1469–1472 (2020).
Lippi, G., Plebani, M. & Henry, B. M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin. Chim. Acta 506, 145–148 (2020).
Yin, S., Huang, M., Li, D. & Tang, N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis https://doi.org/10.1007/s11239-020-02105-8 (2020).
Tang, N., Li, D., Wang, X. & Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 18, 844–847 (2020).
Taylor, F. B. Jr et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb. Haemost. 86, 1327–1330 (2001).
Ranucci, M. et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 18, 1747–1751 (2020).
Panigada, M. et al. Hypercoagulability of COVID-19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 18, 1738–1742 (2020).
Han, H. et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 58, 1116–1120 (2020).
Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 7, e575–e582 (2020).
Leung, C. W. et al. Severe acute respiratory syndrome among children. Pediatrics 113, e535–e543 (2004).
Masi, P. H. G. et al. Systemic inflammatory response syndrome is a major contributor to COVID-19-associated coagulopathy: insights from a prospective single-center cohort study. Circulation 142, 611–614 (2020).
Tang, W. H., Martin, K. A. & Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharmacol. 3, 87 (2012).
Nougier, C. et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J. Thromb. Haemost. 18, 2215–2219 (2020).
Spiezia, L. et al. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb. Haemost. 120, 998–1000 (2020).
Meizlish, M. et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. medRxiv https://doi.org/10.1101/2020.06.29.20140376 (2020).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Jose, R. J. & Manuel, A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir. Med. 8, e46–e47 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629 (2020).
Moore, J. B. & June, C. H. Cytokine release syndrome in severe COVID-19. Science 368, 473–474 (2020).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Merad, M. & Martin, J. C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 20, 355–362 (2020).
Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Zuo, Y. et al. Neutrophil extracellular traps in COVID-19. JCI Insight 5, e138999 (2020).
Java, A. et al. The complement system in COVID-19: friend and foe? JCI Insight 5, 140711 (2020).
Lo, M. W., Kemper, C. & Woodruff, T. M. COVID-19: complement, coagulation, and collateral damage. J. Immunol. 205, 1488–1495 (2020).
Jackson, S. P., Darbousset, R. & Schoenwaelder, S. M. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 133, 906–918 (2019).
Connors, J. M. & Levy, J. H. Thromboinflammation and the hypercoagulability of COVID-19. J. Thromb. Haemost. 18, 1559–1561 (2020).
Escher, R., Breakey, N. & Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 190, 62 (2020).
Xu, X. et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl Acad. Sci. USA 117, 10970–10975 (2020).
Colonna, C. et al. Chilblain-like lesions in children following suspected Covid-19 infection. Pediatr. Dermatol. 37, 437–440 (2020).
Cavalli, G. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2, e325–e331 (2020).
Menter, D. G. et al. Platelet “first responders” in wound response, cancer, and metastasis. Cancer Metastasis Rev. 36, 199–213 (2017).
Lee, S. H. et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol. Med. 8, 779–795 (2016).
Tang, W. H. et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Invest. 121, 4462–4476 (2011).
Leytin, V. Apoptosis in the anucleate platelet. Blood Rev. 26, 51–63 (2012).
Leslie, M. Cell biology. Beyond clotting: the powers of platelets. Science 328, 562–564 (2010).
Feng, W. et al. Dissection of autophagy in human platelets. Autophagy 10, 642–651 (2014).
Ouseph, M. M. et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 126, 1224–1233 (2015).
Mason, K. D. et al. Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 (2007).
Lindemann, S. et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J. Cell Biol. 154, 485–490 (2001).
Weyrich, A. S. et al. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc. Natl Acad. Sci. USA 95, 5556–5561 (1998).
Chae, W. J. et al. The Wnt antagonist Dickkopf-1 promotes pathological type 2 cell-mediated inflammation. Immunity 44, 246–258 (2016).
Koupenova, M., Clancy, L., Corkrey, H. A. & Freedman, J. E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 122, 337–351 (2018).
Koupenova, M. & Freedman, J. E. Platelets and immunity: going viral. Arterioscler. Thromb. Vasc. Biol. 40, 1605–1607 (2020).
Poutanen, S. M. et al. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 348, 1995–2005 (2003).
Tsang, K. W. et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348, 1977–1985 (2003).
Lee, N. et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348, 1986–1994 (2003).
Peiris, J. S. M. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
Liu, C.-L. et al. Clinical and laboratory features of severe acute respiratory syndrome vis-a-vis onset of fever. Chest 126, 509–517 (2004).
Wang, J.-T. et al. Clinical manifestations, laboratory findings, and treatment outcomes of SARS patients. Emerg. Infect. Dis. 10, 818–824 (2004).
Lang, Z.-W. et al. A clinicopathological study of three cases of severe acute respiratory syndrome (SARS). Pathology 35, 526–531 (2003).
Hon, K. L. E. et al. Clinical presentations and outcome of severe acute respiratory syndrome in children. Lancet 361, 1701–1703 (2003).
Wong, R. S. M. et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 326, 1358–1362 (2003).
Choi, K. W. et al. Outcomes and prognostic factors in 267 patients with severe acute respiratory syndrome in Hong Kong. Ann. Intern. Med. 139, 715–723 (2003).
Memish, Z. A., Zumla, A. I., Al-Hakeem, R. F., Al-Rabeeah, A. A. & Stephens, G. M. Family cluster of Middle East respiratory syndrome coronavirus infections. N. Engl. J. Med. 368, 2487–2494 (2013).
Omrani, A. S. et al. A family cluster of Middle East respiratory syndrome coronavirus infections related to a likely unrecognized asymptomatic or mild case. Int. J. Infect. Dis. 17, e668–e672 (2013).
Assiri, A. et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 369, 407–416 (2013).
Assiri, A. et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect. Dis. 13, 752–761 (2013).
Arabi, Y. M. et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann. Int. Med. 160, 389–397 (2014).
Al-Tawfiq, J. A. et al. Hematologic, hepatic, and renal function changes in hospitalized patients with Middle East respiratory syndrome coronavirus. Int. J. Lab. Hematol. 39, 272–278 (2017).
Xu, X.-W. et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ 368, m606 (2020).
Yang, W. et al. Clinical characteristics and imaging manifestations of the 2019 novel coronavirus disease (COVID-19): a multi-center study in Wenzhou City, Zhejiang, China. J. Infect. 80, 388–393 (2020).
Zhang, G. et al. Analysis of clinical characteristics and laboratory findings of 95 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a retrospective analysis. Respir. Res. 21, 74 (2020).
Chan, J. F.-W. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395, 514–523 (2020).
Maquet, J. et al. Thrombocytopenia is independently associated with poor outcome in patients hospitalized for COVID-19. Br. J. Haematol. 190, e276–e279 (2020).
Al-Samkari, H. et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 136, 489–500 (2020).
Manne, B. K. et al. Platelet gene expression and function in COVID-19 patients. Blood 136, 1317–1329 (2020).
Tang, W. H. et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction, and damage in diabetic platelets. Circulation 129, 1598–1609 (2014).
Sinauridze, E. I. et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 97, 425–434 (2007).
Fox, S. E. et al. Pulmonary and cardiac pathology in Covid-19: the first autopsy series from New Orleans. Lancet Respir. Med. 8, 681–686 (2020).
Fitch-Tewfik, J. L. & Flaumenhaft, R. Platelet granule exocytosis: a comparison with chromaffin cells. Front. Endocrinol. 4, 77 (2013).
Sut, C. et al. The non-hemostatic aspects of transfused platelets. Front. Med. 5, 42 (2018).
Ye, Q., Wang, B. & Mao, J. The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J. Infect. 80, 607–613 (2020).
Hottz, E. D. et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 122, 3405–3414 (2013).
Middleton, E. A. et al. Neutrophil extracellular Traps (NETs) contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 136, 1169–1179 (2020).
Zarbock, A., Polanowska-Grabowska, R. K. & Ley, K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. 21, 99–111 (2007).
Wang, T. et al. Attention should be paid to venous thromboembolism prophylaxis in the management of COVID-19. Lancet Haematol. 7, e362–e363 (2020).
Melchinger, H., Jain, K., Tyagi, T. & Hwa, J. Role of platelet mitochondria: life in a nucleus-free zone. Front. Cardiovasc. Med. 6, 153 (2019).
Grozovsky, R., Giannini, S., Falet, H. & Hoffmeister, K. M. Regulating billions of blood platelets: glycans and beyond. Blood 126, 1877–1884 (2015).
Zulfiqar, A. A., Lorenzo-Villalba, N., Hassler, P. & Andres, E. Immune thrombocytopenic purpura in a patient with Covid-19. N. Engl. J. Med. 382, e43 (2020).
Tyagi, T. et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood 123, 1250–1260 (2014).
Maclay, J. D. et al. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax 66, 769–774 (2011).
Burns, G. P. et al. Improved survival following ward-based non-invasive pressure support for severe hypoxia in a cohort of frail patients with COVID-19: retrospective analysis from a UK teaching hospital. BMJ Open Respir. Res. 7, e000621 (2020).
Campbell, C. M. & Kahwash, R. Will complement inhibition be the new target in treating COVID-19 related systemic thrombosis? Circulation 141, 1739–1741 (2020).
Mourad, J. J. & Levy, B. I. Interaction between RAAS inhibitors and ACE2 in the context of COVID-19. Nat. Rev. Cardiol. 17, 313 (2020).
South, A. M., Diz, D. I. & Chappell, M. C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 318, H1084–H1090 (2020).
Youssefian, T., Drouin, A., Masse, J. M., Guichard, J. & Cramer, E. M. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 99, 4021–4029 (2002).
Loria, G. D., Romagnoli, P. A., Moseley, N. B., Rucavado, A. & Altman, J. D. Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood 121, 940–950 (2013).
Rondina, M. T. et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A (H1N1). Chest 141, 1490–1495 (2012).
Gomez-Casado, C. et al. Understanding platelets in infectious and allergic lung diseases. Int. J. Mol. Sci. 20, 1730 (2019).
Middleton, E. A., Weyrich, A. S. & Zimmerman, G. A. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol. Rev. 96, 1211–1259 (2016).
Zhang, Z. et al. Structural analyses of Toll-like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. 25, 3371–3381.e5 (2018).
Koupenova, M. et al. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 35, 1030–1037 (2015).
Koupenova, M. et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 124, 791–802 (2014).
Yang, M., Ng, M. H. & Li, C. K. Thrombocytopenia in patients with severe acute respiratory syndrome (review). Hematology 10, 101–105 (2005).
Subramaniam, S. et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 129, 2291–2302 (2017).
Magro, C. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl. Res. 220, 1–13 (2020).
Coppinger, J. A. et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 103, 2096–2104 (2004).
Perdomo, J. et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 10, 1322 (2019).
Monsalvo, A. C. et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 17, 195–199 (2011).
Boilard, E. et al. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 123, 2854–2863 (2014).
Flaujac, C., Boukour, S. & Cramer-Borde, E. Platelets and viruses: an ambivalent relationship. Cell Mol. Life Sci. 67, 545–556 (2010).
Lefrançais, E. et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544, 105–109 (2017).
Lefrancais, E. & Looney, M. R. Platelet biogenesis in the lung circulation. Physiology 34, 392–401 (2019).
Hottz, E. D., Bozza, F. A. & Bozza, P. T. Platelets in immune response to virus and immunopathology of viral infections. Front. Med. 5, 121 (2018).
Rahman, N. T. et al. MRTFA augments megakaryocyte maturation by enhancing the SRF regulatory axis. Blood Adv. 2, 2691–2703 (2018).
Cunin, P. et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. eLife 8, e44031 (2019).
Teuwen, L. A., Geldhof, V., Pasut, A. & Carmeliet, P. COVID-19: the vasculature unleashed. Nat. Rev. Immunol. 20, 389–391 (2020).
O’Sullivan, J. M., Gonagle, D. M., Ward, S. E., Preston, R. J. S. & O’Donnell, J. S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 7, e553–e555 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035.e19 (2020).
Zhao, Y. et al. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 202, 756–759 (2020).
Pober, J. S. & Sessa, W. C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815 (2007).
Risitano, A. M. et al. Complement as a target in COVID-19? Nat. Rev. Immunol. 20, 343–344 (2020).
Garcia, D. & Erkan, D. Diagnosis and management of the antiphospholipid syndrome. N. Engl. J. Med. 378, 2010–2021 (2018).
Bowles, L. et al. Lupus anticoagulant and abnormal coagulation tests in patients with Covid-19. N. Engl. J. Med. 383, 288–290 (2020).
Harzallah, I., Debliquis, A. & Drenou, B. Lupus anticoagulant is frequent in patients with Covid-19. J. Thromb. Haemost. 18, 2064–2065 (2020).
Proulle, V., Furie, R. A., Merrill-Skoloff, G., Furie, B. C. & Furie, B. Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood 124, 611–622 (2014).
Giannakopoulos, B. & Krilis, S. A. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 368, 1033–1044 (2013).
Boilard, E., Blanco, P. & Nigrovic, P. A. Platelets: active players in the pathogenesis of arthritis and SLE. Nat. Rev. Rheumatol. 8, 534–542 (2012).
Sacharidou, A. et al. Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood 131, 2097–2110 (2018).
Chaturvedi, S. et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 135, 239–251 (2019).
Uthman, I. W. & Gharavi, A. E. Viral infections and antiphospholipid antibodies. Semin. Arthritis Rheum. 31, 256–263 (2002).
Lakos, G. Interference in antiphospholipid antibody assays. Semin. Thromb. Hemost. 38, 353–359 (2012).
Pengo, V. et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J. Thromb. Haemost. 7, 1737–1740 (2009).
de Masson, A. et al. Chilblains are a common cutaneous finding during the COVID-19 pandemic: a retrospective nationwide study from France. J. Am. Acad. Dermatol. 83, 667–670 (2020).
Wiersinga, W. J., Rhodes, A., Cheng, A. C., Peacock, S. J. & Prescott, H. C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA 324, 782–793 (2020).
Csiszar, A. et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 90, 1159–1166 (2002).
Donato, A. J. et al. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 589, 4545–4554 (2011).
Lakatta, E. G. & Levy, D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation 107, 346–354 (2003).
Widlansky, M. E., Gokce, N., Keaney, J. F. Jr & Vita, J. A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 42, 1149–1160 (2003).
Ungvari, Z. et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 15, 555–565 (2018).
Donato, A. J. et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ. Res. 100, 1659–1666 (2007).
Adler, A. et al. NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am. J. Physiol. Heart Circ. Physiol. 285, H1015–H1022 (2003).
Lerman, A. & Zeiher, A. M. Endothelial function: cardiac events. Circulation 111, 363–368 (2005).
Dai, D. F., Rabinovitch, P. S. & Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 110, 1109–1124 (2012).
Csiszar, A., Ungvari, Z., Koller, A., Edwards, J. G. & Kaley, G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genomics 17, 21–30 (2004).
Asai, K. et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler. Thromb. Vasc. Biol. 20, 1493–1499 (2000).
Kiss, T. et al. Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: a potential mechanism for the prevention of vascular cognitive impairment. Geroscience 41, 619–630 (2019).
Tarantini, S. et al. Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+ depletion hypothesis of neurovascular aging. Geroscience 41, 533–542 (2019).
Ungvari, Z. et al. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: from increased cellular senescence to the pathogenesis of age-related vascular diseases. Geroscience 41, 727–738 (2019).
McCord, J. M., Hybertson, B. M., Cota-Gomez, A., Geraci, K. P. & Gao, B. Nrf2 activator PB125((R)) as a potential therapeutic agent against COVID-19. Antioxidants 9, 518 (2020).
Riphagen, S., Gomez, X., Gonzalez-Martinez, C., Wilkinson, N. & Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 395, 1607–1608 (2020).
Verdoni, L. et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 395, 1771–1778 (2020).
Viner, R. M. & Whittaker, E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet 395, 1741–1743 (2020).
Cheung, E. W. et al. Multisystem inflammatory syndrome related to COVID-19 in previously healthy children and adolescents in New York City. JAMA 8, e2010374 (2020).
Feldstein, L. R. et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N. Engl. J. Med. 383, 334–346 (2020).
Dufort, E. M. et al. Multisystem inflammatory syndrome in children in New York State. N. Engl. J. Med. 383, 347–358 (2020).
CDC. Emergency preparedness and response. Multisystem inflammatory syndrome in children (MIS-C) associated with coronavirus disease 2019 (COVID-19). CDC https://emergency.cdc.gov/han/2020/han00432.asp (2020).
Cao, X. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol. 20, 269–270 (2020).
Orenstein, J. M. et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS ONE 7, e38998 (2012).
Zhang, Y. et al. Reduced platelet miR-223 induction in Kawasaki disease leads to severe coronary artery pathology through a miR-223/PDGFRβ vascular smooth muscle cell axis. Circ. Res. 127, 855–873 (2020).
Dhillon, R. et al. Endothelial dysfunction late after Kawasaki disease. Circulation 94, 2103–2106 (1996).
Murphy, D. J. Jr. & Huhta, J. C. Treatment of Kawasaki syndrome with intravenous gamma globulin. N. Engl. J. Med. 316, 881 (1987).
Newburger, J. W. et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N. Engl. J. Med. 324, 1633–1639 (1991).
Newburger, J. W. et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N. Engl. J. Med. 315, 341–347 (1986).
Furusho, K. et al. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet 2, 1055–1058 (1984).
Furusho, K. et al. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet 2, 1359 (1983).
Furchgott, R. F. & Zawadzki, J. V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376 (1980).
Bunting, S., Gryglewski, R., Moncada, S. & Vane, J. R. Arterial walls generate from prostaglandin endoperoxides a substance (prostaglandin X) which relaxes strips of mesenteric and coeliac ateries and inhibits platelet aggregation. Prostaglandins 12, 897–913 (1976).
Vane, J. R. Nobel lecture, 8th December 1982. Adventures and excursions in bioassay: the stepping stones to prostacyclin. Br. J. Pharmacol. 79, 821–838 (1983).
Ware, J. A. & Heistad, D. D. Seminars in medicine of the Beth Israel Hospital, Boston. Platelet-endothelium interactions. N. Engl. J. Med. 328, 628–635 (1993).
Penny, W. F. & Ware, J. A. Platelet activation and subsequent inhibition by plasmin and recombinant tissue-type plasminogen activator. Blood 79, 91–98 (1992).
Loscalzo, J. & Vaughan, D. E. Tissue plasminogen activator promotes platelet disaggregation in plasma. J. Clin. Invest. 79, 1749–1755 (1987).
Albiol, N., Awol, R. & Martino, R. Autoimmune thrombotic thrombocytopenic purpura (TTP) associated with COVID-19. Ann. Hematol. 99, 1673–1674 (2020).
Hindilerden, F., Yonal-Hindilerden, I., Akar, E. & Kart-Yasar, K. Covid-19 associated autoimmune thrombotic thrombocytopenic purpura: report of a case. Thromb. Res. 195, 136–138 (2020).
Coenen, D. M., Mastenbroek, T. G. & Cosemans, J. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood 130, 2819–2828 (2017).
Grossman, E. Does increased oxidative stress cause hypertension? Diabetes Care 31, S185–S189 (2008).
Rodrigo, R., Gonzalez, J. & Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 34, 431–440 (2011).
Jain, K. et al. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: implications for CVD risk patients. EBioMedicine 44, 28–40 (2019).
Jain, K., Tyagi, T., Ionescu, C. N. & Hwa, J. Author’s response to “platelet antioxidants: A conundrum in aging”. EBioMedicine 47, 31–32 (2019).
Aaron, C. P. et al. A longitudinal cohort study of aspirin use and progression of emphysema-like lung characteristics on CT imaging: the MESA lung study. Chest 154, 41–50 (2018).
Mitchell, S. et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 13, 2497–2507 (2002).
Xiang, Y. et al. Hyperglycemia repression of miR-24 coordinately upregulates endothelial cell expression and secretion of von Willebrand factor. Blood 125, 3377–3387 (2015).
Xiang, Y. & Hwa, J. Regulation of VWF expression, and secretion in health and disease. Curr. Opin. Hematol. 23, 288–293 (2016).
Rask-Madsen, C. & King, G. L. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat. Clin. Pract. Endocrinol. Metab. 3, 46–56 (2007).
Meigs, J. B., Hu, F. B., Rifai, N. & Manson, J. E. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 291, 1978–1986 (2004).
NIH. Coronavirus disease 2019 (COVID-19) treatment guidelines. NIH https://www.covid19treatmentguidelines.nih.gov/ (2020).
CDC. Interim clinical guidance for management of patients with coronavirus disease 2019 (COVID-19). CDC https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html (2020).
Beigel, J. H. et al. Remdesivir for the treatment of Covid-19 – final report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2007764 (2020).
Shen, C. et al. Treatment of 5 critically Ill patients with COVID-19 with convalescent plasma. JAMA 323, 1582–1589 (2020).
The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with Covid-19 – preliminary report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2021436 (2020).
Connors, J. M. & Levy, J. H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 135, 2033–2040 (2020).
Bikdeli, B. et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up. J. Am. Coll. Cardiol. 75, 2950–2973 (2020).
McLaughlin, V. V. et al. Results of an expert consensus survey on the treatment of pulmonary arterial hypertension with oral prostacyclin pathway agents. Chest 157, 955–965 (2020).
Arehart, E. et al. Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: potential implications for cyclooxygenase-2 inhibition. Circ. Res. 102, 986–993 (2008).
Stitham, J., Midgett, C., Martin, K. A. & Hwa, J. Prostacyclin: an inflammatory paradox. Front. Pharmacol. 2, 24 (2011).
Veyradier, A. et al. Improvement of von Willebrand factor proteolysis after prostacyclin infusion in severe pulmonary arterial hypertension. Circulation 102, 2460–2462 (2000).
Walford, G. & Loscalzo, J. Nitric oxide in vascular biology. J. Thromb. Haemost. 1, 2112–2118 (2003).
Forstermann, U. & Munzel, T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113, 1708–1714 (2006).
Lei, C. et al. Protocol for a randomized controlled trial testing inhaled nitric oxide therapy in spontaneously breathing patients with COVID-19. medRxiv https://doi.org/10.1101/2020.03.10.20033522 (2020).
DiNicolantonio, J. J. & Barroso-Aranda, J. Harnessing adenosine A2A receptors as a strategy for suppressing the lung inflammation and thrombotic complications of COVID-19: potential of pentoxifylline and dipyridamole. Med. Hypotheses 143, 110051 (2020).
Liu, X. et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 10, 1205–1215 (2020).
Yasmeen, S., Akram, B. H., Hainsworth, A. H. & Kruuse, C. Cyclic nucleotide phosphodiesterases (PDEs) and endothelial function in ischaemic stroke. A review. Cell Signal. 61, 108–119 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04365309?term=NCT04365309&draw=2&rank=1 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04363840 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04410328 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04382755 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04288713 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04453527 (2020).
Kow, C. S. & Hasan, S. S. Meta-analysis of effect of statins in patients with COVID-19. Am. J. Cardiol. 134, 153–155 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04407273 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04472611 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04486508 (2020).
Acknowledgements
The authors thank the scientific research community, the biomedical community and the frontline health-care workers for the amazing progress, in just a few months, that has been made in deciphering COVID-19 disease mechanisms, developing novel therapeutics and improving outcomes, and for the range of epidemiological studies that have been performed. The authors’ work is in part supported by the DeLuca Center for Innovation in Hematology Research at Yale Cancer Center and The Frederick A. DeLuca Foundation, the Yale Cooperative Center of Excellence in Hematology (NIDDK U54DK106857), NIH/NIDDK R01DK102792 (S.H.); NIH/NCI R01CA222518 (D.S.K. and S.H.); NIH NHLBI R01HL142090 and R01HL146101 (K.A.M.); NIH NHLBI R01HL142818 and an AHA Transformational Project Award (H.J.C.); and NHLBI R01HL115247, R01HL122815 and R01HL150515 (J.H.).
Author information
Authors and Affiliations
Contributions
All authors researched data for article. S.X.G., T.T., H.J.C. and J.H. performed the major literature search and wrote the first draft. All the authors substantially contributed to their individual area of expertise, reviewed and edited the manuscript before submission and constructed the graphics for the submitted figure drafts.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Acute respiratory distress syndrome
-
(ARDS). A syndrome characterized by severe acute respiratory failure arising from inflammation and fluid build-up in the lungs.
- Coagulopathy
-
A variety of functional disorders of the coagulation system leading to increased or decreased coagulation.
- Thrombocytopathy
-
A variety of functional disorders of platelets, leading to increased or decreased platelet activity and function.
- Endotheliopathy
-
A variety of functional disorders of the endothelium, leading to increased or decreased endothelial activity and function.
- Disseminated intravascular coagulation
-
(DIC). Coagulopathy characterized by excessive formation of small clots throughout the body.
- Cytokine storm
-
Uncontrolled and excessive release of cytokines, leading to organ damage.
- Macrophage activation syndrome
-
A syndrome characterized by uncontrolled activation and proliferation of macrophages and T cells, leading to a markedly increased release of cytokines.
- NETosis
-
Form of neutrophil cell death associated with the extrusion of a web of chromatin, histones and granule proteins, known as neutrophil extracellular traps, which can capture and kill pathogens and can also form a scaffold for thrombosis.
- Megakaryocytopathy
-
A variety of functional disorders of megakaryocytes leading to increased or decreased megakaryocyte function.
- Kawasaki disease
-
A systemic, febrile illness in young children that can lead to vasculitis and coronary artery aneurysms.
Rights and permissions
About this article

Cite this article
Gu, S.X., Tyagi, T., Jain, K. et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol 18, 194–209 (2021). https://doi.org/10.1038/s41569-020-00469-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-020-00469-1
This article is cited by
-
SARS-CoV-2 S protein activates NLRP3 inflammasome and deregulates coagulation factors in endothelial and immune cells
Cell Communication and Signaling (2024)
-
SARS-CoV-2’s brain impact: revealing cortical and cerebellar differences via cluster analysis in COVID-19 recovered patients
Neurological Sciences (2024)
-
Predictors and outcomes of acute pulmonary embolism in COVID-19; insights from US National COVID cohort collaborative
Respiratory Research (2023)
-
Genomic communication via circulating extracellular vesicles and long-term health consequences of COVID-19
Journal of Translational Medicine (2023)
-
Metabolic alterations upon SARS-CoV-2 infection and potential therapeutic targets against coronavirus infection
Signal Transduction and Targeted Therapy (2023)
