
Abstract
Antimicrobial resistance in neonatal sepsis is rising, yet mechanisms of resistance that often spread between species via mobile genetic elements, ultimately limiting treatments in low- and middle-income countries (LMICs), are poorly characterized. The Burden of Antibiotic Resistance in Neonates from Developing Societies (BARNARDS) network was initiated to characterize the cause and burden of antimicrobial resistance in neonatal sepsis for seven LMICs in Africa and South Asia. A total of 36,285 neonates were enrolled in the BARNARDS study between November 2015 and December 2017, of whom 2,483 were diagnosed with culture-confirmed sepsis. Klebsiella pneumoniae (n = 258) was the main cause of neonatal sepsis, with Serratia marcescens (n = 151), Klebsiella michiganensis (n = 117), Escherichia coli (n = 75) and Enterobacter cloacae complex (n = 57) also detected. We present whole-genome sequencing, antimicrobial susceptibility and clinical data for 916 out of 1,038 neonatal sepsis isolates (97 isolates were not recovered from initial isolation at local sites). Enterobacterales (K. pneumoniae, E. coli and E. cloacae) harboured multiple cephalosporin and carbapenem resistance genes. All isolated pathogens were resistant to multiple antibiotic classes, including those used to treat neonatal sepsis. Intraspecies diversity of K. pneumoniae and E. coli indicated that multiple antibiotic-resistant lineages cause neonatal sepsis. Our results will underpin research towards better treatments for neonatal sepsis in LMICs.
Similar content being viewed by others


Main
Although there has been a substantial decrease in infant mortality over the past 20 years1, the burden remains substantial, with the mortality rate of children under 5 years of age at 38 per 1,000 live births in 20192, and 98% of recorded neonatal deaths now occurring in LMICs3. The World Health Organization (WHO) has declared neonatal sepsis a global concern4 and the burden of neonatal infectious diseases a major challenge. Effective management of sepsis is not always possible when resources are limited3, and the steady increase of antimicrobial resistance (AMR) worldwide further compromises sepsis management5,6.
Despite the burden of neonatal sepsis, accurate information on the causes and consequences of neonatal sepsis in LMICs is scarce4,6. Most studies in LMICs are from a single site, are of limited sample size, or lack accurate methods for sepsis diagnosis, pathogen identification and antibiotic susceptibility measurements7,8,9,10. In 2015 and 2016, two multicentre neonatal sepsis studies in LMICs were published11,12. However, neither study combined antimicrobial susceptibility testing and whole-genome sequencing (WGS), making it difficult to determine the extent of genomic diversity (which would usually be done by comparing the lineages across geographical areas) and resistance. The studies that have taken this approach were of single sites and often used WGS to investigate specific outbreaks13,14,15,16.
In LMICs, the epidemiology of early-onset sepsis (EOS) and late-onset sepsis (LOS) is not well defined3, unlike in high-income countries, where group B Streptococcus is usually considered the main cause of EOS17. A systematic review of the causes of blood culture-positive neonatal sepsis in Sub-Saharan Africa by Okomo et al.18 found that Klebsiella species, Escherichia coli, Enterobacter species and Pseudomonas species accounted for 38% of cases. Other single-site reports showed concordant findings9,11,12,19. However, these studies did not specifically determine whether certain species, or sequence type (ST) groups, are more likely to harbour resistance or virulence determinants, how this compares between different geographical areas and whether there is any relation to sepsis onset or outcome.
Burden of Antibiotic Resistance in Neonates from Developing Societies (BARNARDS; www.barnards-group.com) is a network of 12 clinical study sites in four African (Ethiopia, Nigeria, Rwanda and South Africa) and three South Asian countries (Bangladesh, India and Pakistan). The aim of the BARNARDS study is to assess the burden of AMR in neonates in these LMIC. Here, we report on the isolation and characterization of Gram-negative bacteria (GNB) causing neonatal sepsis in seven LMICs, including their AMR profiles. We report associations between phenotypic and genotypic data and sepsis onset and mortality following biological sepsis (MFBS). We also analyse whole-genome sequences from isolates that cause neonatal sepsis.
Results
Enrolment in BARNARDS and isolation of pathogenic bacteria
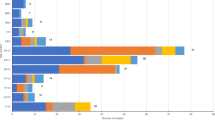
The numbers of neonates recruited, clinically diagnosed with sepsis and with a confirmation of sepsis by positive blood culture are outlined in Fig. 1. Of 36,285 infants ≤60 d old (termed herein as neonates) enrolled in the BARNARDS study from November 2015 to December 2017, 2,483 had culture-confirmed sepsis. All 12 clinical sites used the same criteria for clinical diagnosis of sepsis (Supplementary Fig. 1). We found that cases were mainly EOS for both sites in Pakistan, one site in Bangladesh (BC; see Methods for definitions of all two-letter site abbreviations) and the single site in Ethiopia. In Nigeria, India and South Africa, there were mainly LOS cases. In Rwanda, neonatal sepsis cases were equally split between EOS and LOS (Fig. 2).

The numbers of neonates with missing clinical data for the onset and outcome of sepsis are shown per site. The numbers of isolates collected from neonatal blood cultures are shown per site, with a breakdown by preliminary characterization (as determined by Gram stain) in the flow chart below. The final numbers of isolates included for analysis are highlighted in orange. BB, baby blood; ID, identification (of species); ND, not determined.

The study sites are indicated by coloured squares. The African sites were located in Ethiopia (ES (green)), Nigeria (NK (cyan), NN (light blue) and NW (dark blue)), Rwanda (RK (dark purple) and RU (light purple)) and South Africa (ZAT; olive). The Asian sites were located in Bangladesh (BC (dark pink) and BK (light pink)), India (IN (orange)) and Pakistan (PC (peach) and PP (burgundy)). The numbers next to each clinical site location represent the total number of GNB identified. Inset: the stacked bar graph shows the distribution of the top ten GNB species recovered from blood cultures at the local sites. The onset of neonatal sepsis (EOS, LOS or ND) for GNB per clinical site is represented as a pie chart. The outcome of neonatal sepsis is shown for each continent.
Automated blood culture systems were used to detect microbial growth, with 2,620 microbial isolates recovered. These 2,620 isolates comprised 1,266 Gram-positive bacteria (GPB) isolates, 1,038 GNB isolates, 22 fungal isolates and 294 unassigned isolates (Fig. 1). The methods for collection and identification were standardized across all sites, with equipment and reagents purchased from uniform suppliers. The primary aim of the BARNARDS study was to characterize the extent of β-lactam resistance in GNB causing clinically diagnosed sepsis in infants <60 d old (see Supplementary Figs. 2 and 3). However, at month 17 (during a BARNARDS network event), we anecdotally noted high rates of isolation of Staphylococcus species and therefore collected all of these isolates for analysis (to be reported elsewhere). Of 1,038 isolates, 916 GNB were analysed using WGS. For 122 isolates, identification beyond a Gram stain was not possible because the isolate was lost and/or purification for DNA extraction was unsuccessful (Fig. 1).
In total, 58 different species of GNB were identified across all sites by WGS (Supplementary Table 1), including K. pneumoniae (n = 258), Serratia marcescens (n = 151), Klebsiella michiganensis (n = 117), Enterobacter species (n = 80), E. coli (n = 75), Burkholderia species (n = 61), Acinetobacter species (n = 49), Pseudomonas species (n = 36) and Ralstonia mannitolilytica (n = 21) (Fig. 2). Among the GNB characterized herein, 401 were Klebsiella, with six species identified: K. pneumoniae, K. variicola, K. quasipneumoniae, K. aerogenes, K. oxytoca and K. michiganensis (Supplementary Fig. 4). Burkholderia cenocepacia, K. michiganensis, R. mannitolilytica and S. marcescens were mainly isolated from samples obtained from single sites in Pakistan, Nigeria and Bangladesh (Fig. 2).
Overall, R. mannitolilytica, K. michiganensis, Burkholderia species and Pseudomonas species caused more cases of EOS than other species (Supplementary Table 2 and Supplementary Fig. 5). Of note, similar proportions of fatal sepsis cases were caused by GNB on each continent (92/353 cases in Africa (21%) and 118/475 cases in Asia (20%)). We found that Burkholderia species or K. michiganensis sepsis infections were the most likely to be fatal (Supplementary Table 2 and Supplementary Fig. 5). However, there was a large proportion of missing data from certain clinical sites (due to data collection and entry error; Fig. 1), which limited the strength of conclusions.
Interspecies and intraspecies diversity across clinical sites
To understand the extent of inter- and intraspecies diversity, we aimed to perform WGS on all GNB. Multilocus sequence typing (MLST) was used primarily as an unambiguous tool to examine bacterial relatedness. As MLST can be performed easily via PCR of housekeeping genes, in addition to in silico MLST via WGS, assessing relatedness via this tool (where applicable) provides a dataset that can be extrapolated to existing data on neonatal sepsis where WGS may not be available.
MLST revealed substantial intraspecies diversity, and 40 previously unknown STs were assigned in 12 species (Table 1 and Supplementary Table 2). Fourteen STs were assigned in the Klebsiella genera (Table 1), including all three K. aerogenes STs (all from Africa; n = 2 from Nigeria and n = 1 from South Africa; ST194–196). K. michiganensis was mainly ST180 (from PP in Pakistan; Table 1). Such low ST diversity and close phylogenetic relatedness, as shown by the core genome phylogeny (Supplementary Fig. 6), warrant further investigation. Similarly, we noticed that a single, previously unknown, B. cenocepacia ST (ST1621), also from PP, was dominant (Table 1 and Supplementary Fig. 7). B. cenocepacia ST1621 from PP and the S. marcescens isolates from BC were indistinguishable during core genome analysis (Supplementary Figs. 7 and 8). All local-level clusters will be studied further.
Enterobacter cloacae complex isolates were identified belonging to E. cloacae, E. hormaechei, E. kobei, E. asburiae and E. ludwigii. In total, seven different Enterobacter species (n = 80) with 28 STs were identified. The majority of Enterobacter species were found in Pakistan (n = 39), Nigeria (n = 19) and Rwanda (n = 14) (Fig. 1 and Supplementary Fig. 9). ST171 was common across sites in Africa and Asia; however, ST346 was only detected in Rwanda, ST523 was only detected in Pakistan and ST850 was only detected in Nigeria. Within Enterobacter species, 13 STs were assigned (Table 1) to E. cloacae, E. hormaechei and E. ludwigii isolates.
Acinetobacter species were recovered from ten out of 12 clinical sites in both Africa and South Asia, and 38 out of 49 (78%) were Acinetobacter baumannii. Of these, 17 out of 38 (45%) belonged to international clones (Pasteur MLST) ST1 and ST2 (Supplementary Tables 1 and 2).
AMR of pathogens causing neonatal sepsis
One aim of the BARNARDS study was to describe the AMR profiles of pathogens causing neonatal sepsis. For this, we performed agar dilution to determine the minimum inhibitory concentrations (MICs) of 19 antibiotics, including the current recommended first-line empirical treatments for neonatal sepsis, as well as carbapenems (the incidence of carbapenem-resistant GNB is increasing at an alarming rate), on 885 GNB (31 isolates were not recovered following storage at −80 °C after genomic DNA extraction for WGS; Supplementary Table 2 lists the isolates recoverable for WGS and/or MIC testing). Current data in LMICs focus on profiling AMR within certain species only, and are therefore not exhaustive across all pathogens causing sepsis.
GNB (n = 885) were resistant to ampicillin (95%), cefotaxime (83%) and ceftriaxone (80%), whereas they were sensitive to meropenem (13%), imipenem (15%) and tigecycline (16%) (Fig. 3a). The MICs required to inhibit the growth of 50% of organisms (that is, MIC50 values) of piperacillin/tazobactam, carbapenems (imipenem, meropenem and ertapenem), amikacin, fosfomycin, quinolones (ciprofloxacin and levofloxacin) and colistin were lower than their resistance breakpoints. The MIC90 values of all antibiotics tested were higher than their resistance breakpoints, with the exception of tigecycline, for which the MIC90 was lower than the epidemiological cut-off value for Providencia and Proteus species20,21.

a, Percentages of antimicrobial-resistant aetiological agents of neonatal sepsis, coloured according to bacterial species/group (n = 885 isolates of GNB). The MICs of the antibiotics were determined by agar dilution and the results were interpreted according to EUCAST guidelines and documents20,21. AMC, amoxicillin/clavulanate; AMK, amikacin; AMP, ampicillin; ATM, aztreonam; CAZ, ceftazidime; CIP, ciprofloxacin; CRO, ceftriaxone; CST, colistin; CTX, cefotaxime; ETP, ertapenem; FEP, cefepime; FOF, fosfomycin; GEN, gentamicin; IPM, imipenem; LVX, levofloxacin; MEM, meropenem; TGC, tigecycline; TOB, tobramycin; TZP, piperacillin/tazobactam. b, Sunburst diagram detailing the class A (red), B (yellow) and D (green) carbapenemase resistance genes detected. The second ring from the centre shows the carbapenemase genes identified. The distributions across species and clinical sites are shown in the outer rings. ABU, Acinetobacter baumannii; CFI, Citrobacter freundii; ECO, Escherichia coli; ENT, Enterobacter cloacae complex; KMI, Klebsiella michiganensis; KPN, Klebsiella pneumoniae; KQI, Klebsiella quasipneumoniae; PRO, Providencia rettgeri; SER, Serratia marcescens.
Overall, GNB isolates resistant to at least one of the cephalosporins tested were less likely to cause LOS than EOS (P = 0.017; odds ratio (OR) = 0.63; 95% confidence interval (CI) = 0.43–0.92; Supplementary Table 3). For the statistical analysis, the outcome measurement was MFBS (deceased as response, alive as reference). Concomitant resistance to the three cephalosporins tested versus isolate susceptibility to all produced an odds ratio of 0.626; 95% CI 0.426–0.918; P = 0.017. In this way, concomitant resistance to the three cephalosporins tested among GNB isolates was less likely among infants who stayed alive compared to those who were deceased.
As a marker of extended-spectrum β-lactamase antibiotic resistance gene (ARG), blaCTX-M-15 was inspected. It was detected in at least nine species (n = 523 isolates) and found in isolates from all study sites. We also screened genomes for genes coding for carbapenem resistance (blaNDM, blaOXA-48-like variants and blaKPC). There were 146 single carbapenemase genes in ten species (n = 128 isolates), and two carbapenem resistance gene homologues were present in 24 isolates. blaNDM-1 (n = 90; Bangladesh, n = 23; India, n = 6; Nigeria, n = 16; Pakistan, n = 43; Rwanda, n = 1; South Africa, n = 1) and blaNDM-5 (n = 3; Bangladesh, n = 1; India, n = 1; Nigeria, n = 1) were mainly detected in clinical sites in South Asia, whereas blaNDM-7 was predominantly recovered from Nigeria and Pakistan (n = 19; Bangladesh, n = 1; Nigeria, n = 11; Pakistan, n = 7) (Fig. 3b). In 79 GNB, blaCTX-M-15 plus blaNDM and/or a blaOXA-48-like variant were found. In total, 30 GNB carried a variant of the blaOXA-48-like family, with blaOXA-181 being the most frequent (Pakistan, n = 22; India, n = 1; Fig. 3b). blaOXA-232 was only found in Bangladesh (n = 5), and the two isolates carrying blaOXA-48 (one K. michiganensis and one S. marcescens) were from PP. blaVIM was found in three Pseudomonas aeruginosa isolates (Bangladesh, n = 2 (BC, n = 1; BK, n = 1); India, n = 1). Both blaVIM variants from Bangladesh were blaVIM-2, whereas the variant detected in India was blaVIM-6.
In Enterobacter species, blaCTX-M-15 was found in isolates recovered from both Africa and South Asia (n = 19; Africa, n = 16; South Asia, n = 3); however, Enterobacter containing carbapenemase genes (n = 18) were largely recovered from South Asia (South Asia, n = 16; Africa, n = 2). Five Acinetobacter (A. baumannii, n = 3; Acinetobacter bereziniae, n = 1; Acinetobacter nosocomialis, n = 1) were found to have blaNDM-1 (Africa, n = 2; South Asia, n = 3) (Fig. 3b). Additionally, we found blaOXA-23 within 20 A. baumannii isolates.
The total number of ARGs possessed by each bacterial isolate is shown in Supplementary Table 4. For each ARG increase among E. cloacae, we observed a 13.2% decrease in the likelihood of neonates having LOS (P = 0.016; 95% CI = 0.77–0.97). No other associations between ARG and onset were found (Supplementary Table 3).
Worryingly, we found that 529 (60%) of the GNB isolates tested were resistant to the first-line empirical treatment for neonatal sepsis (both ampicillin and gentamicin).
Plasmids and carbapenemase genes
As many ARGs are carried on mobile genetic elements such as plasmids, we searched for plasmid replicon types in isolates from the different geographical areas and, where possible, analysed linkages between plasmid type and the carriage of specific carbapenemase genes. We detected 1,124 plasmids with 45 inc gene variants, which we categorized into 18 plasmid groups. From these, 1,093 were found within E. coli (n = 169), K. pneumoniae (n = 623), K. michiganensis (n = 142), K. quasipneumoniae (n = 28), Enterobacter species (n = 87) and S. marcescens (n = 44) (Fig. 4). There were 12 plasmid types found within the seven S. enterica isolates, seven among Citrobacter species and five among K. variicola isolates.

Plasmid types (left) found to carry carbapenemase AMR genes are colour coded and linked to the GNB species (right) in which the plasmid type was identified. Particular carbapenemase genes are shown on the far right.
Within the six dominant plasmid carriers (Fig. 4), the most frequently detected inc type was IncFIB, with 255 out of 440 hits within K. pneumoniae genomes. blaNDM-1 was found in IncA/C2 plasmids from K. pneumoniae and Enterobacter species from PP, as well as in IncFIB plasmids from K. pneumoniae from India (Fig. 4 and Supplementary Fig. 10). We found IncX3 plasmids carrying blaNDM-5 in K. pneumoniae from NK, Nigeria, and multiple blaNDM-7 in K. pneumoniae and Enterobacter species from Nigeria and Pakistan (Fig. 4 and Supplementary Fig. 10). Col plasmid types were identified within 82 genomes. We found ColKP3 plasmids carrying blaOXA-181 or blaOXA-232 in isolates of three different species: E. coli, K. pneumoniae and Enterobacter species (Fig. 4 and Supplementary Fig. 10).
Our bioinformatics analysis relied on the interrogation of short-read sequencing data; therefore, it was not possible to analyse the genetic context to link carbapenemase genes and inc type for all genomes. Instead, a representative genome of each species/ST with the largest contig carrying the carbapenemase gene was chosen to maximize the analysis of other genetic material present, including the inc gene (n = 9; Supplementary Fig. 10). This analysis demonstrated cases where the same carbapenemase gene variant was detected in the same plasmid type across different GNB, suggesting that successful dissemination and acquisition within multiple species may be occurring. We also found cases where the same carbapenemase ARG was detected in multiple different plasmids, furthermore evidencing the spread of AMR.
Characterization of K. pneumoniae
K. pneumoniae is an important cause of neonatal sepsis in LMICs; however, there are few data analysing this species beyond antimicrobial susceptibility testing. Here, we have shown that K. pneumoniae was the most frequently identified GNB; therefore, the genomic diversity of this collection was scrutinized to contextualize these isolates, both within this study collection and within previously known collections15,16,22,23. K. pneumoniae (n = 258) was found at all clinical sites (Figs. 1 and 5, Supplementary Table 2 and Supplementary Fig. 4)—predominantly, Ethiopia (n = 95), Nigeria (n = 57) and Pakistan (n = 44).

a, Five-hundred-and-fifty-nine isolates incorporating a global collection23. Blue shading indicates K. pneumoniae isolates from the BARNARDS collection. The branch labels are coloured according to country of origin. b, Detailed core genome characterization of 309 K. pneumoniae isolates (n = 258 BARNARDS). Yellow shading indicates isolates from other studies15,22 causing neonatal sepsis. The outermost rings represent infant outcome (orange) and onset of sepsis (green), followed by the ST, where asterisks represent previously unknown STs. The leaf labels are the code names (coloured according to the study site) of isolates. The branch symbols in the centre denote the carriage of carbapenemase ARGs (blaNDM variants (circles) and blaOXA-48 group variants (squares)). NA, not applicable.
Genomics analysis within both the global15,16,22 (Fig. 5a; see Supplementary Table 5 for literature search inclusion criteria) and neonatal sepsis context23 (Fig. 5b) revealed high diversity of K. pneumoniae, with 156 STs from 17 countries spanning five continents. BARNARDS isolates clustered with previously reported neonatal isolates, including ST45, ST48 and ST348 (refs. 15,22,23), but we also revealed distinct and new genetic lineages. The major AMR-related K. pneumoniae clades in Asia (ST11) and Europe (ST147 and ST307)24 were also identified in this study. We only found ST307 in Rwanda (n = 6) and Nigeria (n = 2). However, ST258, a North American clade frequently associated with blaKPC25,26, was absent, which accords with the absence of blaKPC in this study. We did, however, detect one blaKPC-2 gene in a K. quasipneumoniae from Bangladesh (Supplementary Fig. 10). While other studies have suggested that blaKPC K. pneumoniae causes neonatal sepsis, especially during nosocomial outbreaks25, there is currently little evidence from countries in Africa or South Asia.
BARNARDS’ K. pneumoniae were disseminated throughout the global phylogeny, with 57 STs (Table 1 and Fig. 5a). ST35 and ST37 were predominantly found in Ethiopia (n = 38/39 and n = 29/30, respectively). We found four ST35 K. pneumoniae from other neonatal sepsis publications; however, these sit on a distinct branch in the core genome phylogeny (Fig. 5b) and were more closely related to the single ST35 isolated from RU, Rwanda. ST15 isolates were largely isolated from Pakistan and all carried both blaNDM-1 and blaOXA-181 (n = 22/27; Fig. 5b). ST15 was almost exclusively found at the South Asian clinical sites, with a single ST15 found at NN, Nigeria. ST442 (n = 6) and ST464 (n = 8) were only found in NN and all isolates contained either blaNDM-1 (ST442) or blaNDM-7 (ST464).
Multiple different capsule types (n = 47 KL loci and n = 12 O loci) were identified in silico. ST15 isolates in Pakistan (n = 23) and India (n = 1) were all the O1v1:KL112 serotype, whereas single ST15 isolates in Bangladesh and Nigeria had different serotype combinations of O3b:KL38 and O1v1:KL48, respectively. Similarly, the ST35 isolates from Ethiopia were all O1v2:KL108, whereas K. pneumoniae from RU in Rwanda were O2v1:KL113. Of the eight ST348 isolates, of which seven were from South Africa and one was from NW (Nigeria), all were O1v1:KL62 and all contained the yersiniabactin virulence gene.
In total, 115 isolates had a virulence score of 1, 3 or 4, indicating the presence of yersiniabactin and/or aerobactin/salmochelin virulence genes (see the Kleborate repository27 and Source Data Fig. 5). The odds of LOS were 89% lower for infants with sepsis due to K. pneumoniae who had a virulence score of 3 or 4 compared with those with a score of 0 (P = 0.04; 95% CI = 0.014–0.90). Additionally, the odds of the outcome deceased were 14 times higher for infants with sepsis due to K. pneumoniae who had a virulence score of 3 or 4 compared with those with a virulence score of 0 (P = 0.001; 95% CI = 2.76–68.77; Supplementary Table 3). These results suggest that these genes may be involved in quicker onset and MFBS. Alternatively, they may reflect transmission of distinct isolates from the mother’s microbiota6 and from the clinical environment.
K. pneumoniae harboured multiple β-lactamase genes (Source Data Fig. 5). blaCTX-M-15 was found in 220 out of 258 isolates across diverse STs and at all clinical sites, representing 42% (220/523) of total blaCTX-M-15-positive GNB. Over one-quarter (26%; 69/258) harboured a variant of blaNDM (Figs. 4b and 5b). blaNDM-1 (n = 15), blaNDM-5 (n = 1) and blaNDM-7 (n = 9) were found in Nigeria (mainly NN), whereas blaNDM-1 was the dominant variant in South Asia (Fig. 4b).
All K. pneumoniae were resistant to ampicillin, cefotaxime, ceftriaxone and ceftazidime. K. pneumoniae concomitantly resistant to the three cephalosporins tested (n = 255) were significantly more likely to cause EOS than LOS (P = 0.045; OR = 0.41; 95% CI = 0.17–0.98; Supplementary Table 3). Also, 144 isolates had >15 ARGs. For each additional ARG, we observed a 5.7% (P = 0.028; 95% CI = 0.89–0.99) decrease in the odds of neonates having LOS compared to EOS.
Collectively, this analysis reveals a large degree of intraspecies diversity within K. pneumoniae pathogens causing neonatal sepsis in LMICs. While certain ST groups previously shown to be dominant in particular geographic regions were found during the BARNARDS study, we also detected several different ST groups carrying different carbapenemase ARGs and virulence determinants.
Characterization of E. coli
E. coli has previously been reported as a dominant GNB cause of neonatal sepsis across many different LMICs18. In light of this, we aimed to further characterize the BARNARDS E. coli isolates and to compare the results with existing WGS datasets (Supplementary Table 5 displays the literature search inclusion criteria). The 2014 enterotoxigenic E. coli collection28 displayed large diversity and, when analysed with the BARNARDS dataset, 90 STs across four continents and 21 countries were detected (Fig. 6a). The E. coli analysed herein fell within four main clades of the extended phylogeny. The greatest numbers of E. coli in the BARNARDS study (n = 75) were in Nigeria (n = 23; 31%) and Rwanda (n = 17; 23%), although E. coli neonatal sepsis was identified in 11 of the 12 clinical sites (excluding BK, Bangladesh). A phylogenetic analysis of BARNARDS E. coli revealed four main groups, each containing multiple clades (Fig. 6b), with 37 STs detected. ST10 (n = 9), ST131 (n = 6), ST410 (n = 5) and ST69 (n = 4) were the most common. In the Mentzer et al.28 collection, ST10 was found in five countries across Africa, Asia and South America. Of the nine ST10 E. coli characterized here, seven were from clinical sites within Africa (Ethiopia, n = 1; Nigeria, n = 5; Rwanda, n = 1) and two were from PP, Pakistan. Although all ST10 E. coli belonged to the same phylotype (that is, group A), each isolate had a different O:H serotype profile and the phylogenetic tree (Fig. 6b) shows variability in the branch length, indicating genomic diversity within this ST group. Generally, we found highly variable O:H serotype classification (n = 57 O:H combinations), irrespective of the ST or phylogroup (A–F) (Source Data Fig. 6). Of the phylotypes, A (n = 22), B1 (n = 15) and B2 (n = 18) were the most common.

a, Three-hundred-and-sixty isolates incorporating a global collection28. Blue shading indicates E. coli isolates from the BARNARDS collection (n = 75). The branch labels are coloured according to country of origin. b, Detailed core genome characterization of 87 E. coli isolates (n = 75 BARNARDS). Yellow shading represents isolates from other studies16,22 causing neonatal sepsis. The colours on the right represent infant outcome (orange) and onset of sepsis (green), followed by the ST. The numbers and code names (coloured according to study site) of isolates are also given. The branch symbols denote the carriage of carbapenemase ARGs (blaNDM-5 (circles) and blaOXA-181 (squares). NF, not found.
β-lactam and aminoglycoside ARGs were most commonly found in the South Asian isolates, with the exception of blaCTX-M-15, which was also detected in E. coli from Africa. In total, 21 E. coli harboured blaCTX-M-15 and belonged to a variety of STs, including ST131, ST405, ST410, ST10 and ST167. Carbapenemase genes were detected in only three out of 75 E. coli. The three isolates were from South Asia, and all concomitantly carried blaCTX-M-15 plus blaNDM-5 (ST167; n = 2) or blaOXA-181 (ST410; n = 1).
No significant associations were found between phenotypic or genotypic AMR-related traits of E. coli and the clinical data assessed herein (Supplementary Table 3).
E. coli isolates were extremely diverse, suggesting that there are probably several important and worrisome lineages.
Discussion
In the BARNARDS study, we established a methodological framework to capture and extensively characterize GNB species causing neonatal sepsis in LMICs. We isolated 916 isolates of GNB, characterized them to species level, used WGS to probe genome composition and MLST to assess intra- and interspecies diversity, and documented extremely high rates of AMR.
Most of the Gram-negative isolates from neonates with sepsis were resistant to at least one β-lactam and one aminoglycoside (597/885; 67%), as has been reported previously for cohorts in India29 and 26 countries in Africa18. World Health Organization guidelines30 stipulate ampicillin plus gentamicin as the first line of empirical treatment for neonatal sepsis and third-generation cephalosporins as the second line of treatment. Of note, many of the blood culture isolates from our study were resistant to both lines of treatment, meaning that treatment options are unlikely to be curative.
The identification of 58 different GNB species suggests that the aetiology of neonatal sepsis is complex. We report multiple different lineages causing infection within single species, many of which carry either resistant or putatively virulent mechanisms and several of which have previously been shown to cause neonatal sepsis (for example, ST35 and ST37 K. pneumoniae)23,31. The identification of high-risk clones, such as ST15 in K. pneumoniae32 and the global clones ST1 and ST2 in A. baumannii, which are notorious for nosocomial infection33, indicates the spread and persistence of problematic lineages in LMICs. In addition, through our comprehensive analysis, we identified 40 previously unknown STs, suggesting that well-known and previously unidentified lineages/ST groups are both co-existing and evolving.
A limitation of our study was the inability to follow up all neonates to 60 d (necessitating the exclusion of neonates who were lost to follow-up), which impacted our outcome data (Fig. 1). It is likely that additional local factors (such as the management of sepsis) contributed to mortality; therefore, we cannot attribute MFBS singularly to the presence/absence of genomic traits. Our statistical analyses were exploratory and should be interpreted as hypothesis generating only.
In summary, Klebsiella, E. coli and Enterobacter were the main GNB species responsible for sepsis in neonates. We report that 54% of isolated bacteria were resistant to at least one antibiotic within four to six classes of antibiotics, and observed widespread carriage of both resistance genes and virulence factors in GNB causing neonatal sepsis in LMICs. This large, observational study will inform future research into effective antimicrobial therapies for neonatal sepsis, and may underpin improved infection control practices and could be useful in the development of vaccines for neonatal sepsis in LMICs.
Methods
Study design and processing of blood cultures at clinical sites
A prospective cohort study was conducted through the BARNARDS network consisting of 12 clinical sites in seven countries in Africa and South Asia (Chattogram Maa-O-Shishu Hospital, Chattogram (BC) and Kumudini Women’s Medical College, Mirzapur (BK) in Bangladesh; St. Paul’s Hospital Millennium Medical College, Addis Ababa (ES) in Ethiopia; the Division of Bacteriology, ICMR-National Institute of Cholera and Enteric Diseases, Kolkata (IN) in India; National Hospital Abuja, Abuja (NN), Wuse District Hospital, Abuja (NW) and Murtala Muhammad Specialist Hospital, Kano (NK) in Nigeria; Pakistan Institute of Medical Sciences, Islamabad (PP) and Bhara Kahu Rural Health Centre, Bhara Kahu (PC) in Pakistan; University Central Hospital of Kigali, Kigali (RU) and Kabgayi Hospital, Kabgayi (RK) in Rwanda; and Tygerberg Hospital, Cape Town (ZAT) in South Africa). Ethical approval was obtained from the local ethics committee at each site before the start of the study (Supplementary Table 6). Between November 2015 and December 2017, women in labour or immediately postpartum were recruited prospectively following consent, and their neonates were followed up for the first 60 d of life (at 3, 7, 14, 28 and 60 d of life) or until study withdrawal/death. Neonates admitted to clinical sites showing signs of sepsis were also enrolled. The BARNARDS sample collection workflow is shown in Supplementary Fig. 11. Although it was not strictly a neonatal population, during this study, we employed the term neonate for all enrolments, including those between 30 and 60 d post-birth.
Documentation detailing the parameters for clinically diagnosing sepsis is available in Supplementary Fig. 1. The standard operating procedures for the laboratory processing of blood cultures and subsequent identification of bacteria, which were followed by all clinical sites (following agreement between clinical partners before the start of enrolment), are shown in Supplementary Fig. 2. Laboratory reagents (Liofilchem)—importantly, both agar media and antibiotic discs (used at the clinical sites)—were standardized throughout the network. Bacterial identification performed at each site was confirmed by WGS at Cardiff University. Antimicrobial susceptibility testing was performed twice: initially, at the local sites to guide treatment using antibiotic discs; and then at Cardiff University using the agar dilution method to establish the MICs (details below). The collected clinical data included onset of sepsis (EOS or LOS) and patient outcome following biological sepsis. Neonates that were lost to follow up were categorized along with neonates confirmed alive as ‘not reported deceased’. For the purpose of this study, EOS and LOS were defined as sepsis occurring ≤72 h and >72 h after birth, respectively. If neonates showed clinical signs of sepsis at multiple time points within the first 60 d of life, additional blood cultures were analysed. All viable bacterial species were stored on charcoal swabs (Deltalab) for transport under UN3373 regulations to Cardiff University.
At Cardiff University, GNB isolates were plated onto chromogenic urinary tract infection media supplemented with vancomycin at 10 mg l−1 (Liofilchem) and incubated aerobically overnight at 37 °C. Isolates were identified using a Microflex LT MALDI-TOF MS (Bruker Daltonik) with α-cyano-4-hydroxycinnamic acid matrix (Sigma–Aldrich). Bacterial isolates were stored in TS/72 beads (Technical Service Consultants) at −80 °C and the original swabs were stored at 4 °C. MICs were determined by agar dilution for a panel of 19 antibiotics and interpreted according to the EUCAST guidelines20,21. E. coli ATCC 25922 and P. aeruginosa ATCC 27853 strains were used as quality controls for GNB tests. Supplementary Table 7 depicts the panels of antibiotics and additional control strains used. Supplementary Table 8 defines the EUCAST interpretations used. The MIC50 and MIC90 values for each antibiotic was determined. The phenotypic metadata included the following AMR-related counts: carbapenem resistance (taken as resistance to ertapenem); methicillin resistance (taken as an oxacillin MIC > 2) and an AMR score (the number of antibiotics to which an isolate was resistant) (Supplementary Table 7).
WGS
A single bacterial colony was transferred into 1.8 ml LB broth and incubated at 37 °C and 180 r.p.m. for 18 h. Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen), with an additional RNAse step, on a QIAcube (Qiagen), and quantified using a Qubit Fluorometer 3.0. Genomic libraries were prepared using a Nextera XT V2 kit (Illumina) with bead-based normalization. A total of 48 isolates were multiplexed per sequencing run to provide a depth of coverage of >15×. Paired-end WGS was performed on an Illumina MiSeq using the V3 chemistry to generate fragment lengths of up to 300 base pairs (600 cycles).
Bioinformatics analyses
Bioinformatics analyses were performed using a high-performance computing cluster at Cardiff University (Advanced Research Computing at Cardiff (ARCCA)) and CLIMB (version 1.0)34. Paired-end reads (FASTQ) were subjected to quality control checks before downstream analysis. Trim Galore (version 0.4.3)35 was used to remove the Nextera adapter sequences and low-quality bases. Reports before and after read trimming were generated using FastQC (version 0.11.2)36 and collated using MultiQC (version 1.7)37. The mean read length and number of sequences provided on the MultiQC reports were used to determine the sequencing coverage. Paired-end reads were overlapped using Flash (version 1.2.11)38 and assembled into contigs using SPAdes (version 3.9.0)39. The trimmed FASTQ reads were mapped to the contigs using BWA (version 0.7.15)40 and SAMtools (version 1.3.1)41. Pilon (version 1.22)42 was used to assess any misassemblies/errors in base calling in the resulting mapped BAM file. Final genome assembly metrics were generated using QUAST (version 2.1)43. Bacterial species were identified using both BLAST nt (version 2.2.25; https://blast.ncbi.nlm.nih.gov/Blast.cgi; input = contigs)44 and PathogenWatch (version 3.13.10; https://pathogen.watch; input = contigs). MLST, virulence and plasmid genomic profiles were characterized using SRST2 (version 0.2.0)45 and the associated databases PlasmidFinder46 and VFDB47. Genomes were screened for ARGs using ABRicate (version 0.9.7)48 (databases NCBI49 and ResFinder50).
Novel alleles and novel ST profiles were submitted to BIGSdb (version 1.25.1)51. The O:K locus profiles for all Klebsiella species were determined using Kaptive (version 0.7.0)52 and Kleborate (version 0.2.0; https://github.com/katholt/Kleborate)27. The O:H serotype profiles for all E. coli isolates were determined using SerotypeFinder (version 2.0)53, and SeqSero (version 1.0)54 was used to determine serotypes for Salmonella. In silico E. coli phylotyping was performed using ClermonTyping (version 1.3.0)55. Genomes were annotated using Prokka (version 1.12)30. Strain relatedness analysis was performed using Roary (version 3.12.0)56 to create a core genome alignment and FastTree (version 2.1.11) to generate maximum likelihood phylogenetic trees. Phylogenetic trees were mid-rooted, visualized and annotated using iTOL (version 5.7)57. The plasmid Sankey diagram was generated using the networkD3 package in R version 3.6.2. The immediate genetic context around carbapenemase genes was performed aligning outputs from ResFinder and PlasmidFinder (in ABRicate) with Mobile Element Finder (version 1.0.1) hosted by the Center for Genomic Epidemiology (https://cge.cbs.dtu.dk/services/MobileElementFinder/). The GBK annotation file from Prokka was then analysed for image production in Geneious Prime version 2020.1.2.
Statistical analyses
Statistical associations between clinical outcomes (onset of sepsis (EOS/LOS) and MFBS (alive/deceased)) and phenotypical and genomic traits were explored using univariable logistic regression models with the Wald test in SPSS version 26. The outcomes for the analyses regarding sepsis onset were EOS and LOS and those for MFBS were alive and deceased. Depending on whether predictor variables were continuous (AMR or ARG) or categorical (resistance versus non-resistance to ampicillin and gentamicin, concomitant resistance versus non-concomitant resistance to the three cephalosporins tested, resistance to at least one of the three cephalosporins tested versus to none, resistance versus non-resistance to ertapenem (a marker for carbapenems resistance) or Klebsiella species virulence scores), they were treated as covariates or factors, respectively. For species group analyses, only groups with n = ≥50 isolates were included, except for B. cenocepacia isolates, which did not carry ARGs. Statistical significance was taken at P ≤ 0.05, and estimated ORs are presented along with 95% CIs.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequence reads have been submitted to the European Nucleotide Archive under project number PRJEB33565. Individual accession numbers and additional genomics data can be accessed in Supplementary Table 4 and the source data. The databases used for this study included VFDB (http://www.mgc.ac.cn/VFs/download.htm), NCBI (https://github.com/tseemann/abricate/tree/master/db/ncbi), ResFinder (https://github.com/tseemann/abricate/tree/master/db/resfinder), PlasmidFinder (https://bitbucket.org/genomicepidemiology/plasmidfinder/src/master), MLST (https://github.com/tseemann/mlst/tree/master/db/pubmlst), MGE (https://bitbucket.org/mhkj/mge_finder/src/master/me_finder/), SerotypeFinder (https://bitbucket.org/genomicepidemiology/serotypefinder/src/master) and SeqSero (http://www.denglab.info/SeqSero). Previously published datasets downloaded from the European Nucleotide Archive repository and used for comparative genomics analysis have the identifiers PRJEB2111, PRJEB2581 and PRJEB20875. The following genomes were downloaded from NCBI: PHGE01000000–PHGR01000000, ATNW00000000 and ATNV00000000. Source data are provided with this paper.
References
United Nations Inter-agency Group for Child Mortality Estimation. Levels & Trends in Child Mortality: Report 2018: Estimates Developed by the UN Inter-agency Group for Child Mortality Estimation (United Nations Children’s Fund, 2018).
World Mortality 2019 (Department of Economic and Social Affairs, United Nations, 2019).
Shukla, V., Mwenechanya, M. & Carlo, W. A. Dealing with neonatal emergencies in low-resource settings. Semin. Fetal Neonatal Med. 24, 101028 (2019).
Popescu, C. R. et al. Neonatal sepsis in low-income countries: epidemiology, diagnosis and prevention. Expert Rev. Anti. Infect. Ther. 18, 443–452 (2020).
Folgori, L., Bielicki, J., Heath, P. T. & Sharland, M. Antimicrobial-resistant Gram-negative infections in neonates: burden of disease and challenges in treatment. Curr. Opin. Infect. Dis. 30, 281–288 (2017).
Sankar, M. J. Neonatal sepsis in South Asia: huge burden and spiralling antimicrobial resistance. BMJ 1, k5314 (2019).
Darmstadt, G. L. et al. Population-based incidence and etiology of community-acquired neonatal bacteremia in Mirzapur, Bangladesh: an observational study. J. Infect. Dis. 200, 906–915 (2010).
Peterside, O., Pondei, K. & Akinbami, F. O. Bacteriological profile and antibiotic susceptibility pattern of neonatal sepsis at a teaching hospital in Bayelsa State, Nigeria. Trop. Med. Health 43, 183–190 (2015).
Ullah, O. et al. Antibiotic sensitivity pattern of bacterial isolates of neonatal septicemia in Peshawar, Pakistan. Arch. Iran. Med. 19, 866–869 (2016).
Sorsa, A., Früh, J., Stötter, L. & Abdissa, S. Blood culture result profile and antimicrobial resistance pattern: a report from neonatal intensive care unit (NICU), Asella teaching and referral hospital, Asella, south East Ethiopia. Antimicrob. Resist. Infect. Control 8, 42 (2019).
Hamer, D. H. et al. Etiology of bacteremia in young infants in six countries. Pediatr. Infect. Dis. J. 34, 1–8 (2015).
Infection, N. & Denis, S. Characterisation and antimicrobial resistance of sepsis pathogens in neonates born in tertiary care centres in Delhi, India: a cohort study. Lancet Glob. Health 4, e752–e760 (2016).
Farzana, R., Jones, L. S., Rahman, A. & Andrey, D. O. Outbreak of hypervirulent multidrug-resistant Klebsiella variicola causing high mortality in neonates in Bangladesh. Clin. Infect. Dis. 68, 1225–1227 (2019).
Rohit, A. et al. Whole-genome-based analysis reveals multiclone Serratia marcescens outbreaks in a non-neonatal intensive care unit setting in a tertiary care hospital in India. J. Med. Microbiol. 68, 616–621 (2019).
Wisgrill, L. et al. Outbreak of yersiniabactin-producing Klebsiella pneumoniae in a neonatal intensive care unit. Pediatr. Infect. Dis. J. 36, 638–642 (2019).
Carl, M. A. et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Class. Infect. Dis. 58, 1211–1218 (2014).
Braye, K. et al. Effectiveness of intrapartum antibiotic prophylaxis for early-onset group B streptococcal infection: an integrative review. Women Birth 31, 244–253 (2018).
Okomo, U. et al. Aetiology of invasive bacterial infection and antimicrobial resistance in neonates in sub-Saharan Africa: a systematic review and meta-analysis in line with the STROBE-NI reporting guidelines. Lancet Infect. Dis. 19, 1219–1234 (2019).
Dramowski, A., Madide, A. & Bekker, A. Neonatal nosocomial bloodstream infections at a referral hospital in a middle-income country: burden, pathogens, antimicrobial resistance and mortality. Paediatr. Int. Child Health 35, 265–272 (2015).
Breakpoint Tables for Interpretation of MICs and Zone Diameters Version 9.0 (European Committee on Antimicrobial Susceptibility Testing, 2019).
Guidance Document on Tigecycline Dosing in association with Revision of Breakpoints for Enterobacterales and Other Species With an “Intermediate” Category (European Committee on Antimicrobial Susceptibility Testing, 2018).
Marando, R. et al. Predictors of the extended-spectrum-beta lactamases producing Enterobacteriaceae neonatal sepsis at a tertiary hospital, Tanzania. Int. J. Med. Microbiol. 308, 803–811 (2018).
Holt, K. E. et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl Acad. Sci. USA 112, E3574–E3581 (2015).
Dunn, S. J., Connor, C. & Mcnally, A. The evolution and transmission of multi-drug resistant Escherichia coli and Klebsiella pneumoniae: the complexity of clones and plasmids. Curr. Opin. Microbiol. 51, 51–56 (2019).
Yu, J. et al. Nosocomial outbreak of KPC-2- and NDM-1-producing Klebsiella pneumoniae in a neonatal ward: a retrospective study. BMC Infect. Dis. 16, 563 (2016).
Battikh, H. et al. Clonal spread of colistin-resistant Klebsiella pneumoniae coproducing KPC and VIM carbapenemases in neonates at a Tunisian university hospital. Microb. Drug Resist. 43, 468–472 (2016).
Lam, M. M. C. et al. Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb. Genom. 4, e000196 (2018).
Mentzer, A. et al. Identification of enterotoxigenic Escherichia coli (ETEC) clades with long-term global distribution. Nat. Genet. 46, 1321–1326 (2014).
Infection, N. & Denis, S. Characterisation and antimicrobial resistance of sepsis pathogens in neonates born in tertiary care centres in Delhi, India: a cohort study. Lancet Glob. Health 4, e752–e760 (2016).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Wang, C. Resistance phenotype and clinical molecular epidemiology of carbapenem-resistant Klebsiella pneumoniae among pediatric patients in Shanghai. Infect. Drug Resist. 11, 1935–1943 (2018).
Breurec, S. et al. Klebsiella pneumoniae resistant to third-generation cephalosporins in five African and two Vietnamese major towns: multiclonal population structure with two major international clonal groups, CG15 and CG258. Clin. Microbiol. Infect. 19, 349–355 (2013).
Hamidian, M. & Nigro, S. J.Emergence, molecular mechanisms and global spread of carbapenem-resistant Acinetobacter baumannii. Microb. Genom. 5, e000306 (2019).
Connor, T. R. et al. CLIMB (the Cloud Infrastructure for Microbial Bioinformatics): an online resource for the medical microbiology community. Microb. Genom. 2, e000086 (2016).
Krueger, F. Trim Galore v.0.4.3 https://github.com/FelixKrueger/TrimGalore (The Babraham Institute, 2017).
Andrews, S. FastQC: a quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2009).
Ewels, P., Lundin, S. & Max, K. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Magoč, T. & Salzberg, S. L.FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. Bioinformatics applications note genome analysis QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Mcginnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, 20–25 (2004).
Inouye, M. et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6, 90 (2014).
Carattoli, A. et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903 (2014).
Chen, L., Zheng, D., Liu, B., Yang, J. & Jin, Q. VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 44, D694–D697 (2016).
Seemann, T. ABRicate v.0.9.7 https://github.com/tseemann/abricate (The University of Melbourne, 2019).
Feldgarden, M. et al. Validating the AMRFINder tool and resistance gene database by using antimicrobial resistance genotype–phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63, e00483-19 (2019).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644 (2012).
Jolley, K. A. & Maiden, M. C. J.BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595 (2010).
Wick, R. R., Heinz, E., Holt, K. E. & Wyres, K. L.Kaptive Web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella genomes. J. Clin. Microbiol. 56, e00197-18 (2018).
Joensen, K. G., Tetzschner, A. M. M., Iguchi, A. & Aarestrup, F. M. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426 (2015).
Zhang, S. et al. Salmonella serotype determination utilizing high-throughput genome sequencing data. J. Clin. Microbiol. 53, 1685–1692 (2015).
Beghain, J., Bridier-Nahmias, A., Le Nagard, H., Denamur, E. & Clermont, O.ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 4, e000192 (2018).
Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, 256–259 (2019).
Acknowledgements
We acknowledge Liofilchem for continued support in the distribution of microbiology products to enable standard operating procedures across the clinical sites. We acknowledge J. Parkhill for advice and guidance regarding the phylogenetic analyses. We acknowledge Wales Gene Park and ARCCA for continued bioinformatics support and infrastructure availability. Bioinformatics analysis was largely undertaken using the supercomputing facilities at Cardiff University, which were operated by Advanced Research Computing at Cardiff (ARCCA) on behalf of the Cardiff Supercomputing Facility and HPC Wales and Supercomputing Wales projects. We acknowledge support from Supercomputing Wales, which is partly funded by the European Regional Development Fund via the Welsh Government. We thank the team of curators for the databases hosted on PubMLST (https://pubmlst.org/databases/). We also thank the curators of the Institut Pasteur MLST and Whole-Genome MLST databases for curating the Klebsiella data and making them publicly available at http://bigsdb.pasteur.fr. We thank M. Islam for providing access to the clinical sites and epidemiology data in Bangladesh. We acknowledge R. Kamran, the microbiologist from the Padmashree Institute of Management and Sciences, who sadly passed away in 2018. We thank the team at the Bill & Melinda Gates Foundation; namely, P. Srikantiah, R. Izadnegahdar, K. Klugman and S. Vernam. The BARNARDS study was funded by two awards (US$4.28 million (OPP1119772) and US$849,000 (OP1191522)) from the Bill & Melinda Gates Foundation.
Author information
Authors and Affiliations
Consortia
Contributions
K.S. and M.J.C. designed and guided the study, performed the analysis and wrote the manuscript. K.S., E.P. and J.M. performed the WGS experiments. K.S. and R.A. performed the bioinformatics analysis. M.J.C. contributed to the bioinformatics analysis. E.P. and K. Thomson contributed equally. K. Thomson performed the MIC experiments. K. Thomson and M.J.C. analysed the MIC dataset. K.S. and M.J.C. produced the figures. C.D., R.M., D.G., K. Taiyari and K.H. provided the finalized epidemiological and clinical dataset. M.J.C., D.G. and K. Taiyari performed the statistical analysis. K.S., M.J.C., E.P., K. Thomson, C.D., C.A., A.F., T.H., J.M. and M.N. performed the microbiology culture and sample processing at Cardiff University. G.J.C., D.B., S.S., S.B., P.C., S.M., K.I., F.M., S.U., R.Z., H.S., A.M., J.-B.M., A.R., L.G., S.M., A.N.H.B. and A.W. collected and processed the blood cultures and collected clinical data at the clinical sites. T.R.W., M.J.C., R.M., G.J.C., S.B., K.I., R.Z., J.-B.M. and S.M. designed the BARNARDS study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Microbiology thanks Stephen Baker and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–11 and Tables 1–3 and 5–9.
Supplementary Table 4
Supplementary genomics data per isolate (n = 916).
Source data
Source Data Fig. 5
Raw genomics data and clinical data for 258 K. pneumoniae isolates used for the production of Fig. 5a,b.
Source Data Fig. 6
Raw genomics data and clinical data for 75 E. coli isolates used for the production of Fig. 6a,b.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sands, K., Carvalho, M.J., Portal, E. et al. Characterization of antimicrobial-resistant Gram-negative bacteria that cause neonatal sepsis in seven low- and middle-income countries. Nat Microbiol 6, 512–523 (2021). https://doi.org/10.1038/s41564-021-00870-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-021-00870-7
This article is cited by
-
Longitudinal analysis within one hospital in sub-Saharan Africa over 20 years reveals repeated replacements of dominant clones of Klebsiella pneumoniae and stresses the importance to include temporal patterns for vaccine design considerations
Genome Medicine (2024)
-
Trends in the antimicrobial susceptibility among Chinese neonates from 2012 to 2021: a multicenter study
Antimicrobial Resistance & Infection Control (2024)
-
Complete genome sequence, phenotypic correlation and pangenome analysis of uropathogenic Klebsiella spp
AMB Express (2024)
-
Isolation of four carbapenem-resistant gram-negative species from a single fly
Animal Diseases (2024)
-
Prevalence of carbapenem-resistant gram-negative bacteria among neonates suspected for sepsis in Africa: a systematic review and meta-analysis
BMC Infectious Diseases (2024)
