
Abstract
The coronavirus disease 2019 (COVID-19) pandemic has claimed millions of lives and caused a global economic crisis. No effective antiviral drugs are currently available to treat infections of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The medical need imposed by the pandemic has spurred unprecedented research efforts to study coronavirus biology. Every virus depends on cellular host factors and pathways for successful replication. These proviral host factors represent attractive targets for antiviral therapy as they are genetically more stable than viral targets and may be shared among related viruses. The application of various ‘omics’ technologies has led to the rapid discovery of proviral host factors that are required for the completion of the SARS-CoV-2 life cycle. In this Review, we summarize insights into the proviral host factors that are required for SARS-CoV-2 infection that were mainly obtained using functional genetic and interactome screens. We discuss cellular processes that are important for the SARS-CoV-2 life cycle, as well as parallels with non-coronaviruses. Finally, we highlight host factors that could be targeted by clinically approved molecules and molecules in clinical trials as potential antiviral therapies for COVID-19.
Similar content being viewed by others


Main
Coronaviruses are positive-strand RNA viruses belonging to the subfamily Orthocoronavirinae within the family Coronaviridae (International Committee on Taxonomy of Viruses) and are subdivided into four genera—Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. Coronaviruses cause intestinal and respiratory infections in a variety of birds and mammals, including livestock and domestic animals.
Seven human coronaviruses (HCoVs) have been characterized, four of which cause mild respiratory infections (HCoV-229E, HCoV-NL63, HCoV-OC43 and HCoV-HKU1). The emergence of two highly pathogenic betacoronaviruses in 2002 (severe acute respiratory syndrome coronavirus (SARS-CoV)) and 2012 (Middle East respiratory syndrome coronavirus (MERS-CoV)) revealed that new pathogenic coronaviruses can emerge in the human population by zoonotic transmission (reviewed in ref. 1). In late 2019, a pathogenic coronavirus SARS-CoV-2 was first detected in Wuhan, China, causing an outbreak of severe pneumonia. This human pathogen is thought to have originated in horseshoe bats and was probably transmitted to humans through an intermediate host that remains to be identified2. Owing to its high contagiousness and the occurrence of asymptomatic carriers, SARS-CoV-2 rapidly spread across the globe and continues to claim human lives and obstruct social and economic activity, while vaccination programs are ongoing. The pandemic has prompted countless efforts to develop vaccines and antiviral therapies, but also many fundamental studies to better understand coronavirus biology. As cellular proviral host factors are potential antiviral drug targets, numerous studies have analysed host factor dependencies of coronaviruses, in particular SARS-CoV-2.
In this Review, we provide an overview of proviral host factors for SARS-CoV-2. After explaining the coronavirus life cycle, we discuss the cellular receptors and proteases that are required for SARS-CoV-2 entry. As only few of the identified proviral host factors have been assigned to specific post-entry stages of the life-cycle, these stages are not discussed separately. Instead, we summarize the wealth of information on proviral SARS-CoV-2 host factors that has been produced by genome-wide functional genetic screens and interactome analyses and discuss their roles in cellular processes. Finally, we highlight host factors that may serve as targets for antiviral therapies against COVID-19.
The coronavirus life cycle
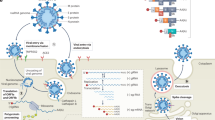
The SARS-CoV-2 virion is an enveloped and pleomorphic particle with a diameter of around 60–100 nm (ref. 3). The helical nucleocapsid contains the ~30 kb single-stranded RNA genome packaged by the nucleocapsid (N) protein4. It is surrounded by the viral envelope, which contains the spike (S), membrane (M) and envelope (E) proteins. Coronavirus entry into cells is mediated by S, a homotrimeric class-I fusion protein (reviewed in ref. 5). The dual role of S is to bind to cellular receptors on target cells and to fuse viral and cellular membranes, triggered by proteolytic cleavage of S by host proteases. Coronaviruses enter cells either through endocytic pathways or by directly fusing with the plasma membrane, depending on the availability of cellular proteases (reviewed in refs. 6,7) (Fig. 1). After fusion, the nucleocapsid is released into the cytoplasm, after which the genomic RNA uncoats by dissociation from N and is subsequently translated.

After SARS-CoV-2 particles attach to the target cell by interacting with receptor(s), cleavage of the S protein by cell-surface proteases (such as TMPRSS2) is thought to trigger fusion with the plasma membrane. Alternatively, SARS-CoV-2 can enter cells through endocytosis, after which fusion is induced by low pH and S cleavage by endosomal/lysosomal proteases (cathepsins). The N protein dissociates from the viral positive-strand (+) RNA genome, which is directly translated to form polyproteins pp1a and pp1ab. These polyproteins are autocatalytically processed into the non-structural proteins nsp1–16, which establish RTCs and remodel cellular membranes to form replication organelles. These organelles are continuous with the ER and provide an optimal environment for viral RNA replication, which mainly occurs inside DMVs. Genome replication starts with the synthesis of a negative-strand (−) copy that functions as a template for the synthesis of new positive-strand RNA genomes, which may enter more rounds of translation or are incorporated into new virions. Discontinuous transcription of the positive-strand genomic RNA yields subgenomic negative-strand RNAs, which function as templates for the synthesis of subgenomic positive-strand RNAs that encode structural and accessory proteins. Nascent viral RNAs exit DMVs through a transmembrane pore to reach sites of translation or virion assembly. Genomic positive-strand RNA, encapsidated by N, as well as the structural proteins S, M and E, assemble at the ERGIC, at which new virions form by budding into the lumen. Finally, progeny virions are released from the host cells. The yellow boxes indicate the different steps of the viral life cycle. The genes that were both identified as proviral SARS-CoV-2 host factors in at least two independent studies (either through pooled functional genetic screening or through individual genetic validation) and individually validated in at least one study (Supplementary Table 1) are shown. Genes that were individually validated in multiple studies and in different cell lines are highlighted in green.
Coronaviruses have capped and polyadenylated genomes that contain multiple open reading frames (ORFs), flanked by 5′ and 3′ untranslated regions. The SARS-CoV-2 genome contains at least 11 ORFs8, encoding 16 non-structural proteins (nsp1–16), 4 structural proteins (S, E, M and N) and a set of putative accessory proteins (ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8 and ORF9b)9. Although coronavirus accessory proteins are non-essential for replication in vitro, these proteins are believed to be required for virulence in vivo (reviewed in ref. 10). Viral gene expression starts with the translation of orf1a and orf1b directly from the positive-strand RNA genome pp1a is encoded by ORF1a, whereas pp1ab is formed by continuous translation of ORF1a and ORF1b through a programmed −1 ribosomal frameshift (regulated by host proteins such as SHFL), which determines the stoichiometry between pp1a and pp1ab. These polyproteins are processed by viral proteases that are located within nsp3 (PLpro) and nsp5 (Mpro or 3CLpro), yielding the 16 replication proteins nsp1–16 (ref. 11). nsp1 shuts down host translation and promotes host mRNA degradation, whereas nsp2–16 establish the viral replication–transcription complex (RTC). nsp2–11 modulate the intracellular environment to favour viral replication, whereas nsp12–16 contain the core enzymatic functions required for RNA synthesis, including the RNA-dependent RNA polymerase (RdRp; nsp12) (reviewed in ref. 12).
RNA replication starts with the synthesis of full-length negative-strand copies of the viral genome. These serve as templates for the production of new positive-strand genomes, which are translated to form more non-structural proteins or are packaged into newly formed virions. Moreover, discontinuous transcription of the genome generates a set of negative-strand subgenomic RNAs, which serve as templates for the synthesis of positive-strand subgenomic RNAs (reviewed in ref. 13). These are translated to form the structural proteins and accessory proteins. RNA replication occurs in replication organelles (formed by nsp3, nsp4 and nsp6), which provide an optimal environment for replication and are thought to shield viral RNA from detection by cytosolic innate immune sensors14. These organelles are endoplasmic reticulum (ER)-derived perinuclear interconnected membrane structures that contain double-membrane vesicles (DMVs), in which RNA synthesis occurs15. Nascent viral RNA molecules escape from these DMVs through a transmembrane pore16 to reach sites of translation or virion assembly, which occurs at the ER–Golgi intermediate compartment (ERGIC)17. Here, newly synthesized viral genomes—coated with N protein—bud into the lumen of the ERGIC to form enveloped particles containing M, E and S proteins. Finally, progeny virions are trafficked to the cell surface for release. Although this process is poorly understood, a recent study showed that the Betacoronavirus mouse hepatitis virus (MHV) hijacks the lysosomal pathway for non-lytic egress of new infectious virions18. Each step of the viral life cycle depends on the interplay between viral components and cellular host factors, which is discussed below for SARS-CoV-2.
SARS-CoV-2 receptors
ACE2
The coronavirus S protein consists of two subunits, S1 and S2. S1 mediates receptor binding, usually through a receptor-binding domain (RBD) within S1, whereas S2 guides membrane fusion. As angiotensin-converting enzyme 2 (ACE2) was previously shown to be a crucial receptor for SARS-CoV infection in vivo19, ACE2 was rapidly identified as a receptor for SARS-CoV-2. Structural studies revealed that the peptidase domain of ACE2 binds to the SARS-CoV-2 S RBD20,21. ACE2 was confirmed to be a functional receptor by showing that overexpression of human ACE2 enabled SARS-CoV-2 infection of poorly susceptible cell lines22,23 and mice24, which are naturally non-susceptible. ACE2 depletion inhibited SARS-CoV-2 infection of Vero E6 (ref. 25), Huh7.5 (refs. 26,27), Caco-2 (ref. 28) and Calu-3 (refs. 29,30) cells. By contrast, other studies reported that SARS-CoV-2 could infect ACE2-deficient cells31, although this may be due to mutations in S32. ACE2 exists in a membrane-bound form and a soluble form, released after cleavage by ADAM17. A recent study suggested that soluble ACE2 can also promote infection by forming a complex with SARS-CoV-2 S and vasopressin33.
Viral tropism is partly determined by the repertoire of receptors that a virus can engage. SARS-CoV-2 enters the body primarily through the respiratory tract34 and is most abundantly detected in the airways, but also resides in the kidneys, liver, heart, brain, blood and intestines35,36. ACE2 expression in specific airway cells is increased in patients with COVID-19 (ref. 37), most likely because ACE2 is an interferon-stimulated gene38. Although SARS-CoV-2 RNA was predominantly detected in airway cell types that also express ACE2 (refs. 37,39), a strict correlation at the single-cell level between ACE2 expression and SARS-CoV-2 infection has not yet been demonstrated. As SARS-CoV-2 infects many organs, it is possible that other receptors contribute to viral dissemination in vivo. Recently identified SARS-CoV-2 candidate receptors that may cooperate with or act as an alternative to ACE2 are discussed below and summarized in Table 1.
Auxiliary SARS-CoV-2 receptors
Several SARS-CoV-2 receptors most likely function as cofactors that enhance entry through other receptors. It was shown that heparan sulfate (HS) binds to SARS-CoV-2 S40,41,42 and that HS depletion decreases SARS-CoV-2 infection in vitro42,43. Thus, analogous to other viruses, SARS-CoV-2 may engage HS to facilitate initial cell attachment, increasing the likelihood of subsequent interactions with entry receptors44. As many viruses bind to HS as a consequence of adaptation to in vitro cultured cells, future studies should establish whether HS binding is a natural ability of SARS-CoV-2. Scavenger receptor class B member 1 (SRB1) is a cell-surface receptor that is involved in the uptake of lipids, including high-density lipoprotein (HDL). SARS-CoV-2 S1 binds to HDL, and the addition of HDL to cells enhances infection45. In the presence of HDL, overexpression of SCARB1 (which encodes SRB1) stimulated infection, whereas knockdown of SCARB1 reduced SARS-CoV-2 infection, suggesting that SRB1 mediates the cellular uptake of HDL-bound virus. A unique feature of SARS-CoV-2 that is absent in SARS-CoV is a polybasic motif (RRAR) at the S1/S2 boundary, which can be cleaved by furin46, resulting in a C-terminally exposed RRAR peptide. Two independent studies showed that this peptide directly binds to neuropilin-1 (NRP1) and that NRP1 promotes SARS-CoV-2 infection47,48 (Table 1). Although SRB1 and NRP1 may be able to independently support infection, the fact that these receptors enhance SARS-CoV-2 entry in the presence of ACE2 overexpression45,47,48 suggests that they serve as cofactors that potentiate infection through ACE2 or other receptors.
Alternatives to ACE2
Several candidate receptors were shown to enable SARS-CoV-2 infection in the absence of ACE2. The cell-surface proteins tyrosine-protein kinase receptor UFO (Axl)31, low-density lipoprotein receptor class A domain–containing protein 3 (LDLRAD3) and C-type lectin domain family 4 member G (CLEC4G)49 were all shown to bind to the N-terminal domain (NTD) of SARS-CoV-2 S. Moreover, their depletion in cell lines reduced SARS-CoV-2 infection and their overexpression in ACE2-knockout cells promoted infection, showing that these proteins are probably alternative receptors to ACE2. Basigin (also known as CD147/EMMPRIN), a broadly expressed protein that binds to various ligands, was shown to bind to the S RBD50, although another study did not confirm this finding51. Notably, expression of human basigin enabled SARS-CoV-2 infection in mice. As SARS-CoV-2 cannot use mouse ACE2 (ref. 23), these findings implicate basigin as an ACE2-independent host factor.
Other receptors
Several other candidate receptors were shown to bind to SARS-CoV-2 S and to facilitate SARS-CoV-2 infection when overexpressed, including asialoglycoprotein receptor 1 (ASGPR1), Kremen protein 1 (ref. 52) and transferrin receptor53. The innate immune receptors CD209 (also known as DC-SIGN) and C-type lectin domain family 4 member M (CLEC4M; also known as L-SIGN or CD209L) bind to numerous viruses, including SARS-CoV and SARS-CoV-2 (refs. 54,55,56,57). These receptors have been suggested to enable virus transport between target cells (trans-infection)58. Whether depletion of the above receptors in cells affects SARS-CoV-2 infection remains to be shown. Finally, several other SARS-CoV-2 receptor candidates were suggested on the basis of their direct interaction with S (Table 1).
In conclusion, several SARS-CoV-2 candidate receptors have been identified in addition to ACE2, although it remains to be established whether these receptor interactions are biologically relevant or have resulted from cell-culture-induced virus mutations. Future studies will need to evaluate whether their expression levels in SARS-CoV-2 target cells is sufficient to be relevant for infection. At present, none of the SARS-CoV-2 receptors identified, including ACE2, has been validated by genetic depletion in an animal model. Such studies will be required to establish the relevance of each receptor for SARS-CoV-2 in vivo.
Proteases required for SARS-CoV-2 entry
After receptor binding, the viral envelope fuses with the host cell membrane, a process guided by host protease–mediated cleavage of S. SARS-CoV-2 has remarkable flexibility in protease requirements, and the local protease availability and temperature59 influence the viral entry route and cell tropism (reviewed in refs. 6,7). Proteolytic activation of S occurs in two sequential steps.
The first cleavage (priming) occurs at the S1/S2 boundary for some, but not all, coronaviruses, typically during S biosynthesis at the trans-Golgi network of infected cells. After this cleavage, the two subunits remain bound together by non-covalent bonds and are incorporated together into assembled virions. Priming generally facilitates receptor binding and can expose hidden cleavage sites. The unique polybasic motif at the S1/S2 boundary in SARS-CoV-2 S forms a minimal furin-like cleavage site60. Indeed, it was shown that SARS-CoV-2 particles harbour cleaved S protein and that furin inhibition substantially reduces the amount of cleavage46,60,61. Furin is a ubiquitously expressed endoprotease that is mainly localized in the trans-Golgi. Thus, the polybasic furin cleavage site is believed to be a gain-of-function that enables systemic spread of SARS-CoV-2. In support of this, loss of the furin cleavage site attenuates replication in respiratory cells and reduces pathogenesis in animal models, but does not completely abolish infection61,62,63,64. MERS-CoV S also harbours a multibasic site that can be primed by furin, in contrast to S in the more closely related SARS-CoV, which is not primed60.
The second cleavage (activation) occurs at the S2′ site immediately downstream of the fusion peptide and is crucial for infection of all coronaviruses. It induces conformational changes that liberate the fusion peptide, which then penetrates the host cell membrane, leading directly to membrane fusion. SARS-CoV-2 S activation at S2′ can be accomplished by transmembrane protease serine 2 (TMPRSS2) on the plasma membrane, but also by endosomal cathepsin proteases, of which cathepsin L is probably the most important22,65. S1/S2 priming is a prerequisite for subsequent TMPRSS2-mediated activation at the S2′ site, but not for S2′ activation by cathepsin L in TMPRSS2-negative cells60,62,66. These observations are consistent with the concept of early and late entry routes for coronaviruses. For SARS-CoV-2, priming by furin and activation by TMPRSS2 enable the more efficient early route through fusion at the plasma membrane. The gain of an S1/S2 multibasic site may explain the expanded tropism of SARS-CoV-2 to epithelial cells of the aerodigestive tract, which highly express TMPRSS2 (refs. 67,68). In cells lacking TMPRSS2, S1/S2 priming is redundant and SARS-CoV-2 is endocytosed, with fusion occurring late in acidified endo/lysosomal compartments after activation by cathepsins66.
Other proteases, such as PC1, trypsin-like proteases and cathepsins, can cleave peptides mimicking the multibasic S1/S2 site of SARS-CoV-2 in vitro69. Moreover, studies using pseudoviruses and selective protease inhibitors suggest that alternative proteases can cleave at S1/S2 in cells66. However, the biological relevance of alternative proteases in the pathogenesis of SARS-CoV-2 needs to be determined. Finally, plasmin, which is commonly elevated in people with hypertension or diabetes, may cleave at the multibasic site70. Similarly, S2′ cleavage can be accomplished by alternative transmembrane serine proteases, as was demonstrated for transmembrane protease serine 4 (TMPRSS4) in small intestinal enterocytes67.
Functional genetic screens for SARS-CoV-2 host factors
Several independent CRISPR-based functional genetic screens have been carried out (Box 1) to uncover SARS-CoV-2 proviral host factors. Most were performed on a genome-wide scale25,26,29,30,43,64,71,72, but some of the screens probed a subset of genes derived from interactome analyses27,73 (Fig. 2). All of the screens were based on cell survival, which has the limitation that genes that are essential for cell proliferation are unlikely to be detected. Moreover, such functional screens are biased towards genes that are required for early stages of the viral life cycle and fail to detect genes that are involved in virion assembly or release. Of the many experimental parameters that differed between screens (Supplementary Table 1), the choice of cell line seems to be crucial because gene overlap was mainly observed between screens carried out in the same cell type (Fig. 2). Although lung tissue is the primary site of SARS-CoV-2 infection, multiple cell lines from different origins have been used to define host factor dependencies, mainly due to lack of a virus-induced cytopathic effect in lung-derived cell lines. Vero E6 cells are non-human African green monkey kidney-derived cells that are permissive to a wide range of viruses in vitro. Whereas the human hepatocellular carcinoma Huh7 or colorectal adenocarcinoma Caco-2 cells are permissive to SARS-CoV-2 infection, the lung adenocarcinoma A549 cell line requires ectopic overexpression of the ACE2 receptor to support SARS-CoV-2 replication, while infection and cytopathic effect in the lung cell line Calu-3 is low. Here, we discuss genes that were consistently identified using functional genetic screens and were individually validated as proviral SARS-CoV-2 host factors (Fig. 1 and Supplementary Table 2). Although the precise role in the SARS-CoV-2 life cycle remains unknown for many of these genes, some have known functions in the life cycle of other viruses (Table 2).

a–c, Overview of human genes identified as SARS-CoV-2 proviral host factors in functional genetic screens (a) and genes encoding proteins identified in interactome screens as binding partners for viral RNA (b) or viral proteins (c). a, The number of top-ranked hits or validated proviral genes found in CRISPR-based loss-of-function studies (Zhu et al.64, Daniloski et al.71, Biering et al.29, Rebendenne et al.30, Hoffmann et al.27, Schneider et al.26, Baggen et al.43, Wang et al.72 and Wei et al.25). b, The number of proteins identified in viral RNA interactomes (Lee et al.99, Flynn et al.73, Schmidt et al.100, Kamel et al.101 and Labeau et al.102). Gene names are listed for proteins that meet one of the following three criteria: identified in five out of the six screens; identified in all four cell types; or identified in three cell types and functionally validated. Genes are clustered according to molecular function. c, The total number of cellular proteins identified in viral protein interactomes for each publication (Laurent et al.108, St-Germain et al.107, Li et al.104, Gordon et al.78, Davies et al.103, Stukalov et al.105 and Samavarchi-Tehrani et al.106) (top). Bottom, the number of publications in which a specific virus–host protein–protein interaction was detected. Genes encoding human proteins identified either in four or more publications or identified with the highest frequency for a specific SARS-CoV-2 protein (and in more than 50% of conducted screens) are shown. For viral proteins with >20 cellular interaction partners identified as such, only partners with a specificity index >2 are included (that is, the total detection number of the cellular protein divided by the number of unique viral proteins with which it was found to interact). Different isoforms were not taken into account. Genes and proteins detected in Vero cells were converted to their human orthologue. The complete gene lists are provided in Supplementary Table 2. The central Venn diagram represents the total number of genes or proteins identified in at least two screens by each approach. Genes identified by multiple orthogonal approaches are listed and clustered according to molecular function. Genes that have been individually validated as proviral factors by knockdown or knockout are shown in blue.
The only gene identified by screens in all five cell lines tested is ACE2 (Fig. 2). Of the other receptor candidates identified, transmembrane protein 30A (TMEM30A) was identified in one screen43, whereas the HS biosynthesis gene EXT1 was independently identified in three Huh7 screens26,43,72. Importantly, screens carried out in Huh7, A549 and Vero E6 cells identified CTSL (encoding cathepsin L), but not TMPRSS2, suggesting that viral entry occurred through the late endosomal route in these models. By contrast, screens in Calu-3 and Caco-2 cells29,30 identified TMPRSS2, but not CTSL, implying that these screens probably identified factors that are involved in the early entry route.

Vesicle trafficking
Besides receptors and proteases, members of different complexes involved in vesicle biology were identified as essential for SARS-CoV-2 infection, including Rab GTPases (RAB7A, RAB10 and RAB14). In particular, RAB7A was extensively validated and found in multiple screens, as were components of its dedicated Rab guanine nucleotide exchange factor, the Mon1–Ccz1 complex74 (CCZ1, CCZ1B and C18orf8). RAB7A and CCZ1B were also found to support HCoV-229E and HCoV-OC43 infection26,27,72. Among other functions, Rab7 regulates endosomal maturation and interacts with the homotypic fusion and protein sorting (HOPS) complex, of which a subunit, the Vam6/Vps39-like protein (VPS39), was found to support SARS-CoV-2 infection. The HOPS complex mediates fusion of lysosomes with late endosomes or autophagosomes and, as shown for MHV75 and Ebola virus76 (Table 2), may cooperate with Rab7 to traffic incoming SARS-CoV-2 virions to lysosomal fusion sites. Rab7 was also found to promote cell-surface expression of ACE2 (ref. 71). Moreover, SARS-CoV-2 ORF3A binds to the HOPS subunit Vam6/Vps39-like protein to block autophagosome–lysosome fusion, preventing lysosomal destruction of viral components in the later stages of its life cycle77. Genetic screens also identified the S1 (ATP6AP1) and A (ATP6V1A) subunits of the vacuolar-ATPase proton pump71, which were found to bind to SARS-CoV-2 nsp6 and M, respectively78. These factors are required for luminal acidification of vesicles and also facilitate influenza A virus entry79,80. TMEM106B, which encodes another endo-lysosomal protein, was shown in multiple studies to be required for SARS-CoV-2 but not HCoV-229E26,43,72. TMEM106B regulates lysosome function and interacts with the V-ATPase S1 subunit81. TMEM106B facilitates SARS-CoV-2 pseudovirus entry43, further suggesting a role of the lysosomal compartment in SARS-CoV-2 entry. Furthermore, TMEM106B expression is elevated in SARS-CoV-2-infected airway cells from patients with COVID-19 (ref. 43), but whether this is the cause or consequence of infection needs to be determined.
Many genes uncovered by genetic screens are involved in retrograde transport82. These include members of the retromer and retriever complexes (VPS29, VPS35 and VPS35L), the retromer-associated protein sorting nexin-27 (SNX27) and components of the COMMD–CCDC22–CCDC93 (CCC) complex (Fig. 1). These complexes facilitate cargo enrichment and budding from the endosomal membrane to generate recycling vesicles. This budding process is driven by branched actin polymerization initiated by the Arp2/3 and WASH complexes83, of which members were found in multiple SARS-CoV-2 screens64,71 (ACTR2, ACTR3, ARPC3, ARPC4 and WASHC4). Finally, multiple screens identified a member of the exocyst complex26,72 (EXOC2), which interacts with the Arp2/3 complex84 and mediates tethering of recycling and secretory vesicles to the plasma membrane. As knockout of retrograde transport genes inhibited SARS-CoV-2 pseudovirus entry and reduced cell-surface expression of ACE2 (ref. 64), SARS-CoV-2 may require this process for receptor recycling, as shown for human T-cell leukaemia–lymphoma virus85. Alternatively, retrograde transport may facilitate virus trafficking to replication sites, as shown for human papillomavirus86, or may serve to deliver cargoes to replication sites, as suggested for hepatitis C virus87 (Table 2).
Notably, screens in Calu-3 cells did not identify any of the above-mentioned vesicle-trafficking genes, but revealed two subunits of clathrin-associated adaptor protein complex 1 (AP1B1 and AP1G1)29,30, which mediates transport between the trans-Golgi network and endosomes (Fig. 1). These genes were shown to support SARS-CoV-2 entry without affecting ACE2 expression on the cell surface30 and may be required for the correct localization of other proviral factors that are involved in TMPRSS2-mediated entry.
PI3K signalling
Genetic screens also identified members of the phosphatidylinositol 3-kinase (PI3K) pathway (Fig. 1) as host factors for SARS-CoV-2, as well as for HCoV-229E and HCoV-OC43 (refs. 26,43,64,71,72). These include PI3K type 3 (PIK3C3) and PI3K regulatory subunit 4 (PIK3R4), core subunits of a complex that produces phosphatidylinositol 3-phosphate (PI3P) and regulators of this complex (WDR81 and WDR91)64,71. Screens also uncovered protein VAC14 homologue (VAC14)26,72, which regulates the conversion of PI3P to phosphatidylinositol 3,5-bisphosphate. This signalling pathway is involved in a wide range of cellular functions, including endosomal maturation, retrograde transport and autophagy initiation (reviewed in ref. 88). PI3K signalling may therefore facilitate SARS-CoV-2 infection by initiating the vesicle-trafficking processes discussed above, or by activating the autophagy machinery. Indeed, multiple studies showed that SARS-CoV-2 infection depends on transmembrane protein 41B (TMEM41B)26,43,89, which is involved in the early stages of autophagosome formation90. TMEM41B is also required for HCoV-229E, HCoV-OC43 and HCoV-NL63 (refs. 26,43); SARS-CoV; and MERS-CoV89, and was recently shown to facilitate flavivirus replication complex formation91 (Table 2). As genes required for later stages of autophagosome formation were not identified, it is possible that SARS-CoV-2 specifically hijacks early components of the autophagy machinery, including PI3K signalling, to support its life cycle.
Lipid homeostasis
Several genes identified in genetic screens with SARS-CoV-2 control cholesterol synthesis26,72. Sterol-regulatory-element-binding protein 1 and 2 (SREBF1 and SREBF2) are transcription factors that upregulate enzymes required for fatty acid and cholesterol synthesis. Their activity is regulated by sterol-regulatory-element-binding protein cleavage-activating protein (SCAP) and endopeptidase S1P and S2P (MBPTS1 and MBPTS2)92, which were all identified in genetic screens. SREBF2 was also found to support the replication of HCoV-229E, HCoV-OC43 and HCoV-NL63 (ref. 26). Moreover, SARS-CoV-2 was found to require the late endosomal and lysosomal proteins Niemann–Pick C1 and C2 (NPC1 and NPC2), which transport cholesterol from the lysosomal lumen into the lysosomal membrane93. Although these genes probably have indirect roles in infection, NPC1 can also directly function as a viral receptor, as shown for Ebola virus76 (Table 2). Recently, 25-hydrocholesterol—which broadly inhibits the fusion of enveloped viruses by depleting cholesterol from the plasma membrane94—was shown to inhibit SARS-CoV-2 S-mediated fusion, suggesting a role of cholesterol in viral entry. Genetic screens also identified two components (TMEM30A and ATP8B1) of a P4-ATPase flippase complex that transports aminophospholipids from the outer to the inner leaflet of various membranes29,30,43,64. Moreover, the lipid transport protein sigma non-opioid intracellular receptor 1 (SIGMAR1) was found to bind to nsp6 in an interactomic screen78. As all steps of the viral life cycle are membrane-associated, cholesterol and other lipids could have various roles in SARS-CoV-2 infection, including innate immune system suppression, as shown for other viruses95.
Other proviral host factors
Other identified SARS-CoV-2 host factors are involved in transcriptional regulation, including transcription factors, histone-modifying enzymes and members of the SWI/SNF complex25,26,64,71 (Fig. 1). Moreover, screens identified several components of E3 ubiquitin ligases25,71. These genes may indirectly affect virus replication by regulating expression levels of other proviral or antiviral factors. For example, EP300 and HMGB1 enhance the expression of ACE2 on the cell surface25,30,96. E3 ubiquitin–protein ligase SIAH1 (SIAH1) and JmjC-domain-containing protein 6 (JMJD6) were previously shown to support infection of dengue virus97 and vesicular stomatitis Indiana virus98, respectively, by promoting the degradation of antiviral proteins (Table 2).
Together, functional genetic screens uncovered numerous proviral genes that are probably required for SARS-CoV-2 entry, translation or replication (Box 1). Notably, viruses used in two screens contained deletions near the polybasic cleavage site in S43,64, whereas the sequences of the passaged viruses used in screens in the other studies were not specified25,26,29,30,71,72. Such deletions prevent TMPRSS2-mediated S activation and force virus entry through the endocytic route64. Thus, future studies with wild-type SARS-CoV-2 may be useful to extend the compendium of SARS-CoV-2 proviral host factors.
Viral RNA and protein interactomes
Interactome screens probe for cellular factors interacting with viral RNA and/or proteins and can therefore provide valuable insights into pathways that facilitate or inhibit the viral life cycle. These screens are complementary to functional genetic screens as they can also detect cell-essential factors. In brief, RNA interactome screens involve SARS-CoV-2 infection of cells, followed by RNA–protein cross-linking and identification of interacting proteins73,99,100,101,102. The SARS-CoV-2 RNA interactome was determined in multiple cell lines (Fig. 2) using different cross-linking techniques: formaldehyde and ultraviolet light. Ultraviolet light at 254 nm (RNA antisense purification coupled with mass spectrometry (RAP–MS))99,100 is a more specific RNA–protein cross-linker than formaldehyde (comprehensive identification of RNA-binding proteins (chIRP-MS))73,102, as it does not efficiently cross-link protein–protein interactions. An even more specific cross-linking technique (viral RNA interactome capture (vRIC)-MS)101 applies 365 nm ultraviolet light after treatment with a transcription inhibitor and the photoactivatable nucleotide analogue 4-thiouridine to ensure exclusive cross-linking of 4-thiouridine-containing viral RNA. The SARS-CoV-2 protein interactome was mapped in HEK293T/17 and A549 cells using affinity-tag purification (affinity purification (AP)–MS)78,103,104,105 or pull-down of tagged proteins using proximity biotinylation (BioID)106,107,108. In contrast to RNA interactomes, protein interactomes were determined by expressing individual viral proteins in uninfected cells. This approach requires prudence during interpretation, as well as validation, before making claims about virus–host interactions, as individually expressed viral proteins may localize differently and lack the context of true infection. Although these screens vary in the set of probed SARS-CoV-2 proteins, integrating the results of these studies generates a robust cellular interactome for 21 SARS-CoV-2 proteins (Fig. 2 and Supplementary Table 2).
Viral RNA interactome
When the viral RNA is released into the cytoplasm, it hijacks cellular RNA-binding proteins involved in all stages of the mRNA life cycle to achieve the arduous task of regulating translation and replication in the hostile environment of the host cell. A total of 95 proteins, representing a broad range of RNA-related functionalities, were found to bind to SARS-CoV-2 RNA in at least three different cell types, whereas 34 proteins were identified in all four cell types (Fig. 2). Flynn et al. reported a significant bias towards antiviral factors in their viral RNA interactome73, which may explain the limited overlap between RNA interactomes and proviral factors identified in functional screens. Nevertheless, 17 of these 95 frequently detected interactors were functionally validated as proviral host factors for SARS-CoV-2 (Fig. 2).
A strong interplay was detected between SARS-CoV-2 RNA and ribonucleoprotein granules such as paraspeckles, P-bodies and stress granules. Similarly, protein families such as cold-shock domain proteins, eukaryotic translation initiation factors (EIF3/4) and heterogeneous nuclear ribonucleoproteins showed extensive viral RNA interaction. Several heterogeneous nuclear ribonucleoproteins have known functions in the life cycle of coronaviruses and other positive-strand RNA viruses, facilitating RNA synthesis and translation109. This overlapping SARS-CoV-2 RNA interactome further consisted of proteins that are involved in a plethora of cellular functions, such as transcriptional regulation, mRNA processing, stress response, fatty acid metabolism and the tRNA-splicing ligase complex101 (Fig. 2).
SARS-CoV-2 relies completely on the host translation machinery to translate its capped and poly-adenylated genome. Thus, unsurprisingly, many RNA interactome studies contained factors that are involved in the recognition of the poly(A) tail (PABPC1, PABPC4 and PABPCN1) and components of eukaryotic initiation factors (eIF) responsible for cap binding (eIF4F subunits) or subsequent connection to the ribosome (eIF3), as well as some 40S ribosome subunits. Remarkably, eIF4E was not retrieved in any screen, suggesting that SARS-CoV-2 translation is eIF4E-independent, similar to other capped positive-strand RNA viruses such as Sindbis virus110. In this regard, the identification of La-related protein 1 (LARP1) in the interactome of both SARS-CoV-2 RNA73,99,100 and the N protein78 is of special interest. LARP1 regulates the stability and translation of cellular mRNAs containing an oligopyrimidine (TOP) motif by binding to this motif and the adjacent cap, blocking the access of eIF4E. As LARP1 was validated as an antiviral factor binding to the 5′-leader of SARS-CoV-2, which contains a TOP-like motif, this may explain why eIF4E is absent from the viral RNA interactome.
Viral protein interactome
Of the cellular proteins found to interact with a specific SARS-CoV-2 protein (Fig. 2), a handful were identified in all of the screens conducted by at least five different research groups for a specific viral protein (G3BP1, G3BP2, TOMM70, RAE1, VPS39, MARK2 and MARK3). G3BP1, G3BP2, TOMM70 and RAE1 encode factors that are involved in host antiviral responses and are discussed in more detail in Box 2. The HOPS subunit Vam6/Vps39-like protein (VPS39) interacted consistently with ORF3a, as discussed above. ORF9b bound to microtubule affinity regulating kinase (MARK) 2 and 3, of which the latter was shown to affect the monocyte count in the blood of patients with COVID-19 (ref. 111). Although MARK2 is upregulated in SARS-CoV- and MERS-CoV-infected cells112, its kinase activity was found to be decreased in SARS-CoV-2-infected cells113. Interestingly, MARK2 is also hijacked by HIV-1 to facilitate microtubule-associated particle uncoating114.
Several other interesting proteins identified with a high frequency are HERC1, CCDC22, ALG11 and ZDHHC5. Probable E3 ubiquitin–protein ligase HERC1 (encoded by HERC1) was picked up as a nsp8 binder and is a guanine nucleotide exchange factor that activates Rab proteins, several of which were functionally validated as proviral host factors. ORF6 showed robust interaction with the proviral coiled-coil-domain-containing protein 22 (CCDC22), a component of the CCC complex and regulator of NF-κB signalling. For glycolipid alpha-1,2-mannosyltransferase (ALG11), a direct link with coronavirus infection was lacking until now. However, the high-confidence interaction of ALG11 with nsp4 is an interesting topic for further research, as ALG11 is an ER-localized N-glycosylation protein, and N-glycosylation of MHV nsp4 has been shown to affect DMV morphology and RNA replication115. The most frequent interaction partner for SARS-CoV-2 S was the plasma-membrane-localized palmitoyltransferase ZDHHC5. Coronaviruses rely on post-translational palmitoylation of S for efficient virion production and S-mediated membrane fusion116. ZDHHC5 drives S palmitoylation and the consequent infectivity of HCoV-229E, and may be of equal importance for the palmitoylation of SARS-CoV-2 S117.
Host factors as SARS-CoV-2 drug targets?
Agents that target proviral host factors may be attractive drug candidates because of a reduced chance of resistance development and the potential for broad action against viral variants. Several efforts to identify agents that inhibit SARS-CoV-2 by targeting a host protein have been undertaken28,118,119,120,121,122. Drug repurposing approaches that bypass costly and time-consuming drug discovery processes are of particular interest. Here, we summarize agents that are directed against rigorously validated host factors and that are currently in clinical trials for COVID-19 treatment, or that are approved drugs that are provisionally eligible for drug repurposing (Table 3).
SARS-CoV-2 receptors
Multiple strategies to hamper the interaction between SARS-CoV-2 and ACE2 are being pursued (Table 3; reviewed in ref. 123). The use of excessive soluble ACE2 as a decoy to trap SARS-CoV-2 virus is under investigation. Other strategies aim to block the binding of the S RBD to ACE2 using pseudoligands or blocking antibodies, or to interfere with ACE2 expression. As such, multiple clinical studies are evaluating the use of the anti-acne drug isotretinoin, which acts, at least partly, by downregulating ACE2 (ref. 122). Notably, ACE2 is a key enzyme of the renin–angiotensin–aldosterone system (RAAS), which regulates blood pressure. RAAS inhibitors (angiotensin converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARBs)) are commonly prescribed hypertension drugs. Although ACEi do not directly block ACE2, studies suggested that they increase ACE2 expression and may therefore worsen COVID-19 severity124, raising concerns about the continuation of these drugs in patients with COVID-19 who are hypertensive. However, clinical investigations revealed no adverse effects125,126, probably because ACEi/ARBs also fulfil anti-inflammatory and anti-oxidative functions. Thus, international recommendations are now against the discontinuation of these drugs in patients with COVID-19.
Blocking the accessibility of alternative receptors basigin (CD147) and HS is also under clinical investigation for COVID-19 treatment (Table 3).
Host proteases
TMPRSS2-mediated S activation at the plasma membrane is an attractive therapeutic intervention point. As TMPRSS2 is not a cell-essential gene, inhibition is expected to cause negligible side effects. The inhibitor camostat mesylate, which has been approved for non-COVID-19 indications in Japan, is being investigated in multiple ongoing clinical trials, as well as alternative approved serine protease inhibitors (namostat, aprotinin). The protein α1-antitrypsin (α1AT) is a highly abundant circulating serine protease inhibitor that is part of the human innate immune system and has been shown to inhibit TMPRSS2 and SARS-CoV-2 entry127. Clinical trials with α1AT purified from donor blood, available as a pharmaceutical product (prolastin), are underway. As TMPRSS2 expression is androgen regulated128, several studies also investigated androgen-directed therapy (bicalutamide, enzulatamide, isotretinoin). Isotretinoin reduces dihydrotestosterone levels and, as such, downregulates TMPRSS2. As it also downregulates ACE2, isotretinoin may have a multifaceted impact on SARS-CoV-2 infectivity.
Inhibition of cathepsins, the main proteases required for pH-dependent endocytic entry, has also been examined as COVID-19 therapy129. However, the anti-malaria drugs chloroquine and hydroxychloroquine, which inhibit endosomal acidification required for cathepsin activity, were found to be unsuccessful in the clinic, possibly due to the existence of multiple entry routes, despite having proven antiviral activity in cell culture130. Similarly, studies on chlorpromazine, an anti-psychotic drug that inhibits clathrin-dependent endocytosis, and tamoxifen, an anti-cancer drug that inhibits endosomal acidification, were planned but put on hold. The glycopeptide antibiotic teicoplanin, a cathepsin L inhibitor that has been shown to inhibit the entry of SARS-CoV, MERS-CoV and Ebola virus in cell culture131, was suggested as a complementary treatment option for COVID-19 (ref. 132) but is not currently under clinical investigation.
The inexpensive, commonly used drug tranexamic acid, which may inhibit SARS-CoV-2 S priming by suppressing plasmin activation, is the subject of a planned clinical trial. Similarly, vigil, a plasmid delivery vehicle that contains a furin-specific short hairpin RNA that is in advanced clinical testing for cancer treatment, has been proposed as a COVID-19 therapy133.
Other host factors
Only a few approved drugs against rigorously validated SARS-CoV-2 host factors involved in post-entry steps are currently available (Table 3). Histone deacetylase inhibitors are a relatively new class of anti-cancer agents that interfere with transcriptional regulation. For example, panobinostat was shown to downregulate ACE2 in cell culture134. However, the use of histone deacetylase inhibitors against SARS-CoV-2 remains unproven. The phosphatidylinositol 3-phosphate 5-kinase (PIKFYVE) inhibitor apilimod was shown to reduce viral replication in primary cells and ex vivo lung cultures and is currently being trialled against COVID-19 (ref. 121). The same applies to niclosamide, an approved antiparasitic agent that targets TMEM16 proteins and inhibits S-mediated syncytium formation and viral replication120, and plitidepsin, an anti-cancer agent (targeting elongation factor 1-alpha 1) with limited approval that shows in vitro and in vivo antiviral activity135.
Outlook
The urgency imposed by the COVID-19 pandemic and the availability of functional genetic and interactomic technologies have led to the discovery of SARS-CoV-2 host factors at an unprecedented rate. Taken together, these studies have identified a large cadre of candidate receptors, genetic dependencies and cellular proteins that interact with SARS-CoV-2 RNA and proteins. Although the biological roles of some factors have been extensively investigated, other proviral host factors that were identified in multiple studies have not been characterized. Future studies should further unravel the roles of these cross-validated factors in SARS-CoV-2 infection. Moreover, studies in animal models will be required to assess the relevance of identified host factors in vivo. As SARS-CoV-2 will probably not be the last zoonotic coronavirus to emerge, it will be crucial to explore which druggable proviral host factors are also required by other pathogenic coronaviruses, to develop broad-spectrum antivirals in preparation for possible future epidemics.
References
Cui, J., Li, F. & Shi, Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Wacharapluesadee, S. et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 12, 972 (2021).
Yao, H. et al. Molecular architecture of the SARS-CoV-2 virus. Cell 183, 730–738 (2020).
Neuman, B. W. et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 80, 7918–7928 (2006).
Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261 (2016).
Tang, T., Bidon, M., Jaimes, J. A., Whittaker, G. R. & Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 178, 104792 (2020).
Whittaker, G., Daniel, S. & Millet, J. Coronavirus entry: how we arrived at SARS-CoV-2. Curr. Opin. Virol. 47, 113–120 (2021).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921 (2020).
Chan, J. F. W. et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 9, 221–236 (2020).
Liu, D. X., Fung, T. S., Chong, K. K. L., Shukla, A. & Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 109, 97–109 (2014).
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H. & Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-020-00468-6 (2020).
Fehr, A. R. & Perlman, S. in Coronaviruses. Methods in Molecular Biology Vol. 1282 (eds. Maier, H. et al.) 1–23 (HumanaPress, 2015).
Sawicki, S. G., Sawicki, D. L. & Siddell, S. G. A contemporary view of coronavirus transcription. J. Virol. 81, 20–29 (2007).
de Wilde, A. H., Snijder, E. J., Kikkert, M. & van Hemert, M. J. in Roles of Host Gene and Non-coding RNA Expression in Virus Infection (eds Tripp, R. & Tompkins, S.) 1–42 (Springer, Cham, 2018).
Snijder, E. J. et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. 18, e3000715 (2020).
Wolff, G. et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 369, 1395–1398 (2020).
Stertz, S. et al. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361, 304–315 (2007).
Ghosh, S. et al. β-Coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183, 1520–1535 (2020).
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11, 875–879 (2005).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell https://doi.org/10.1016/j.cell.2020.02.052 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Bao, L. et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583, 830–833 (2020).
Wei, J. et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell https://doi.org/10.1016/j.cell.2020.10.028 (2021).
Schneider, W. M. et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell 184, 120–132 (2021).
Hoffmann, H. et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 29, 267–280 (2020).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Biering, S. B. et al. Genome-wide, bidirectional CRISPR screens identify mucins as critical host factors modulating SARS-CoV-2 infection. Preprint at bioRxiv https://doi.org/10.1101/2021.04.22.440848 (2021).
Rebendenne, A. et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal coronaviruses. Preprint at bioRxiv https://doi.org/10.1101/2021.05.19.444823 (2021).
Wang, S. et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 31, 126–140 (2021).
Puray-Chavez, M. et al. Systematic analysis of SARS-CoV-2 infection of an ACE2-negative human airway cell. Cell Rep. https://doi.org/10.1016/j.celrep.2021.109364 (2021).
Yeung, M. L. et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell https://doi.org/10.1016/j.cell.2021.02.053 (2021).
Harrison, A. G., Lin, T. & Wang, P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 41, 1100–1115 (2020).
Schurink, B. et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1, e290–e299 (2020).
Puelles, V. G. et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 383, 590–592 (2020).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035 (2020).
Bost, P. et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 181, 1475–1488 (2020).
Liu, L. et al. Heparan sulfate proteoglycans as attachment factor for SARS-CoV-2. ACS Cent. Sci. 7, 1009–1018 (2021).
Zhang, Q. et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 6, 80 (2020).
Clausen, T. M. et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 183, 1043–1057 (2020).
Baggen, J. et al. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat. Genet. https://doi.org/10.1038/s41588-021-00805-2 (2021).
Cagno, V., Tseligka, E. D., Jones, S. T. & Tapparel, C. Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses 11, 596 (2019).
Wei, C. et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2, 1391–1400 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 Spike glycoprotein. Cell 181, 281–292 (2020).
Daly, J. L. et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 370, 861–865 (2020).
Cantuti-Castelvetri, L. et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860 (2020).
Zhu, S. et al. Genome-wide CRISPR activation screen identifies novel receptors for SARS-CoV-2 entry. Preprint at bioRxiv https://doi.org/10.1101/2021.04.08.438924 (2021).
Wang, K. et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 5, 283 (2020).
Shilts, J., Crozier, T. W. M., Greenwood, E. J. D., Lehner, P. J. & Wright, G. J. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci. Rep. 11, 413 (2021).
Gu, Y. et al. Interaction network of SARS-CoV-2 with host receptome through spike protein. Preprint at bioRxiv https://doi.org/10.1101/2020.09.09.287508 (2020).
Tang, X. et al. Transferrin receptor is another receptor for SARS-CoV-2 entry. Preprint at bioRxiv https://doi.org/10.1101/2020.10.23.350348 (2020).
Soh, W. T. et al. The N-terminal domain of spike glycoprotein mediates SARS-CoV-2 infection by associating with L-SIGN and DC-SIGN. Preprint at bioRxiv https://doi.org/10.1101/2020.11.05.369264 (2020).
Gao, C. et al. SARS-CoV-2 spike protein interacts with multiple innate immune receptors. Preprint at bioRxiv https://doi.org/10.1101/2020.07.29.227462 (2020).
Amraei, R. et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent. Sci. 7, 1156–1165 (2021).
Thépaut, M. et al. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. PLoS Pathog. 17, e1009576 (2021).
Rahimi, N. C-type lectin CD209L/L-SIGN and CD209/DC-SIGN: cell adhesion molecules turned to pathogen recognition receptors. Biology 10, 1 (2021).
Laporte, M. et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLOS Pathogens 17, e1009500 (2021).
Hoffmann, M., Kleine-Weber, H. & Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78, 779–784 (2020).
Papa, G. et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 17, e1009246 (2021).
Johnson, B. A. et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591, 293–299 (2021).
Lau, S. Y. et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg. Microbes Infect. 9, 837–842 (2020).
Zhu, Y. et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat. Commun. https://doi.org/10.1038/s41467-021-21213-4 (2021).
Ou, X. et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 11, 1620 (2020).
Tang, T. et al. Proteolytic activation of SARS-CoV-2 Spike at the S1/S2 boundary: potential role of proteases beyond furin. ACS Infect. Dis. https://doi.org/10.1021/acsinfecdis.0c00701 (2021).
Zang, R. et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 5, eabc3582 (2020).
Sungnak, W. et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687 (2020).
Jaimes, J. A., Millet, J. K. & Whittaker, G. R. Proteolytic cleavage of the SARS-CoV-2 Spike protein and the role of the novel S1/S2 site. Iscience 23, 101212 (2020).
Ji, H. L., Zhao, R., Matalon, S. & Matthay, M. A. Elevated plasmin(Ogen) as a common risk factor for COVID-19 susceptibility. Physiol. Rev. 100, 1065–1075 (2020).
Daniloski, Z. et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184, 92–105 (2021).
Wang, R. et al. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell https://doi.org/10.1016/j.cell.2020.12.004 (2020).
Flynn, R. A. et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell https://doi.org/10.1016/j.cell.2021.03.012 (2021).
van den Boomen, D. J. H. et al. A trimeric Rab7 GEF controls NPC1-dependent lysosomal cholesterol export. Nat. Commun. 11, 5559 (2020).
Burkard, C. et al. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 10, e1004502 (2014).
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011).
Miao, G. et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 56, 427–442 (2021).
Gordon, D. E. et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370, eabe9403 (2020).
Peng, S. et al. Endogenous cellular microRNAs mediate antiviral defense against influenza A virus. Mol. Ther. Nucleic Acids 10, 361–375 (2018).
Estrin, M. A. et al. Host-directed combinatorial RNAi improves inhibition of diverse strains of influenza A virus in human respiratory epithelial cells. PLoS ONE 13, e0197246 (2018).
Klein, Z. A. et al. Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in article loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95, 281–296 (2017).
McNally, K. E. & Cullen, P. J. Endosomal retrieval of cargo: retromer is not alone. Trends Cell Biol. 28, 807–822 (2018).
Derivery, E. et al. The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev. Cell 17, 712–723 (2009).
Zuo, X. et al. Exo70 interacts with the Arp2/3 complex and regulates cell migration. Nat. Cell Biol. 8, 1383–1388 (2006).
Al-Saleem, J. et al. HTLV-1 Tax-1 interacts with SNX27 to regulate cellular localization of the HTLV-1 receptor molecule, GLUT1. PLoS ONE 14, e0214059 (2019).
Lipovsky, A. et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl Acad. Sci. USA 110, 7452–7457 (2013).
Yin, P., Hong, Z., Yang, X., Chung, R. T. & Zhang, L. A role for retromer in hepatitis C virus replication. Cell. Mol. Life Sci. 73, 869–881 (2016).
Backer, J. M. The intricate regulation and complex functions of the class III phosphoinositide 3-kinase Vps34. Biochem. J. 473, 2251–2271 (2016).
Kratzel, A. et al. A genome-wide CRISPR screen identifies interactors of the autophagy pathway as conserved coronavirus targets. Preprint at bioRxiv https://doi.org/10.1101/2021.02.24.432634 (2021).
Moretti, F. et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 19, e45889 (2018).
Hoffmann, H. et al. TMEM41B is a pan-flavivirus host factor. Cell https://doi.org/10.1016/j.cell.2020.12.005 (2020).
Brown, M. S. & Goldstein, J. L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl Acad. Sci. USA 96, 11041–11048 (1999).
Meng, Y., Heybrock, S., Neculai, D. & Saftig, P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 30, 452–466 (2020).
Wang, S. et al. Cholesterol 25-hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 39, e106057 (2020).
Osuna-Ramos, J. F., Reyes-Ruiz, J. M. & Del Ángel, R. M. The role of host cholesterol during flavivirus infection. Front. Cell. Infect. Microbiol. 8, 388 (2018).
Sherman, E. J. et al. Identification of ACE2 modifiers by CRISPR screening. Preprint at bioRxiv https://doi.org/10.1101/2021.06.10.447768 (2021).
Murphy Schafer, A. R., Smith, J. L., Pryke, K. M., DeFilippis, V. R. & Hirsch, A. J. The E3 ubiquitin ligase SIAH1 targets MyD88 for proteasomal degradation during dengue virus infection. Front. Microbiol. 11, 24 (2020).
Zhang, W. et al. JMJD6 negatively regulates cytosolic RNA induced antiviral signaling by recruiting RNF5 to promote activated IRF3 K48 ubiquitination. PLOS Pathog. 17, e1009366 (2021).
Lee, S. et al. The SARS-CoV-2 RNA interactome. Mol. Cell 81, 2838–2850 (2021).
Schmidt, N. et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat. Microbiol. 6, 339–353 (2020).
Kamel, W. et al. Global analysis of protein-RNA interactions in SARS-CoV-2-infected cells reveals key regulators of infection. Mol. Cell 81, 2851–2867 (2021).
Labeau, A. et al. Characterization and functional interrogation of SARS-CoV-2 RNA interactome. Preprint at bioRxiv https://doi.org/10.1101/2021.03.23.436611 (2021).
Davies, J. P., Almasy, K. M., McDonald, E. F. & Plate, L. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus nonstructural proteins identifies unique and shared host-cell dependencies. ACS Infect. Dis. 6, 3174–3189 (2020).
Li, J. et al. Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med 2, 99–112 (2021).
Stukalov, A. et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature https://doi.org/10.1038/s41586-021-03493-4 (2021).
Samavarchi-Tehrani, P. et al. A SARS-CoV-2–host proximity interactome. Preprint at bioRxiv https://doi.org/10.1101/2020.09.03.282103 (2020).
St-Germain, J. R. et al. A SARS-CoV-2 BioID-based virus-host membrane protein interactome and virus peptide compendium: new proteomics resources for COVID-19 research. Preprint at bioRxiv https://doi.org/10.1101/2020.08.28.269175 (2020).
Laurent, E. M. N. et al. Global BioID-based SARS-CoV-2 proteins proximal interactome unveils novel ties between viral polypeptides and host factors involved in multiple COVID19-associated mechanisms. Preprint at bioRxiv https://doi.org/10.1101/2020.08.28.272955 (2020).
Lloyd, R. E. Nuclear proteins hijacked by mammalian cytoplasmic plus strand RNA viruses. Virology 479, 457–474 (2015).
Carrasco, L., Sanz, M. A. & González-Almela, E. The regulation of translation in alphavirus-infected cells. Viruses 10, 70 (2018).
Pietzner, M. et al. Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun. 11, 6397 (2020).
Guzzi, P. H., Mercatelli, D., Ceraolo, C. & Giorgi, F. M. Master regulator analysis of the SARS-CoV-2/human interactome. J. Clin. Med. 9, 982 (2020).
Bouhaddou, M. et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182, 685–712 (2020).
Malikov, V. & Naghavi, M. H. Localized phosphorylation of a kinesin-1 adaptor by a capsid-associated kinase regulates HIV-1 motility and uncoating. Cell Rep. 20, 2792–2799 (2017).
Gadlage, M. J. et al. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 84, 280–290 (2010).
Petit, C. M. et al. Palmitoylation of the cysteine-rich endodomain of the SARS–coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 360, 264–274 (2007).
Lee, M. et al. Fatty acid synthase inhibition prevents palmitoylation of SARS-CoV2 Spike protein and improves survival of mice infected with murine hepatitis virus. Preprint at bioRxiv https://doi.org/10.1101/2020.12.20.423603 (2020).
Chen, C. Z. et al. Drug repurposing screen for compounds inhibiting the cytopathic effect of SARS-CoV-2. Front. Pharmacol. 11, 592737 (2021).
Hashimoto, K. Repurposing of CNS drugs to treat COVID-19 infection: targeting the sigma-1 receptor. Eur. Arch. Psychiatry Clin. Neurosci. 271, 249–258 (2021).
Braga, L. et al. Drugs inhibiting TMEM16 proteins block SARS-CoV-2 Spike-induced syncytia. Nature https://doi.org/10.1038/s41586-021-03491-6 (2021).
Riva, L. et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586, 113–119 (2020).
Sinha, S. et al. In vitro and in vivo identification of clinically approved drugs that modify ACE 2 expression. Mol. Syst. Biol. 16, e9628 (2020).
Jia, H., Neptune, E. & Cui, H. Targeting ACE2 for COVID-19 therapy: opportunities and challenges. Am. J. Respir. Cell Mol. Biol. https://doi.org/10.1165/rcmb.2020-0322ps (2020).
Ferrario, C. M. et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111, 2605–2610 (2005).
Cohen, J. B. et al. Continuation versus discontinuation of renin–angiotensin system inhibitors in patients admitted to hospital with COVID-19: a prospective, randomised, open-label trial. Lancet Respir. Med. 9, 275–284 (2021).
Williams, B. Renin-angiotensin system inhibitors in hospitalised patients with COVID-19. Lancet Respir. Med. 9, 221–222 (2021).
Wettstein, L. et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 12, 1726 (2021).
Clinckemalie, L. et al. Androgen regulation of the TMPRSS2 gene and the effect of a SNP in an androgen response element. Mol. Endocrinol. 27, 2028–2040 (2013).
Yang, N. & Shen, H. M. Targeting the endocytic pathway and autophagy process as a novel therapeutic strategy in COVID-19. Int. J. Biol. Sci. 16, 1724–1731 (2020).
Hoffmann, M. et al. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature 585, 588–590 (2020).
Zhou, N. et al. Glycopeptide antibiotics potently inhibit cathepsin l in the late endosome/lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV). J. Biol. Chem. 291, 9218–9232 (2016).
Baron, S. A., Devaux, C., Colson, P., Raoult, D. & Rolain, J. Teicoplanin: an alternative drug for the treatment of COVID-19? Int. J. Antimicrob. Agents 55, 105944 (2020).
Nemunaitis, J., Stanbery, L. & Senzer, N. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection: let the virus be its own demise. Future Virol. 15, 381–395 (2020).
Takahashi, Y. et al. Histone deacetylase inhibitors suppress ACE2 and ABO simultaneously, suggesting a preventive potential against COVID-19. Sci. Rep. 11, 3379 (2021).
White, K. M. et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 371, 926–931 (2021).
Sun, S. H. et al. A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe 28, 124–133 (2020).
Ichimura, T. et al. KIM-1/TIM-1 is a receptor for SARS-CoV-2 in lung and kidney. Preprint at medRxiv https://doi.org/10.1101/2020.09.16.20190694 (2020).
Yang, C. et al. Kidney injury molecule-1 is a potential receptor for SARS-CoV-2. J. Mol. Cell Biol. https://doi.org/10.1093/jmcb/mjab003 (2021).
Carlos, A. J. et al. The chaperone GRP78 is a host auxiliary factor for SARS-CoV- 2 and GRP78 depleting antibody blocks viral entry and infection. J. Biol. Chem. 296, 100759 (2021).
Baker, A. N. et al. The SARS-COV-2 Spike protein binds sialic acids and enables rapid detection in a lateral flow point of care diagnostic device. ACS Cent. Sci. 6, 2046–2052 (2020).
Xiao, F. et al. Interactions between the hepatitis C virus nonstructural 2 protein and host adaptor proteins 1 and 4 orchestrate virus release. Mbio 9, e02233–17 (2018).
Camus, G. et al. The clathrin adaptor complex AP-1 binds HIV-1 and MLV gag and facilitates their budding. Mol. Biol. Cell 18, 986–994 (2007).
Caillet, M. et al. Rab7A is required for efficient production of infectious HIV-1. PLoS Pathog. 7, e1002347 (2011).
Qi, M. et al. Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog. 9, e1003278 (2013).
Mehedi, M. et al. Actin-related protein 2 (ARP2) and virus-induced filopodia facilitate human respiratory syncytial virus spread. PLoS Pathog. 12, e1006062 (2016).
Petersen, J. et al. The major cellular sterol regulatory pathway is required for Andes virus infection. PLoS Pathog. 10, e1003911 (2014).
Yuan, S. et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 10, 120 (2019).
Wichit, S. et al. Imipramine inhibits chikungunya virus replication in human skin fibroblasts through interference with intracellular cholesterol trafficking. Sci. Rep. 7, 3145 (2017).
Friesland, M., Mingorance, L., Chung, J., Chisari, F. V. & Gastaminza, P. Sigma-1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection. J. Virol. 87, 6377–6390 (2013).
Sir, D. et al. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl Acad. Sci. USA 107, 4383–4388 (2010).
Lin, Y. C., Jeng, K. S. & Lai, M. M. C. CNOT4-mediated ubiquitination of influenza A virus nucleoprotein promotes viral RNA replication. Mbio 8, e00597–17 (2017).
Oakland, T. E., Haselton, K. J. & Randall, G. EWSR1 binds the hepatitis C virus cis-acting replication element and is required for efficient viral replication. J. Virol. 87, 6625–6634 (2013).
Kim, H. S. et al. Arrayed CRISPR screen with image-based assay reliably uncovers host genes required for coxsackievirus infection. Genome Res. 28, 859–868 (2018).
Bagchi, P., Inoue, T. & Tsai, B. EMC1-dependent stabilization drives membrane penetration of a partially destabilized non-enveloped virus. Elife 5, e21470 (2016).
Maric, M. et al. A functional role for torsina in herpes simplex virus 1 nuclear egress. J. Virol. 85, 9667–9679 (2011).
Hölper, J. E., Klupp, B. G., Luxton, G. W. G., Franzke, K. & Mettenleiter, T. C. Function of torsin AAA+ ATPases in pseudorabies virus nuclear egress. Cells 9, 738 (2020).
Puschnik, A. S., Majzoub, K., Ooi, Y. S. & Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 15, 351–364 (2017).
Li, B. et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 11, 164 (2020).
Heaton, B. E. et al. A CRISPR activation screen identifies a pan-avian influenza virus inhibitory host factor. Cell Rep. 20, 1503–1512 (2017).
Meylan, E. & Tschopp, J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell 22, 561–569 (2006).
Loo, Y.-M. & Gale, M. Immune signaling by RIG-I-like receptors. Immunity 34, 680–692 (2011).
Vazquez, C. & Horner, S. M. MAVS coordination of antiviral innate immunity. J. Virol. 89, 6974–7 (2015).
White, J. P., Cardenas, A. M., Marissen, W. E. & Lloyd, R. E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2, 295–305 (2007).
Lu, S. et al. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 12, 502 (2021).
Liu, X.-Y., Wei, B., Shi, H.-X., Shan, Y.-F. & Wang, C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 20, 994–1011 (2010).
Jiang, H. et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 17, 998–1000 (2020).
Miorin, L. et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl Acad. Sci. USA 117, 28344–28354 (2020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Microbiology thanks Valeria Cagno, Alfredo Castello and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Table 1
Overview of technical parameters of CRISPR-based functional genetic screens with SARS-CoV-2.
Supplementary Table 2
Overview of genes displayed in Fig. 1.
Supplementary Table 3
Overview of genes identified as SARS-CoV-2 host factors in functional genetic screens by individual functional validation, and genes encoding proteins found in viral RNA or viral protein interactomes.
Supplementary Table 4
Overview of overlapping genes shown in Fig. 2.
Rights and permissions
About this article

Cite this article
Baggen, J., Vanstreels, E., Jansen, S. et al. Cellular host factors for SARS-CoV-2 infection. Nat Microbiol 6, 1219–1232 (2021). https://doi.org/10.1038/s41564-021-00958-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-021-00958-0
This article is cited by
-
SARS-CoV-2 biology and host interactions
Nature Reviews Microbiology (2024)
-
ACE2-dependent and -independent SARS-CoV-2 entries dictate viral replication and inflammatory response during infection
Nature Cell Biology (2024)
-
Proteomic analysis of SARS-CoV-2 particles unveils a key role of G3BP proteins in viral assembly
Nature Communications (2024)
-
SARS-CoV-2 infects epithelial cells of the blood-cerebrospinal fluid barrier rather than endothelial cells or pericytes of the blood-brain barrier
Fluids and Barriers of the CNS (2023)
-
Oligonucleotide usage in coronavirus genomes mimics that in exon regions in host genomes
Virology Journal (2023)
