
Abstract
ATPases are a group of enzymes that can cyclically convert the free energy of ATP hydrolysis into mechanical work. GTPases are another class of enzymes that are predominantly associated with signal transduction processes, but their role in mechanotransduction is less established. It was previously shown that the binding of the GTPase Rab5 to the tethering protein EEA1 induces a large conformational change in EEA1 from a rigid, extended to a flexible, collapsed state. This entropic collapse of EEA1 gives rise to an effective force that can pull tethered membranes closer. It currently remains unclear if EEA1 can return from the collapsed to the extended conformation without the aid of chaperone proteins. Here we show that EEA1 in a bulk solution can undergo multiple flexibility transition cycles driven by the energetics of Rab5 binding and unbinding as well as GTP hydrolysis. Each cycle can perform up to 20kBT of mechanical work. Hence, Rab5 and EEA1 constitute a two-component molecular motor driven by the chemical energy derived from the Rab5 GTPase cycle. We conclude that tethering proteins and their small GTPase partners can have active mechanical roles in membrane trafficking.
Similar content being viewed by others


Main
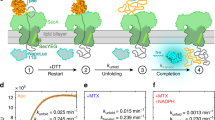
Intracellular traffic involves a complex choreography of mechanical and chemical steps, from the formation of vesicles and their movement along cytoskeletal tracks up to the tethering and fusion with their appropriate target membrane compartments1,2,3. Molecular motors that cyclically transduce chemical energy (ATP) to transport vesicles over long distances are a canonical example of the coupling of chemistry with mechanics. However, such a coupling is less understood for membrane tethering, driven by the pairing of small GTPases with either multi-subunit complexes4,5 or long dimeric coiled-coil tether molecules6. Recently, conformational changes in the early endosomal tether EEA1, caused by its binding to the small GTPase Rab5, have been shown to result in mechanical forces pulling membranes in close proximity to each other7. The prevalence of dimeric coiled-coil motifs in tethering molecules suggests that these long molecules can play generic mechanical roles in regulating and overcoming distance barriers that physically separate membranes7,8, thus facilitating fusion. EEA1 is a coiled-coil dimeric molecule with a contour length of 222 ± 26 nm (ref. 7) and binds to the small GTPase in its ‘active’ GTP-bound form, Rab5(GTP), but not in its ‘inactive’ GDP-bound form, Rab5(GDP) (Fig. 1a). On binding to Rab5(GTP) via a Rab5-binding domain located near the N terminus, EEA1 undergoes a general change in conformation, from a more rigid ‘extended’ state to a more flexible ‘collapsed’ state7 (Fig. 1a). The flexibility transition of EEA1 causes it to collapse, that is, adopt lower end-to-end distance configurations due to entropic reasons, thus generating an effective force than can bring tethered membranes closer (Fig. 1a). It is noteworthy that such an entropic collapse associated with a flexibility transition along the length of the molecule could be shared by other coiled-coil tethers such as GCC185, whose flexibility is mediated by a local unwinding of specific sequences in the central region3.

Binding of the Rab5(GTP) to EEA1 triggers a transition of the EEA1 molecule from a rigid, extended state to a more flexible, collapsed state. a, Sketch depicting the collapsed/extended states of EEA1 on binding/unbinding to the active/inactive forms of Rab5. b, Doubly tagged (Alexa 488 and 647) EEA1 molecules are used in the dcFCCS experimental setup to measure the dynamics of a single end of the molecules in solution. The time series of the fluorescence intensity fluctuations within the confocal volume is used to quantify the dynamics of molecular motion. c,d, Dynamics in solution for EEA1 alone (blue) and when mixed with Rab5(GTP) (red) are quantified by the MSD plotted over lag time τ (c) and the local scaling exponent α of the MSD (d). The black solid and dashed lines represent scaling exponents α = 3/4 and α = 2/3, respectively, corresponding to regimes where the persistence length of EEA1 is comparable with its contour length, that is, extended and smaller than the contour length that is collapsed. The solid blue (EEA1) and red (EEA1+Rab5(GTP)) lines represent fits to the experimental data using equation (1).
A high flux of vesicles in the endosomal system (~1,000 clathrin-coated vesicles per minute9) necessitates the recycling of the EEA1 molecules, following the collapse mechanism, for new rounds of vesicle tethering and fusion. Thus, after the collapse to a more flexible configuration, EEA1 must regain its extended conformation, that is, switch its flexibility back to a stiffer state. However, it remains unclear whether completing a cycle of EEA1 collapse and extension requires chaperones, or if the transition back to an extended configuration can be supported by the energy of the GTPase cycle alone (Fig. 1a). The latter scenario would make the system similar to molecular motors like kinesin or myosin10, with the key difference that the hydrolysis cycle would not drive the cyclic movement of a lever arm to perform mechanical work against an opposing force10, but instead supports a molecular flexibility transition that results in a possibly reversible entropic collapse. Here we resolved this question by combining dual-colour fluorescence cross-correlation spectroscopy (dcFCCS)11,12,13 and semiflexible polymer theory14,15 to measure and interpret the conformational dynamics of EEA1 on interaction with its GTPase partner Rab5. We found that no external chaperones are required for the conformational cycling of EEA1. Further, this reversible process results in a mechanochemical work cycle establishing EEA1 and Rab5 as a two-component molecular motor system.
FCS measurements of flexibility changes in EEA1
The dynamics of a long molecule, such as EEA1, in solution—comprising intramolecular motions, namely, bending, rotation and centre-of-mass displacement of the entire macromolecule—are similar to that of a randomly moving polymer. In addition to the effects of hydrodynamics due to the surrounding solvent, these dynamics are predominantly affected by the flexibility of the polymer, which is quantified by persistence length λ in relation to its total length, namely, contour length L (ref. 16). In particular, λ and L affect the crossovers from the dynamics due to bending modes on shorter timescales to rotation and centre-of-mass diffusion on longer timescales. Consequently, the evaluation of the complex crossover dynamics characterizes the stiff (λ ≳ L) or flexible (λ ≪ L) state of the polymer. Following previous experiments and analysis based on the semiflexible polymer theory, these crossovers can be inferred by measuring the mean square displacement (MSD) of one end of the long polymer molecule using fluorescence correlation spectroscopy (FCS)12,17,18.
To perform FCS measurements on EEA1, we fluorescently labelled one of its termini. Owing to its size, the timescales corresponding to the centre-of-mass diffusion of EEA1 through an FCS confocal volume are on the order of a few milliseconds, whereas the dynamics due to the internal molecular bending and rotational motions occur on relatively faster timescales, that is, tens to hundreds of microseconds. Often, in a technique such as FCS, these fast dynamics can be masked due to the photophysical processes of the fluorescent molecule that occur on similar timescales13. Therefore, to mitigate these effects and to access the intramolecular dynamics, we performed dcFCCS on EEA1 molecules that are simultaneously tagged at the same end with fluorescent molecules of two spectrally non-overlapping colours (Alexa 488 and 647), that is, dual-labelled EEA1 (Fig. 1b and Supplementary Sections 1 and 2). To achieve the dual labelling of EEA1, we took advantage of its dimeric nature and engineered its C-terminal sequence to add the recognition sequence for the Staphylococcus aureus sortase A (SrtA) enzyme (Methods, Supplementary Fig. 1 and Supplementary Section 1B)— the C-terminus is specifically chosen to avoid interference with the binding of Rab5(GTP) at the N-terminal end of EEA1 (ref. 19) (Fig. 1a). We then performed the SrtA reaction to obtain a population of EEA1 molecules consisting of monomers labelled at their C-terminal end with either of the two fluorescent tags (Fig. 1b, inset).
The FCS measurements were performed by the continuous recording of a time series of fluorescence intensity fluctuations of the dual-labelled EEA1 at a concentration of 100 nM (that is, total volume of 50 μl in a diluted regime) within a confocal volume of 0.285 μm3 (Fig. 1b). The measurements were performed before and immediately after the addition of 2 μM Rab5(GTP) to the same EEA1 sample, a concentration that ensures the binding of most of the EEA1 molecules to Rab5(GTP) (ref. 19). The time-series data are used to compute the time cross-correlations across the two colours G(τ) from which the MSD 〈r2(τ)〉 of one end of EEA1 with and without binding to Rab5(GTP) was extracted using a suitable mathematical transformation (Methods and Supplementary Section 2).
The MSD data corresponding to EEA1 before (Fig. 1c, blue data points) and after (Fig. 1c, red data points) binding to Rab5(GTP) show a marked difference in scaling behaviours characterized by the exponent α(τ), where 〈r2(τ)〉 ~ τα. At long-enough timescales, due to the diffusive motion of the molecular centre of mass, the MSD scales linearly with τ, that is, the exponent α(τ) ≈ 1 for both these EEA1 states. Strikingly, at intermediate timescales, corresponding to the dynamics due to internal polymer motions, this exponent is different for the unbound and Rab5(GTP)-bound forms of EEA1. Although α ≈ 3/4 for the unbound EEA1, we find that it is substantially lowered, that is, α ≈ 2/3, on the addition and thereby binding of Rab5(GTP) to EEA1 (Fig. 1d). The scaling behaviour of α ≈ 2/3 is what is expected for long, flexible polymers, that is, λ ≪ L, the so-called Zimm-scaling regime for semiflexible polymers, whereas the exponent α ≈ 3/4 corresponds to the so-called rigid-rod limit, that is, λ ≳ L (refs. 12,14,17).
Given that the contour length of EEA1 remains mostly unchanged on Rab5(GTP) binding7, the measured change in the scaling exponent strongly suggests a reduction in the persistence length λ of EEA1 on binding to Rab5(GTP). We extracted the persistence length λ of EEA1 in the bound and unbound states by fitting the MSD to
where the quantities D, Δn and τn, namely, the long-time centre-of-mass diffusivity, length and timescales corresponding to the rotation and bending motions, respectively, depend on persistence length λ and contour length L. Indeed, from this analysis, we find that the persistence length of EEA1 undergoes a reduction on Rab5(GTP) binding—\({\lambda }_{\mathrm{l}}\approx 6{9}_{-1}^{+9}\,{{{\rm{nm}}}}\)—compared with the persistence length of free EEA1, namely, \({\lambda }_{\mathrm{l}}\approx 18{4}_{-2}^{+2}\,{{{\rm{nm}}}}\); such a reduction is consistent with the stiffness transition previously characterized7. The scaling behaviour that we have measured here highlights two key aspects: (1) the softening of EEA1 on its binding to Rab5(GTP) probably occurs throughout its length, that is, EEA1 undergoes a global mechanical switch from a rigid-rod like polymer to a flexible one; and (2) a continuous measurement of scaling opens a window into the long-term tracking of the EEA1 polymer mechanics.
EEA1 flexibility switches reversibly
We next asked whether the flexibility switch of EEA1 induced by Rab5(GTP) binding can be reversed and if it can occur cyclically without the aid of additional factors such as chaperones. To evaluate this, we tracked the long-term behaviour of a population of EEA1 molecules after the addition of 2 μM Rab5(GTP), under the aforementioned experimental conditions. By measuring the changes in the scaling exponent α, we found that the population of EEA1 molecules recovers to a rigid state similar to free EEA1 (Fig. 2a). The kinetics of this recovery, of the population of EEA1 molecules to their original state, are consistent with the intrinsic (low) bulk GTPase activity of Rab5 at 25 °C (khydrolysis ≈ 5.5 × 10−4 s−1) (ref. 20), suggesting that the recovery is coupled to the hydrolysis of GTP. The kinetics of Rab5 GTP hydrolysis (intrinsic rates) would imply that after 70 min, little (<10%) active Rab5 remains in the solution20.

a, EEA1 alone (blue) in the extended state undergoes an entropic collapse (red) on Rab5(GTP) addition followed by a recovery to the original extended state over time. A second round of Rab5(GTP) addition results in another EEA1 collapse–extension cycle. The filled coloured circles correspond to experimental data and the solid black lines are fit to equation (1). b,c, Collapse–extension cycles are quantified from changes over time in the local slope minima αmin (b) and persistence length λ (c). The quantified results in (b) and (c) show that EEA1 in the extended state (blue; αmin ≈ 3/4 and λ ≈ 170 nm) undergoes a collapse (red; αmin ≈ 2/3 and λ ≈ 70 nm) on binding to Rab5(GTP), and recovers over time to the original extended state (red; αmin ≈ 3/4 and λ ≈ 190 nm). A second collapse–extension cycle occurs on the re-addition of Rab5(GTP). The blue and red solid lines correspond to αmin = 3/4 (extended state) and αmin = 2/3 (collapsed state). Flexibility increase is only attributed to the binding of Rab5(GTP), as confirmed by the addition of Rab5(GDP) and GST, which do not interact with EEA1. d, Scaling exponent α(τ) of EEA1 alone (blue) in the extended state and on the sequential addition of 1, 2 and 10 μM of Rab5(GDP) (grey). The results from the addition of 2 μM GST (yellow) to EEA1 (right). e,f, The αmin (e) and persistence length λ (f) values, quantified from the data shown in d for different concentrations of Rab5(GDP) and GST added to EEA1. The red horizontal region marks the corresponding values for the EEA1collapsed state on its interaction with Rab5(GTP). The bootstrap αmin (b,e) and λ (c,f) are obtained via 104 and 105 bootstrap samplings of the data, respectively; the open circles and error bars are mean and error of the mean for αmin, and the peak value and error of the peak for λ, respectively, as shown in Supplementary Section 2F3.
The ‘recovered’ state of EEA1 is similar in terms of its mechanical properties to the initial unbound EEA1, with a persistence length of λ ≈ 190 ± 5 nm (Fig. 2b,c and Supplementary Section 2F). These results suggest that EEA1 has regained its extended conformation. If EEA1 has indeed been fully recycled to its initial extended state, it should be able to undergo new cycles of collapse and extension on the re-addition of Rab5(GTP). Such a recycling would not be possible if the EEA1 molecule enters an energetically proximate yet inactive intermediate state that requires other inputs, such as the activity of chaperones, to be restored to its fully ‘active’ state. To test this, we measured a second cycle of entropic collapse by adding a fresh aliquot of 2 μM active Rab5(GTP) to a solution of EEA1 that had undergone a cycle of collapse and re-extension. Remarkably, the recovered EEA1 collapsed once again into the flexible state and then recovered to the extended form (Fig. 2a, second cycle). Note that the values of persistence length λ are similar in both cycles. These results indicate that EEA1 can reversibly undergo multiple stiffness–flexibility transitions (Fig. 2c). These global flexibility transitions of EEA1 are solely triggered by the interaction with active Rab5(GTP) and do not require any additional factors, as a minimal system.
Finally, in all our experiments, the MSD scaling exponent for the population of EEA1 on recovery was measurably higher than that for free EEA1, that is, we measured α > 3/4, concomitantly with a slight increase in the effective persistence length (Fig. 2b,c). We speculated that this might be due to the presence of Rab5(GDP) in the solvent that does not bind to EEA1. To test this hypothesis, we performed separate experiments where we added different concentrations of Rab5(GDP) to free EEA1. Consistent with our expectation, the addition of Rab5(GDP) did not cause the transition of EEA1 to a flexible state. Instead, we found that the scaling exponent for the EEA1 dynamics in the presence of different concentrations of Rab5(GDP) was slightly higher than free EEA1, that is, α > 3/4 (Fig. 2d,e, Supplementary Fig. 5 and Supplementary Section 2F), suggesting that non-specific interactions may slightly reduce the fluctuations of EEA1. Similarly, the presence of a distinct, non-interacting protein, like glutathione S-transferase (GST), did not reduce the scaling exponent of EEA1 (Fig. 2d,e). This increase in the scaling exponent is indeed consistent with what we observed in our recovery experiments, suggesting that Rab5(GDP) has a similar effect of increasing the scaling exponent. These data, together with the recovery kinetics, suggest that although Rab5(GTP) binding could account for the softening transition of EEA1, GTP hydrolysis and Rab5(GDP) unbinding may participate in the recovery of EEA1 back to the extended state.
Altogether, these measurements demonstrate that the long coiled-coil tether EEA1 can repeatedly undergo reversible flexibility transitions on interaction with active Rab5 and that these transitions occur without the aid of external agents. These observations, combined with previous optical tweezers measurements of the force generated during the softening and subsequent collapse of EEA1 (ref. 7), suggest that EEA1 and Rab5 are involved in a mechanochemical work cycle, that is, they form a ‘two-component molecular motor’ system. By a two-component molecular motor, we mean that its mechanical aspects (stiffness changes and force generation) and chemical aspects (binding, GTP hydrolysis, phosphate release and unbinding) are attributed to two separate molecules—EEA1 and Rab5—that act in concert.
Semiflexible polymer model for the EEA1–Rab5 motor
To discuss the thermodynamics of the work cycle of EEA1 and Rab5, we consider a simplified mechanochemical model (Fig. 3). We assume that EEA1 is a semiflexible polymer with bending rigidity κ that is constant along the length of the molecule and that is related to persistence length λ according to λ = κ/kBT, where kBT is thermal energy. The bending rigidity is a mechanical quantity that includes various intra- and intermolecular interactions, such as the pairing of heptad repeats in the EEA1 coiled coil, in a coarse-grained manner. The interaction of EEA1 with Rab5 leads to an instantaneous change in bending rigidity κ that can be followed by a mechanical equilibration of the semiflexible polymer via a change in its end-to-end distance r.

a, Chemical potential differences provided by active or inactive Rab5 interactions (green, binding Rab5(GTP); grey, GTP hydrolysis, phosphate release and unbinding of Rab5(GDP)) affect the effective persistence length of EEA1, coupling chemical transformations of the GTPase to tether mechanics. The state transitions are indicated with colour-coded numbers: 1, unbound and extended EEA1; 2, extended EEA1–Rab5 complex; 3, collapsed EEA1–Rab5 complex; 4, unbound and collapsed EEA1. The transition paths are as follows: 1→2, capture (green); 2→3, collapse (red); 3→4, release (grey); 4→1, recovery (blue). The variables held constant during the transitions are indicated next to the respective transition paths. b, With fixed contour length L, only persistence length λ determines the equilibrium behaviour, like the average extension. The size of the system determines a critical persistence length λc = 2π−3/2L above which the equilibrium extension is greater than zero. The two states of unbound and bound EEA1 toggle around this critical value. c, Probability density of the extension illustrates how the substantial shift between high and low persistence lengths (from λh = 250 nm (blue) to λl = 50 nm (red); L = 210 nm) affects the expected end-to-end tether extension in the constant-length ensemble. The states are colour coded as in a. The arrows designate the direction of the transition paths. For comparison, the probability density for the critical persistence length is also shown (grey dashed curve). d–f, Bending energy U(r, λ) (d) and conformational entropy S(r, λ) (e) contribute to the free energy A(r, λ) (f) of the system. The states and transitions are colour coded in each panel as in a. Together, these panels indicate the asymmetry of the individual motor substeps due to the semiflexible nature of the tether: although the collapse is entropically driven, the recovery is dominated by the energetic contribution. Furthermore, at their respective equilibrium extensions, it costs more free energy to turn a stiff polymer into a more flexible one than the contrary case.
Our theoretical analysis is performed in a ‘fixed-extension ensemble’, where the end-to-end distance of the EEA1 molecule is fixed on shorter timescales but is allowed to slowly vary as the molecule proceeds through the mechanochemical cycle. The key elements of the cyclic protocol involve four processes that comprise a closed path (Fig. 3a; the cycle is indicated by the lines connecting the points 1, 2, 3 and 4): (1) on binding to Rab5(GTP), EEA1 reduces its bending rigidity κ and its persistence length decreases from λh to λl (upward green arrow); (2) the extended but soft semiflexible polymer equilibrates mechanically, resulting in a reduction in its end-to-end distance (red arrow, collapse); (3) triggered via a combination of chemical steps that include GTP hydrolysis by Rab5, phosphate release and unbinding of Rab5(GDP) from EEA1, EEA1 undergoes a switch back from soft to stiff, that is, its persistence length increases from λl to λh (downward grey arrow); (4) the now collapsed but rigid semiflexible polymer equilibrates mechanically, resulting in an extension of its end-to-end distance (blue arrow). Two segments of the path, namely, 2→3 (collapse) and 4→1 (recovery), amount to a change in extension that can proceed against an external opposing force, and EEA1 can perform mechanical work in these segments. Transitions between the two flexibility states of EEA1, namely, 1→2 and 3→4, are mediated by the aforementioned chemical steps. The chemical potential differences Δμh→l and Δμl→h capture the free-energy differences of the corresponding chemical transitions within the EEA1–Rab5 cycle. Taken together, the EEA1–Rab5 system can convert chemical energy into mechanical work during one passage of this coarse-grained mechanochemical cycle.
We next discuss how EEA1–Rab5 can perform mechanical work in the fixed-extension ensemble to which the protocol of slowly varying extension described above is applied. The EEA1 molecule is held in place between two attachment points that are fixed in space, that is, with a fixed end-to-end distance, whereas the force that the molecule exerts on the attachment points fluctuates. The two attachment points exert equal but opposite external forces to keep their positions fixed in space. As the cyclic protocol proceeds, the molecule is subjected to a slow change in attachment-point positions, leading to a slow change in the end-to-end distance of EEA1 concomitant with a change in the average force that the molecule exerts on the attachment points. We note that the switch of bending rigidity κ on Rab5 binding (1→2) and Rab5 release (3→4) causes EEA1 to transition between two regimes: λ ≪ L when flexible and λ ≳ L in the more rigid state. This requires us to use the Blundell–Terentjev model for semiflexible polymers that is valid in both regimes15.
We highlight a key aspect of this specific polymer model that makes it suitable to describe both flexibility regimes of the EEA1–Rab5 system. In an isothermal environment, for a polymer with fixed contour length L, the average equilibrium end-to-end distance \(\tilde{r}\) is determined by the effective persistence length λ. The model identifies a critical persistence length λc = 2π−3/2L, below which \(\tilde{r}\) becomes zero (Fig. 3b) and the polymer behaviour is effectively Gaussian. This critical point, therefore, separates the regimes of high and low flexibility, as identified even for EEA1 in the FCS experiments (Figs. 1 and 2). This transition, therefore, suggests a qualitative difference, that is, a buckling transition between the two regimes for a semiflexible polymer: one in which the filament can withstand thermal fluctuations (λ > λc) to maintain a finite extension and the other one in which it cannot and therefore has an equilibrium end-to-end distance of zero.
Experiments performed here and in previous work7 indicate that EEA1 is indeed poised close to such a critical point. The unbound extended state of EEA1 and the Rab5(GTP)-bound collapsed state indeed lie on either side of the critical persistence length λc ≈ 75 nm for the L ≈ 210 nm molecule (Fig. 3b). Altogether, this suggests a mechanical-state transition in EEA1 mediated by its chemical interaction with Rab5. For our subsequent analysis (and without any loss of generality), we take λh = 250 nm and λl = 50 nm, which are the consensus values from various experiments (dcFCCS here, and optical tweezers and rotary-shadowing electron microscopy7) for the extended and collapsed states, respectively.
To compute the forces exerted by EEA1, we first consider the conformational free energy of a polymer in the absence of chemical transitions. We start from the analytical form for the polymer end-to-end distance distribution, P(r, λ). The Helmholtz conformational free energy of EEA1 is related as A(r, λ) = –kBTlog[P(r, λ, L)], from which the forces exerted by the molecule are computed as 〈F〉(r, λ, L) = −∂rA(r, λ, L). The free energy A(r, λ), particularly the balance between bending energy U(r, λ) and conformational entropy S(r, λ) (Fig. 3d,e), reveal interesting aspects of the EEA1–Rab5 motor. Before discussing the relative contributions of the entropic and bending energy components, it is evident that the mechanical transitions are driven by the minimization of the polymer free energy (Fig. 3f). This minimization can be understood as follows: the persistence-length switch along path 1→2 is associated with a reduction in both conformational entropy S and bending energy U of EEA1 (Fig. 3d,e and Supplementary Section 3). The collapse of EEA1, that is, 2→3 is, therefore, entropic in origin, and the mechanical force generation is akin to that of an entropic spring21. In contrast, the recovery of EEA1 to the extended configuration along path 4→1 is driven by the stored polymer bending energy, which results in a pushing force. In equilibrium, the unbound polymer has a finite extension that exactly balances the entropic and energetic contributions (Supplementary Fig. 8). As expected, all the states of a semiflexible polymer fall on a universal curve that asymptotically approaches the two extremes: a rigid rod (fully elastic) or a flexible freely jointed chain (Supplementary Section 3A).
From the force–extension relation, we estimate the mechanical work performed by the two-component motor during the different stages of the cycle (Supplementary Section 3B). Here we consider two generic scenarios for the work cycle: (1) EEA1 is tethered to a vesicle cargo that it can drag within the dense cytoplasmic milieu and (2) an in vitro situation in which EEA1 is tethered to a colloidal probe (spherical bead) and exerts a force against the optical tweezers that hold the colloid7. We compute the average force at a given extension, namely, 〈F〉(r, λ, L) = −∂rA(r, λ, L), and this force–extension relationship (shown for the two persistence lengths associated with the extended and collapsed states of EEA1; Fig. 4a) corresponds to having an external force Fext = −〈F(r, λ)〉 that maintains the polymer end-to-end extension r fixed. In the fixed-extension ensemble discussed above, the capture and release extensions are variable, and their magnitudes, together with the persistence length corresponding to the two flexibility states of EEA1, determine the amount of work that is extracted. In the first scenario, that is, the context of cellular vesicle trafficking, EEA1 most probably captures a vesicle at the average equilibrium extension of the unbound tether (rcapture ≈ 175.5 nm) and collapses until other molecular layers start to engage with the vesicle (rrelease ≈ 75.0 nm), leading to the docking and completion of membrane fusion. From here, the tether recovers and could potentially push out a load following the blue curve (Fig. 4a) to reach the unbound equilibrium extension, after which the cycle could start again with a new round of vesicle tethering. On the other hand, analogous to the in vitro situation of EEA1 dragging a tethered colloid against optical tweezers, there is a time-dependent tension and drag force22, during the entropic collapse, against which EEA1 performs work (Fig. 4b). We calculated the mechanical work using the calibrated trap stiffness and measured displacements for each experiment corresponding to a pairing of EEA1 with Rab5(GTP)-covered beads. The mechanical work distribution that was measured in optical tweezers single-molecule experiments (Fig. 4c) quantitatively matches the mechanical work computed from our model. Based on the optical tweezers measurements and the different combinations of persistence lengths in the extended and collapsed states, the maximum work that can be obtained by an entropic collapse mechanism ranges between 6kBT and 20kBT. The exact amount depends on the relative differences in flexibility of EEA1 in the two states (Supplementary Section 3 and Supplementary Fig. 9 provide detailed investigations for different values of persistence length).

a, With the switchable semiflexible polymer as the working substance, cyclic motor-like processes can be run by the corresponding protocols. One example is an idealized Stirling-type engine. Here the four steps of the work cycle are as follows: 1→2, isometric softening after active Rab5 capture without performing work; 2→3, isothermal collapse (red) to perform work (red-shaded area); 3→4, isometric stiffening after the release of inactive Rab5 (grey); 4→1, isothermal extension (blue) to perform work during recovery (blue-shaded area). This portion might be harder to extract due to the detachment of GTPase. The indicated capture extension (green) corresponds to the equilibrium extension of the unbound tether, whereas the release extension (grey) is variable and has been chosen for illustrative purposes. b, The EEA1 motor system can perform mechanical work against an opposing external force, and the collapse work can be measured using optical tweezers. Here the collapsing two-component EEA1–Rab5 complex (red curve and arrow) moves microspheres against the opposing restoring force of the optical traps (yellow curve and arrow) until an equilibrium is reached (intersection between the red and yellow curves). This protocol has been experimentally realized in a dual-trap experiment7. c, Relative occurrence of the measured collapse work in single-molecule EEA1–Rab5 optical tweezers experiments described elsewhere7. Note that only the collapse work is probed (red-shaded area in a). Panel c adapted with permission from ref. 7, Springer Nature Ltd.
For this two-component system to operate as a self-sufficient GTP-fuelled motor, the increase in free energy from 1→2 and 3→4 must be accounted for by the chemical energy transitions arising from chemical potential differences and the associated chemical transitions due to Rab5(GTP) binding/unbinding, GTP hydrolysis and phosphate release, as well as the unbinding of Rab5(GDP). These chemical free energies, combined into the effective chemical potentials Δμh→l(r = rcapture) and Δμl→h(r = rrelease), set upper bounds on the mechanical work that the EEA1–Rab5 motor can perform. It is important to note that for EEA1–Rab5 to proceed through the cycle without the aid of external factors, the sum of the two chemical transitions, Δμtotal = Δμh→l(rcapture) + Δμl→h(rrelease), cannot exceed the available chemical energy per cycle derived from GTP hydrolysis, that is, Δμtotal < ΔμGTP hydrolysis. The dissociation constant between Rab5(GTP) and the zinc-finger domain of EEA1 is KD = 2.4 μM (ref. 19), which translates into a free energy of 12.9kBT for Rab5(GTP) binding. Several lines of evidence suggest that the binding between active Rab5 and the full-length dimeric EEA1 is even stronger23, supporting the assumption that the binding step could provide enough free energy to drive the flexibility transition (1→2; Fig. 3a). Meanwhile, the hydrolysis of a single Rab5(GTP) can produce an energy equivalent to 20–25kBT (ref. 24). Since the energy of GTP hydrolysis is larger than the energy obtained from binding, it is feasible that the sum of both chemical potentials (capture and release; even considering that according to the model, release would require ~3kBT; Fig. 3f) are lower than the energy obtained from GTP hydrolysis. Altogether, although the polymer model allows us to compute the mechanical work performed by EEA1 during the entire cycle, the chemical energy obtained from binding, GTP hydrolysis, phosphate release and unbinding sets upper bounds for this work, which can be as high as 20–25kBT (ref. 24). The energy released by either GTP hydrolysis and/or Rab5(GTP) binding would be sufficient to account for the work performed during a single cycle of EEA1 extension and collapse for the persistence length values that we have measured in the various experiments (dcFCCS here and optical tweezers and rotary-shadowing electron microscopy earlier7). Conversely, this limits the allowable transitions in bending rigidity κ of the extended and collapsed configurations of EEA1. Further, since the EEA1–Rab5 system requires two chemical energy transitions (1→2 and 3→4; Figs. 3a and 4a; both free-energy sources (Rab5(GTP) binding and GTP hydrolysis)) can be utilized to complete a full cycle. Finally, the efficiency of this motor (Supplementary Fig. 10) is in the range of other molecular motors such as kinesins10.
Discussion
Although the roles of small GTPases in cellular signal transduction have long been recognized, their function in mediating mechanical processes has, thus far, remained less explored. On the other hand, many ATPases like myosin and kinesin have been recognized as force-generating soft machines for several decades, although they are rarely seen as signalling molecules. Together with our earlier results7, this work demonstrates that the coiled-coil protein EEA1 and the small GTPase signalling molecule Rab5 work together as a two-component molecular motor system that can transfer the chemical energy of GTP hydrolysis into mechanical work, and simultaneously toggling the state of GTPase during the process. Altogether, this sets this GTPase-driven mechanism apart from other ATP-driven motors10.
Our results have important implications for membrane tethering and fusion well beyond the specific case of EEA1 and Rab5. Several long coiled-coil tether proteins that function at distinct stages of the exocytic and endocytic pathways are effectors of small GTPases25,26,27,28—these long molecules have traditionally been described as spacers, rulers29 and rods30, suggesting static and fixed distances between the ends. Accordingly, the observation that the physical length of a coiled-coil domain is much more conserved than its sequence31 has been interpreted along the lines that a static distance between the two ends is functionally important. Yet, contrary to the widely accepted notions of coiled-coils as static and fixed rulers29, our work shows that EEA1—a prominent example of a long coiled-coil tethering molecule—is poised at a critical point close to bistable flexibility states. The switch between these states is mediated by a series of chemical steps that induce large-scale movements on the order of ~100 nm. The mechanism described here could be shared by several other protein tethers. The Golgi coiled-coil tether such as GCC185 exhibits a great degree of flexibility due to the unwinding of the coiled-coil central region3. This supports the idea that long coiled-coil tethers are ‘breathing’ molecules, that is, metastable mechanical structures. Although in that case, the effect of binding to Rab GTPases has not been explored yet, it is possible that it may control the dominant conformation of the tether, as shown for EEA1. This would make the tether–small GTPase coupling a widespread paradigm not only for membrane recognition but also to convert the energy stored in GTP into work in the mechanochemical pathways to membrane fusion. If a work cycle was a general requirement of the membrane tethering-to-fusion process, the multiprotein tethering complexes such as Exocyst and HOPS could use mechanical energy as part of their function in a similar fashion to the long coiled-coil tethers. However, in contrast to flexibility transitions, one could expect this to be an enthalpy-driven transition that is a lever-arm-like action at a single location as opposed to an overall ‘softening’ of the protein.
The model we identified to harness mechanical work from this cyclically acting polymer motor (Fig. 4a) is reminiscent of a classical Stirling engine. Although classical engines use gas as the working substrate and exploit temperature gradients to generate mechanical work via expansion/compression of the gas, the two-component polymer motor that we describe here uses the polymer itself as a working substrate with flexibility changes resulting in force generation. Consequently, this kind of mechanism suggests new avenues for the design of synthetic molecular engines32.
Methods
Cloning, expression and purification of proteins
The cDNA-encoding EEA1 was subcloned into the pOEM1-based vector pOCC151 (PEPC; MPI-CBG), which includes an N-terminal GST tag followed by an HRV-3C cleavable site between the tag and inserted gene. On the EEA1 C-terminus, the amino-acid sequence GGGSGGGGSGGGGSGGGGSLPETGGGG was added. The cDNA-encoding Rab5a was subcloned into the pET11-based vector pOCC9 (PEPC; MPI-CBG), which includes an N-terminal hexahistidine tag followed by an HRV-3C cleavable site between the tag and inserted gene. GST was expressed from the pGST2 vector. The plasmid pET30b-7M SrtA, containing a C-terminal hexahistidine tag, was a gift from Hidde Ploegh (Addgene plasmid 51141).
Rab5, GST and SrtA7m were expressed in Escherichia coli BL21 (DE3) and protein purification was performed in a standard buffer (20.0 mM Tris at pH 7.4, 150.0 mM NaCl, 5.0 mM MgCl2 and 0.5 mM tris(2-carboxyethyl)phosphine) (Supplementary Information). For GTP loading, freshly purified Rab5 was supplemented with 10 mM EDTA and tenfold excess GTP. The mixture was incubated for 15 min at 4 °C before the addition of 10 mM MgCl2, and subsequently ran over a desalting column equilibrated in the standard buffer. EEA1 was expressed in SF9 cells growing in ESF921 media (Expression Systems) (Supplementary Section 1).
Dual-colour labelling of EEA1
The SrtA recognition site (LPETG) was added to the EEA1 C-terminus during subcloning. The SrtA-based reaction was performed in a buffer containing 20.0 mM Tris at pH 7.4, 150.0 mM NaCl and 0.5 mM tris(2-carboxyethyl)phosphine. A mixture of 1.0 μM EEA1, 30.0 μM GGGaWC-A488, 30.0 μM GGGaWC-A647 and 1.5 μM SrtA7m was incubated for 1 h at room temperature on a rotator wheel. The obtained EEA1–fluorophore conjugate was purified by size exclusion chromatography and the purity was evaluated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, laser scanning imaging (Typhoon FLA9500, GE Healthcare; wavelengths, 473 and 635 nm) and Coomassie staining. GGGaWC-A488 and GGGaWC-A647 were produced by the Biomolecular Synthesis facility at B CUBE (Supplementary Section 1B).
Setup and equipment for dcFCCS
The dcFCCS experiments were performed on a ConfoCor 3 setup (Carl Zeiss LSM780 NLO) operated in the photon-counting mode (objective, C-Apochromat 40×/1.2 W autocorr M27; excitation sources, 488 nm (Ar-ion laser, 4.52 μW) and 633 nm (He–Ne laser, 6.23 μW); emission range for detectors, 499–552 and 641–694 nm; pinhole diameter, 58 μm; temperature, 23.0 ± 0.5 °C). Then, 10 nM dsDNA (50 μl), dual labelled with A488 and A647 at the opposite ends, was used to maximize the confocal volume overlap by adjusting the objective collar for the maximum photon counts per molecule. The confocal volume alignments in the x–y plane and z axis were visualized by confocal microscopy of fluorescent-membrane-coated beads at their equatorial plane and by a z scan of the supported lipid bilayers, respectively, where the membranes were doped with fluorescent membrane probes DiO and DiD (Supplementary Section 2).
Dynamics of a single polymer end
The correlation G(τ) of the fluctuating labelled end is related to the MSD (<r2(τ)>) of the end of the polymer:
where <N>, ωxy and ωz correspond to the average number of molecules in the confocal volume and the lateral and axial widths of the confocal volume, respectively. Normalized correlation curves were fitted to equation (2) and the roots (<r2(τ)>) were obtained by the good Broyden method using a custom-written computer code. The local exponent α(τ) was computed by finding the local slope for a moving window of <r2(τ)> corresponding to a decade of lag time τ using the expression
By fitting <r2(τ)> to equation (1) for n ≤ 2, the diffusion coefficient D, timescale pre-factor Δn and timescale (τn) were obtained. The persistence length λ was calculated by fitting to the expanded form (equation (4)) of equation (1), written as a function of the radius of cross-sectional area (a), contour length (L) and persistence length (λ) of the polymer.
The parameters a (1 nm) and L (220 nm) were obtained from the crystal structure of EEA1 coiled-coil region (PDB:1JOC) and rotary-shadowing electron microscopy, respectively. Using a and L as fixed parameters in equation (4), the free parameter λ can be evaluated.
Bootstrapping
The recovery from the collapsed to extended state results in intermediate populations that were probed by performing bootstrapping on the dcFCCS curves. Bootstrapping was performed on 180 curves. The mean correlation (G(τ)avg) curve was obtained from 20 curves selected at random. Here 105 G(τ)avg curves were sampled with replacement to calculate the distribution of λ. For intermediate stages of recovery, multimodal distributions were obtained for the two cycles with peaks corresponding to collapsed and extended states (Supplementary Section 2E4).
Experiments to probe EEA1 flexibility in different states
Experiments were carried out in a 384-well glass-bottom plate with 175 ± 15-μm-thick glass sealed with aluminium foil to prevent drying. Then, 50 μl EEA1 (100 nM) were added to a well and the confocal beams were focused 20 μm above the glass surface to perform dcFCCS. Then, 240 correlation curves of 30 s each were recorded for 2 h. The first 30 min recordings were removed to let the system equilibrate. Two rounds of 2 μM Rab5(GTP) were added to the solution and dcFCCS was evaluated for 2.5 h after each addition. The data were further analysed as described in Supplementary Section 2.
Collapse work estimation from optical tweezers experiments
To estimate the order of magnitude of mechanical work performed during the Rab5-driven collapse of EEA1, we re-analysed the single-molecule optical tweezers data from our earlier study. In brief, for these experiments, glass beads with supported lipid bilayers carrying purified EEA1 or active Rab5(GTP) were trapped in dual-trap optical tweezers. The traps were successively brought closer together until there was a connection between EEA1 on one microsphere and Rab5(GTP) on the other one. We observed transient interactions that pulled both beads together and resulted in sub-piconewton forces (Fig. 3 and refs. 33,34,35,36,37,38,39,40,41,42,43,44). Using the known trap stiffness and displacements for each experiment, here we calculated the mechanical work for each event for the case of pairing EEA1 with Rab5(GTP)-covered beads (N = 38). We interpret the measured work in the optical tweezers as corresponding to transition 1→2→3 (Fig. 4a), which amounts to mechanical work performed during entropic collapse. The histogram of this collapse work is shown in Fig. 4c.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. All other data that support the plots within this paper and other findings of this study are available from the corresponding authors upon reasonable request.
Code availability
All code used to produce these results are available from the corresponding authors upon reasonable request.
References
Zerial, M. & McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117 (2001).
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 (2009).
Cheung, P. Y. P., Limouse, C., Mabuchi, H. & Pfeffer, S. R. Protein flexibility is required for vesicle tethering at the Golgi. eLife 4, e12790 (2015).
Balderhaar, H. J. K. & Ungermann, C. CORVET and HOPS tethering complexes—coordinators of endosome and lysosome fusion. J. Cell Sci. 126, 1307–1316 (2013).
Bröcker, C., Engelbrecht-Vandré, S. & Ungermann, C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. 20, R943–R952 (2010).
Gillingham, A. K. & Munro, S. Long coiled-coil proteins and membrane traffic. Biochim. Biophys. Acta 1641, 71–85 (2003).
Murray, D. H. et al. An endosomal tether undergoes an entropic collapse to bring vesicles together. Nature 537, 107–111 (2016).
Wilson, J. M. et al. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol. Biol. Cell 11, 2657–2671 (2000).
Griffiths, G., Back, R. & Marsh, M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J. Cell Biol. 109, 2703–2720 (1989).
Howard, J. Mechanics of Motor Proteins and the Cytoskeleton Vol. 743 (Oxford Univ. Press, 2001).
Aragon, S. & Pecora, R. Fluorescence correlation spectroscopy as a probe of molecular dynamics. J. Chem. Phys. 64, 1791–1803 (1976).
Petrov, E. P., Ohrt, T., Winkler, R. & Schwille, P. Diffusion and segmental dynamics of double-stranded DNA. Phys. Rev. Lett. 97, 258101 (2006).
Schwille, P., Meyer-Almes, F. J. & Rigler, R. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 72, 1878–1886 (1997).
Hinczewski, M., Schlagberger, X., Rubinstein, M., Krichevsky, O. & Netz, R. R. End-monomer dynamics in semiflexible polymers. Macromolecules 42, 860–875 (2009).
Blundell, J. & Terentjev, E. Buckling of semiflexible filaments under compression. Soft Matter 5, 4015–4020 (2009).
Doi, M. & Edwards, S. F. The Theory of Polymer Dynamics Vol. 73 (Oxford Univ. Press, 1988).
Shusterman, R., Alon, S., Gavrinyov, T. & Krichevsky, O. Monomer dynamics in double-and single-stranded DNA polymers. Phys. Rev. Lett. 92, 048303 (2004).
Hinczewski, M. & Netz, R. R. Global cross-over dynamics of single semiflexible polymers. Europhys. Lett. 88, 18001 (2009).
Mishra, A., Eathiraj, S., Corvera, S. & Lambright, D. G. Structural basis for Rab GTPase recognition and endosome tethering by the C2H2 zinc finger of Early Endosomal Autoantigen 1 (EEA1). Proc. Natl Acad. Sci. USA 107, 10866–10871 (2010).
Simon, I., Zerial, M. & Goody, R. S. Kinetics of interaction of Rab5 and Rab7 with nucleotides and magnesium ions. J. Biol. Chem. 271, 20470–20478 (1996).
Marko, J. F. & Siggia, E. D. Stretching DNA. Macromolecules 28, 8759–8770 (1995).
Bohbot-Raviv, Y., Zhao, W., Feingold, M., Wiggins, C. H. & Granek, R. Relaxation dynamics of semiflexible polymers. Phys. Rev. Lett. 92, 098101 (2004).
Simonsen, A. et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394, 494–498 (1998).
Beard, D. A. & Qian, H. Chemical Biophysics: Quantitative Analysis of Cellular Systems (Cambridge Univ. Press, 2008).
Short, B., Haas, A. & Barr, F. A. Golgins and GTPases, giving identity and structure to the Golgi apparatus. Biochim. Biophys. Acta 1744, 383–395 (2005).
Drin, G., Morello, V., Casella, J.-F., Gounon, P. & Antonny, B. Asymmetric tethering of flat and curved lipid membranes by a golgin. Science 320, 670–673 (2008).
Nyitrai, H., Wang, S. S. H. & Kaeser, P. S. ELKS1 captures Rab6-marked vesicular cargo in presynaptic nerve terminals. Cell Rep. 31, 107712 (2020).
Cheung, P. Y. P. & Pfeffer, S. R. Transport vesicle tethering at the trans Golgi network: coiled coil proteins in action. Front. Cell Dev. Biol. 4, 18 (2016).
Truebestein, L. & Leonard, T. A. Coiled-coils: the long and short of it. BioEssays 38, 903–916 (2016).
White, G. E. & Erickson, H. P. Sequence divergence of coiled coils—structural rods, myosin filament packing, and the extraordinary conservation of cohesins. J. Struct. Biol. 154, 111–121 (2006).
Surkont, J., Diekmann, Y., Ryder, P. V. & Pereira-Leal, J. B. Coiled-coil length: size does matter. Proteins 83, 2162–2169 (2015).
Zhang, L., Marcos, V. & Leigh, DA. Molecular machines with bio-inspired mechanisms. Proc. Natl Acad. Sci. USA 115, 9397–9404 (2018).
Dai, X., Böker, A. & Glebe, U. Broadening the scope of sortagging. RSC Adv. 9, 4700–4721 (2019).
Jeong, H. J., Abhiraman, G. C., Story, C. M., Ingram, J. R. & Dougan, S. K. Generation of Ca2+-independent sortase A mutants with enhanced activity for protein and cell surface labeling. PLoS ONE 12, e0189068 (2017).
Mao, H., Hart, S. A., Schink, A. & Pollok, B. A. Sortase-mediated protein ligation: a new method for protein engineering. J. Am. Chem. Soc. 126, 2670–2671 (2004).
Antos, J. M. et al. Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Protein Sci. 89, 15.3.1–15.3.9 (2017).
Wenger, J. et al. Dual-color fluorescence cross-correlation spectroscopy in a single nanoaperture: towards rapid multicomponent screening at high concentrations. Opt. Express 14, 12206–12216 (2006).
Bacia, K., Petrášek, Z. & Schwille, P. Correcting for spectral cross-talk in dual-color fluorescence cross-correlation spectroscopy. ChemPhysChem 13, 1221–1231 (2012).
Weidemann, T., Wachsmuth, M., Tewes, M., Rippe, K. & Langowski, J. Analysis of ligand binding by two-colour fluorescence cross-correlation spectroscopy. Single Mol. 3, 49–61 (2002).
Krieger, J. W. & Langowski, J. QuickFit 3.0: a data evaluation application for biophysics (2010–2021).
Broyden, C. G. A class of methods for solving nonlinear simultaneous equations. Math. Comput. 19, 577–593 (1965).
Ries, J., Petrášek, Z., García-Sáez, A. J. & Schwille, P. A comprehensive framework for fluorescence cross-correlation spectroscopy. New J. Phys. 12, 113009 (2010).
Resch-Genger, U. Standardization and Quality Assurance in Fluorescence Measurements II: Bioanalytical and Biomedical Applications Vol. 6 (Springer, 2008).
Sengupta, P., Garai, K., Balaji, J., Periasamy, N. & Maiti, S. Measuring size distribution in highly heterogeneous systems with fluorescence correlation spectroscopy. Biophys. J. 84, 1977–1984 (2003).
Acknowledgements
We thank M. Rao and P. Pullarkat for discussions. We thank R. Schäfer for providing the GST protein. S.T. and A.S. acknowledge support from the Department of Atomic Energy, Government of India, under projects RTI4001 and RTI4006; the Simons Foundation (grant no. 287975 to S.T.); and the Max Planck Society through a Max Planck Partner Group at NCBS-TIFR (S.T.). M.J. and S.W.G. were supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC-2068–390729961. J.A.S. and J.L. were financially supported by the Human Frontiers in Science Program (HFSP), grant no. RGP0019/2020. This study was financially supported by the Max Planck Society. We thank the following services and facilities of the MPI-CBG for their support: PEPC and Light Microscopy. We also thank the Central Imaging and Flow Cytometry Facility (CIFF) at the NCBS.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
M.J. conceived the two-component molecular motor idea. S.T., A.S., J.A.S., M.Z. and J.L. conceived and designed the FCS experiments. J.A.S. and J.L. performed the biochemistry and sample preparation. A.S. and J.A.S. performed the FCS experiments. A.S. analysed the FCS data with guidance from S.T. M.J. analysed the optical tweezers data. J.A.S., J.L. and M.Z. contributed regarding knowledge of the GTPase cycle to the development of the physics model. M.J. developed the polymer physics model, with help from S.T. and S.W.G. All the authors interpreted the data and contributed to the writing of the paper. S.T., M.Z. and S.W.G. provided the overall supervision for the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Physics thanks Michael Hinczewski and Yongli Zhang for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10 and Discussion.
Source data
Source Data Fig. 1
Dual color intensity versus time data; average cross-correlation curve to plot MSD(tau) and alpha(tau).
Source Data Fig. 2
a, Average correlation curve to plot MSD(tau) and alpha(tau). b, Minimum of local exponent from bootstrapping for GTP hydrolysis. c, Persistence length from bootstrapping for GTP hydrolysis. d, Average correlation curves for EEA1, Rab5(GDP) and GST. e, Minimum of local exponent from bootstrapping of EEA1, Rab5(GDP) and GST. f, Persistence length from bootstrapping of EEA1, Rab5(GDP) and GST.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, A., Soler, J.A., Lauer, J. et al. Two-component molecular motor driven by a GTPase cycle. Nat. Phys. 19, 1185–1192 (2023). https://doi.org/10.1038/s41567-023-02009-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41567-023-02009-3
This article is cited by
-
Cochaperones convey the energy of ATP hydrolysis for directional action of Hsp90
Nature Communications (2024)
-
Molecular motor may help design tiny machines
Nature India (2023)
-
A molecular motor for cellular delivery and sorting
Nature Physics (2023)
