
Abstract
Bacterial cells in nature are frequently exposed to changes in their chemical environment1,2. The response mechanisms of isolated cells to such stimuli have been investigated in great detail. By contrast, little is known about the emergent multicellular responses to environmental changes, such as antibiotic exposure3,4,5,6,7, which may hold the key to understanding the structure and functions of the most common type of bacterial communities: biofilms. Here, by monitoring all individual cells in Vibrio cholerae biofilms during exposure to antibiotics that are commonly administered for cholera infections, we found that translational inhibitors cause strong effects on cell size and shape, as well as biofilm architectural properties. We identified that single-cell-level responses result from the metabolic consequences of inhibition of protein synthesis and that the community-level responses result from an interplay of matrix composition, matrix dissociation and mechanical interactions between cells. We further observed that the antibiotic-induced changes in biofilm architecture have substantial effects on biofilm population dynamics and community assembly by enabling invasion of biofilms by bacteriophages and intruder cells of different species. These mechanistic causes and ecological consequences of biofilm exposure to antibiotics are an important step towards understanding collective bacterial responses to environmental changes, with implications for the effects of antimicrobial therapy on the ecological succession of biofilm communities.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
Raw and analysed data that support the findings of this study are available from the corresponding author upon request.
Code availability
Our open-source and user-friendly biofilm image analysis software tool BiofilmQ46 is available online (https://drescherlab.org/data/biofilmQ/). The raw developer-level code used to analyse data is available from the corresponding author upon request.
References
D’Costa, V. M. et al. Antibiotic resistance is ancient. Nature 477, 457–461 (2011).
Baym, M. et al. Spatiotemporal microbial evolution on antibiotic landscapes. Science 353, 1147–1151 (2016).
Hoffman, L. R. et al. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 436, 1171–1175 (2005).
Koch, G. et al. Evolution of resistance to a last-resort antibiotic in Staphylococcus aureus via bacterial competition. Cell 158, 1060–1071 (2014).
Jenssen, H., Hamill, P. & Hancock, R. E. W. Peptide antimicrobial agents. Clin. Microbiol. Rev. 19, 491–511 (2006).
Houry, A. et al. Bacterial swimmers that infiltrate and take over the biofilm matrix. Proc. Natl Acad. Sci. USA 109, 13088–13093 (2012).
Andersson, D. I. & Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 12, 465–478 (2014).
O’Neill, J. ed. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations (HM Government, 2014).
Van Acker, H., Van Dijck, P. & Coenye, T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 22, 326–333 (2014).
Lebeaux, D., Ghigo, J.-M. & Beloin, C. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 78, 510–543 (2014).
Koo, H., Allan, R. N., Howlin, R. P., Stoodley, P. & Hall-Stoodley, L. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat. Rev. Microbiol. 15, 740–755 (2017).
Meylan, S., Andrews, I. W. & Collins, J. J. Targeting antibiotic tolerance, pathogen by pathogen. Cell 172, 1228–1238 (2018).
Garcia, L. G. et al. Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J. Antimicrob. Chemother. 68, 1455–1464 (2013).
Kwan, B. W., Valenta, J. A., Benedik, M. J. & Wood, T. K. Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 57, 1468–1473 (2013).
Grant, S. S. & Hung, D. T. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 4, 273–283 (2013).
Brooun, A., Liu, S. & Lewis, K. A Dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 44, 640–646 (2000).
Chua, S. L. et al. Selective labelling and eradication of antibiotic-tolerant bacterial populations in Pseudomonas aeruginosa biofilms. Nat. Commun. 7, 10750 (2016).
Gupta, K., Marques, C. N. H., Petrova, O. E. & Sauer, K. Antimicrobial tolerance of Pseudomonas aeruginosa biofilms is activated during an early developmental stage and requires the two-component hybrid SagS. J. Bacteriol. 195, 4975–4987 (2013).
Okshevsky, M. & Meyer, R. L. The role of extracellular DNA in the establishment, maintenance and perpetuation of bacterial biofilms. Crit. Rev. Microbiol. 41, 341–352 (2015).
Mah, T.-F. et al. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426, 306–310 (2003).
Nguyen, D. et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986 (2011).
Høiby, N., Bjarnsholt, T., Givskov, M., Molin, S. & Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 35, 322–332 (2010).
Tseng, B. S. et al. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 15, 2865–2878 (2013).
Dale, J. L., Nilson, J. L., Barnes, A. M. T. & Dunny, G. M. Restructuring of Enterococcus faecalis biofilm architecture in response to antibiotic-induced stress. NPJ Biofilms Microbiomes 3, 15 (2017).
Wong, K. C., Brown, A. M., Luscombe, G. M., Wong, S. J. & Mendis, K. Antibiotic use for Vibrio infections: important insights from surveillance data. BMC Infect. Dis. 15, 226 (2015).
Drescher, K. et al. Architectural transitions in Vibrio cholerae biofilms at single-cell resolution. Proc. Natl Acad. Sci. USA 113, E2066–E2072 (2016).
Singh, P. K. et al. Vibrio cholerae combines individual and collective sensing to trigger biofilm dispersal. Curr. Biol. 27, 3359–3366 (2017).
Hartmann, R. et al. Emergence of three-dimensional order and structure in growing biofilms. Nat. Phys. 15, 251–256 (2019).
Adams, D. W. & Errington, J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 7, 642–653 (2009).
Stewart, E. J., Satorius, A. E., Younger, J. G. & Solomon, M. J. Role of environmental and antibiotic stress on Staphylococcus epidermidis biofilm microstructure. Langmuir 29, 7017–7024 (2013).
Berk, V. et al. Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science 337, 236–239 (2012).
Fong, J. C. et al. Structural dynamics of RbmA governs plasticity of Vibrio cholerae biofilms. eLife 6, 1–22 (2017).
Smith, D. R. et al. In situ proteolysis of the Vibrio cholerae matrix protein rbma promotes biofilm recruitment. Proc. Natl Acad. Sci. USA 112, 10491–10496 (2015).
Fong, J. C. N. & Yildiz, F. H. The rbmBCDEF gene cluster modulates development of rugose colony morphology and biofilm formation in Vibrio cholerae. J. Bacteriol. 189, 2319–2330 (2007).
Douarche, C., Allain, J.-M. & Raspaud, E. Bacillus subtilis bacteria generate an internal mechanical force within a biofilm. Biophys. J. 109, 2195–2202 (2015).
Yan, J., Nadell, C. D., Stone, H. A., Wingreen, N. S. & Bassler, B. L. Extracellular-matrix-mediated osmotic pressure drives Vibrio cholerae biofilm expansion and cheater exclusion. Nat. Commun. 8, 327 (2017).
Vidakovic, L., Singh, P. K., Hartmann, R., Nadell, C. D. & Drescher, K. Dynamic biofilm architecture confers individual and collective mechanisms of viral protection. Nat. Microbiol. 3, 26–31 (2018).
Nadell, C. D., Drescher, K., Wingreen, N. S. & Bassler, B. L. Extracellular matrix structure governs invasion resistance in bacterial biofilms. ISME J. 9, 1700–1709 (2015).
Jørgensen, B. R. & Huss, H. H. Growth and activity of Shewanella putrefaciens isolated from spoiling fish. Int. J. Food Microbiol. 9, 51–62 (1989).
Kimata, N., Nishino, T., Suzuki, S. & Kogure, K. Pseudomonas aeruginosa isolated from marine environments in Tokyo bay. Microb. Ecol. 47, 41–47 (2004).
Brislawn, C. J. et al. Forfeiting the priority effect: turnover defines biofilm community succession. ISME J. 13, 1865–1877 (2019).
Skorupski, K. & Taylor, R. K. Positive selection vectors for allelic exchange. Gene 169, 47–52 (1996).
Guder, J. C., Schramm, T., Sander, T. & Link, H. Time-optimized isotope ratio LC–MS/MS for high-throughput quantification of primary metabolites. Anal. Chem. 89, 1624–1631 (2017).
Ducret, A., Quardokus, E. M. & Brun, Y. V. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat. Microbiol. 1, 16077 (2016).
O’Toole, G. A. Microtiter dish biofilm formation assay. J. Vis. Exp. 47, e2437 (2011).
Hartmann, R. et al. BiofilmQ, a software tool for quantitative image analysis of microbial biofilm communities. Preprint at bioRxiv https://doi.org/10.1101/735423 (2019).
Beyhan, S. & Yildiz, F. H. Smooth to rugose phase variation in Vibrio cholerae can be mediated by a single nucleotide change that targets c-di-GMP signalling pathway. Mol. Microbiol 63, 995–1007 (2007).
Acknowledgements
We thank P. K. Singh, P. Pearce, S. Vaidya, M. Bayer, K. Neuhaus, E. Jimenez and K. Strenger for strains and support during this project, and thankfully acknowledge grants from the Max Planck Society, Human Frontier Science Program (CDA00084/2015-C), Deutsche Forschungsgemeinschaft (SFB 987), Behrens-Weise-Stiftung, Minna-James-Heineman-Stiftung and European Research Council (StG-716734) to K.D., the MIT-Germany MISTI Seed Fund Program to K.D. and J.D., and the James S. McDonnell Foundation (J.D.). C.D.N. is supported by the Alexander von Humboldt Foundation, National Science Foundation (MCB 1817342), a Burke Award from Dartmouth College, a pilot award from the Cystic Fibrosis Foundation (STANTO15RO) and NIH grant P20-GM113132 to the Dartmouth BioMT COBRE.
Author information
Authors and Affiliations
Contributions
K.D. conceived, supervised and coordinated the project. C.D.N. and K.D. designed experiments. J.D. designed simulations. F.D.-P. and L.V. performed all biofilm experiments. F.D.-P. and L.V. performed liquid culture experiments. F.D.-P., M.L. and H.L. performed metabolite measurements and analysis. R.H. and H.J. developed image and data analysis methods. F.D.-P., R.H. and H.J. performed image analysis. R.H. and B.S. performed simulations. F.D.-P. and L.V. generated strains. F.H.Y. and K.M.T. developed strains and analysis ideas. All authors made important conceptual contributions to the project and interpreted results, often in group discussions. F.D.-P. and R.H. made the figures. F.D.-P., C.D.N. and K.D. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Screening biofilm architecture after antibiotic exposure and identifying the minimum inhibitory concentration (MIC).
(a) Confocal xy-slices of biofilms exposed to different antibiotics for 24 h and stained with the SYTO 9 nucleic acid dye. The conditions tests were the following: untreated control biofilm, tetracycline (Tet; 3 µg/mL, 8x the MIC), chloramphenicol (Cm; 10 µg/mL, 8x the MIC), erythromycin (Ery; 200 µg/mL, 4x the MIC), kanamycin (Kan; 200 µg/mL, 4x the MIC), rifampicin (Rif; 6 µg/mL, 5x the MIC), ciprofloxacin (Cip; 0.5 µg/mL, 6.3x the MIC), ampicillin (Amp; 400 µg/mL, a concentration at which the cell morphology was significantly modified), and ceftibuten (Ctb; 50 µg/mL, a concentration at which the cell morphology was significantly modified). Images are representative of n = 3 independent experiments. (b) Fold-change of cell volume, cell aspect ratio, and cell density (calculated as volume fraction) of biofilms treated with different protein synthesis inhibitors for 6 h, relative to untreated biofilms (mean ± SEM, n = 15 samples for control, n = 9 for Tet, n = 7 for Ery, n = 14 for Kan, and n = 8 for Cm; samples correspond to different biofilms). Statistical significances were calculated using a one-way ANOVA with Bonferroni’s correction. Statistically non-significant differences (NS) correspond to p = 0.93, 0.51, 0.99, 0.99, 0.99, 0.99, 0.46 (left to right). *, ** and **** indicate p < 0.05, p < 0.01 and p < 0.0001 respectively. (c) Batch culture growth curves of wild-type V. cholerae N16961 grown in M9 medium supplemented with glucose and with different antibiotic concentrations. Every line corresponds to the average between 2 technical replicates, and each concentration has been tested on 2 separate days (each resulting in one line). For ampicillin and ceftibuten, the MIC determination was not possible from the concentrations tested, due to the lack of cell lysis. (d) List of antibiotic concentrations used in panel c according to their color-coding.
Extended Data Fig. 2 Nematic order and vertical alignment of cells in biofilms during tetracycline treatment, compared with untreated controls.
(a, b) Spatiotemporal changes of the average nematic order (a), or the vertical alignment (b), as a function of time and position inside the biofilm during tetracycline treatment. Each pixel in the heatmap is coloured according to the average nematic order (a) or vertical alignment (b) at a given time and spatial position in the biofilm. (c, d) Spatiotemporal changes of the average nematic order (c), or the vertical alignment (d), for control biofilms that were not treated with tetracycline. In these kymograph heatmaps, the pixel values correspond to averages over all cells with similar distances from the surface of the biofilm at a particular time. Heatmaps are representative of n = 5 different biofilms.
Extended Data Fig. 3 Effect of tetracycline treatment on biomass measurements using the crystal violet assay.
(a) Fold-changes for the cell volume and cell density of the wild- type (WT) and the vpvCW240R rugose strain (which is a biofilm hyper-producer strain47) for tetracycline-treated biofilms in comparison with untreated biofilms, measured in flow chambers using confocal imaging at the single-cell level. Data are shown as mean ± SE, n = 15, 9, 7, 11, for WT (-Tet), WT (+Tet), rugose (-Tet), and rugose (+Tet), respectively. Each sample corresponds to an independent biofilm. These results show that the rugose strain also displays similar biofilm architecture responses to Tet treatment. Statistical significances were calculated using a one-way ANOVA with Bonferroni’s correction. **** indicates p < 0.0001. (b) Growth curve of rugose and WT biofilms using the crystal violet assay in 96-well plates; mean ± SD, n = 3 independent biological replicates. These results show that only the rugose strain forms substantial biofilms in static 96-well plates. (c) Fold-change in biofilm biomass measured by crystal violet absorbance of rugose biofilms, which were grown for 14 h in 96-well plates followed by 6 h treatment with tetracycline (Tet; 3 µg/mL, 8x the MIC), trimethoprim (Tmp; 10 µg/mL, 4x the MIC), chloramphenicol (Cm; 10 µg/mL, 8x the MIC), erythromycin (Ery; 200 µg/mL, 4x the MIC), or kanamycin (Kan; 200 µg/mL, 4x the MIC); mean ± SE, n = 3 independent biological replicates. For all experiments, each biological replicate is the average of 8 technical replicates from different wells on the same microtiter plate. The translational inhibitors Tet, Cm, Ery, and Kan show the same qualitative behaviour: an increase in crystal violet biofilm signal after 6 h of antibiotic exposure. Only the Tet+Tmp treatment does not show the increase in crystal violet signal, consistent with Fig. 2g of the main text.
Extended Data Fig. 4 Increase in cell volume and decrease in cell density precede cell death.
(a) Confocal xy-slices of a biofilm constitutively expressing mTFP1 (shown in cyan), grown in the presence of propidium iodide. Alive cells are only visible in the cyan fluorescent channel, whereas dead cells are also visible in the red fluorescent channel. (b) Same biofilm as in panel a, now imaged after 6 h of tetracycline (Tet) treatment. (c) Same biofilm as in panel a, after 12 h of Tet treatment. (d) Untreated control biofilm. (e) Percentage of dead cells in the biofilm as a function of treatment time. Centre lines correspond to mean and width of the shaded areas around each line correspond to standard errors, n = 13 samples for Tet, n = 9 for control, n = 4 for Tmp, n = 4 for Tmp+Tet. Each sample corresponds to a different biofilm.
Extended Data Fig. 5 Changes in cell volume and metabolite levels of planktonic cells during exposure to tetracycline.
(a) Cell volume fold-change (comparing each time point to the untreated (-Tet) sample at 0 h (mean ± SEM, n = 5 independent biological replicates. (b) Fold-changes in 57 metabolites after 2 h of Tet treatment in comparison with untreated cells (mean ± SEM, n = 3 independent biological replicates).
Extended Data Fig. 6 The RbmA-His strain phenocopies the wild- type strain.
(a) Fold-change of cell volume and cell density (measured as volume fraction) of wild-type biofilms and biofilms formed by a strain producing His-tagged RbmA (RbmA-His) treated with tetracycline, relative to the biofilms before the treatment. Values are displayed as mean ± SEM (n = 17 different biofilms). Statistical significances were calculated using a one-way ANOVA with Bonferroni’s correction for multiple comparisons. Statistically non-significant differences are labelled NS, which both correspond to p = 0.99. **** indicates p < 0.0001. (b, c) RbmA-His biofilms, shown here, display the same spatiotemporal changes in biofilm architecture as the WT biofilms, for which the corresponding kymograph heatmaps are shown in Fig. 1e–h. Heatmaps show the changes of the average cell volume (b) and cell density (c) as a function of time and spatial location during tetracycline treatment inside the RbmA- His biofilms. Panels on the left correspond to Tet treatment and panels on the right correspond to untreated control conditions. Each pixel in these kymograph heatmaps is coloured according to the average cell volume or cell density at a given time and spatial position in the biofilm. Cell volumes and cell density volume fraction values are averaged over all cells with similar distances from the surface of the biofilm. The RbmA-His strain was grown and imaged using antibodies as described in the methods section. Heatmaps are representative of n = 5 different biofilms.
Extended Data Fig. 7 Antibiotic-induced cell volume increase is independent of RbmA concentration.
Cell volume fold-change (comparing 6 h and 0 h of Tet treatment) in biofilms of a ΔrbmA strain carrying the PBAD:rbmA construct, measured as a function of arabinose concentration (mean ± SEM, n = 7, 12, 13, 17, 10, 18, 14 samples for arabinose concentrations of 0%, 0.2%, 0.3%, 0.5%, 1%, 2%, and 5% respectively). Each sample corresponds to a different biofilm.
Extended Data Fig. 8 Simulated biofilms were subjected to a decrease in cell-cell attraction and an increase in cell volume, revealing the contribution of each effect to the antibiotic-induced biofilm architecture changes.
Biofilm growth was simulated as described in the methods section until the biofilm size reached 1,000 cells, corresponding to the 0 h time point in the heatmaps in this figure. For these 1,000-cell biofilms, tetracycline treatment was simulated by decreasing the attraction potential, or by increasing the cell volume, or by both effects together. (a) Kymograph heatmaps of simulated 1,000-cells biofilms subject to a linear decrease in cell-cell attraction over the course of different times τpot. The value of τpot corresponds to the time for the cell-cell attraction potential to decrease to zero, starting from the value used to simulate biofilm growth. If the attraction potential is set to zero immediately (corresponding to τpot = 0), the resulting biofilm dynamics do not closely resemble the experiments, indicating that the cell-cell attraction decreases over an extended period of time. The control kymograph heatmap corresponds to biofilms were neither the attraction potential or the cell volume were changed. Each heatmap is the average of n = 3 simulations. (b) Heatmaps of simulated 1,000-cells biofilms that were subjected to a decrease in cell-cell attraction over different times (τpot) and simultaneously subjected to a linear increase in cell volume over 6 h (τvol = 6 h). Each heatmap is the average of n = 3 simulations. In Fig. 3h of the main text, a kymograph heatmap is shown for simulations in which only the cell volume is linearly increased with a time scale τvol = 6 h, according to the experimentally determined single-cell volume growth rate during Tet-treatment. (c) Experimental changes of the average cell density as a function of space and time during tetracycline treatment inside the biofilm as shown in Fig. 1g, reproduced here for convenience. Heatmap in panel c is a representative of n = 5 different biofilms.
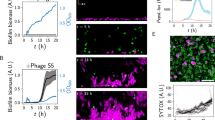
Extended Data Fig. 9 Tetracycline-treated V. cholerae biofilms are susceptible to colonization and population invasion by an isogenic strain and by bacteriophages.
(a-b) Each of the two panels shows an independent replica experiment of a tetracycline-treated V. cholerae biofilm colonized by an isogenic strain, which is expressing a different fluorescent protein. Resident biofilms constitutively express mKOκ (cells shown in yellow), invaders constitutively express sfGFP (cells shown in cyan). Images correspond to confocal xy-slices, acquired 2 µm above the substrate. The resident biofilm underwent tetracycline (Tet) treatment for 6 h or control treatment (medium without Tet), followed by 2 h exposure to invader cells. Following exposure to invader cells, the medium was exchanged to fresh, sterile medium and the imaging was started (labelled as the 0 h time point here). A third independent replica experiment is shown in Fig. 4a, b of the main text. (c) Confocal xy-slice of a resident V. cholerae WT biofilm, expressing mKOκ constitutively (cells shown in yellow), imaged directly above the glass substrate. The resident biofilm underwent exposure to tetracycline (Tet) and fluorescently labelled vibriophage N-4 virions (visible as cyan spots) for 6 h. During 6 h of Tet-treatment cell death is negligible, as shown in Extended Data Fig. 4. (d) Confocal xy- slice of control biofilm (not treated with tetracycline) exposed to vibriophage N-4 virions for 6 h. Images are representative of n = 5 different biofilms.
Extended Data Fig. 10 Biofilm architecture in the lower part of the biofilm is representative of the whole biofilm.
(a) Comparison between the cell volume, cell aspect ratio, and cell density (measured as volume fraction) between the lower part of the biofilm and the whole biofilm. The ratio of these biofilm architecture parameters was calculated using the mean value of these parameters in the part of the biofilm that is bounded by the z = 0 µm and z = 10 µm planes, and the mean value of these parameters in the whole biofilm (mean ± SEM, n = 19 different biofilms). (b, c) Cell volume, cellular aspect ratio, and cell density before tetracycline treatment (panel b) or after 6 hours of tetracycline treatment (panel c) as measured for complete biofilm volumes, or for the cells located in the biofilm volume bounded by the z = 0 µm and z = 10 µm planes (mean ± SD, n = 8 samples in panel b and n = 5 samples in panel c; each sample corresponds to a different biofilm). The cellular location was measured as the shortest distance of each cell to the interface between the biofilm and the liquid growth medium. This interface is termed “biofilm boundary” in this manuscript. For the experiments in this figure, biofilms were stained with the nucleic acid dye SYTO 9 prior to imaging. The cell volume, aspect ratio, and cell density were nearly identical between the whole biofilm and the bottom-most 10 µm of the biofilms.
Supplementary information
Supplementary Information
Supplementary Tables 1–4 and Supplementary References.
Supplementary Video 1
The video shows a confocal xy-slice, imaged at different time points during 6 h of exposure to 3 µg ml−1 tetracycline (8× the MIC). Images were acquired 2 µm above the glass coverslip. The biofilm was formed by V. cholerae WT cells, expressing mKOκ constitutively (cells shown in yellow). Video is representative of n = 5 independent different biofilms.
Supplementary Video 2
The video shows a confocal xy-slice, imaged at different time points during 6 h of growth, as a control for Supplementary Video 1. Images were acquired 2 µm above the glass coverslip. The biofilm was formed by V. cholerae WT cells, expressing mKOκ constitutively (cells shown in yellow). Video is representative of n = 5 independent different biofilms.
Rights and permissions
About this article

Cite this article
Díaz-Pascual, F., Hartmann, R., Lempp, M. et al. Breakdown of Vibrio cholerae biofilm architecture induced by antibiotics disrupts community barrier function. Nat Microbiol 4, 2136–2145 (2019). https://doi.org/10.1038/s41564-019-0579-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-019-0579-2
This article is cited by
-
Understanding the intricacies of microbial biofilm formation and its endurance in chronic infections: a key to advancing biofilm-targeted therapeutic strategies
Archives of Microbiology (2024)
-
The biofilm matrix: multitasking in a shared space
Nature Reviews Microbiology (2023)
-
Effects of chitosan nanoparticles loaded with mesenchymal stem cell conditioned media on gene expression in Vibrio cholerae and Caco-2 cells
Scientific Reports (2022)
-
Biofilm viability checker: An open-source tool for automated biofilm viability analysis from confocal microscopy images
npj Biofilms and Microbiomes (2021)
-
Short-range quorum sensing controls horizontal gene transfer at micron scale in bacterial communities
Nature Communications (2021)
