
Abstract
Zika virus (ZIKV) is an emerging, mosquito-borne RNA virus. The rapid spread of ZIKV within the Americas has unveiled microcephaly1 and Guillain–Barré syndrome2,3 as ZIKV-associated neurological complications. Recent reports have also indicated other neurological manifestations to be associated with ZIKV, including myelitis4, meningoencephalitis5 and fatal encephalitis6. Here, we investigate the neuropathogenesis of ZIKV infection in type I interferon receptor IFNAR knockout (Ifnar1−/−) mice, an infection model that exhibits high viral burden within the central nervous system. We show that systemic spread of ZIKV from the site of infection to the brain requires Ifnar1 deficiency in the haematopoietic compartment. However, spread of ZIKV within the central nervous system is supported by Ifnar1-deficient non-haematopoietic cells. Within this context, ZIKV infection of astrocytes results in breakdown of the blood–brain barrier and a large influx of CD8+ effector T cells. We also find that antiviral activity of CD8+ T cells within the brain markedly limits ZIKV infection of neurons, but, as a consequence, instigates ZIKV-associated paralysis. Taken together, our study uncovers mechanisms underlying ZIKV neuropathogenesis within a susceptible mouse model and suggests blood–brain barrier breakdown and T-cell-mediated neuropathology as potential underpinnings of ZIKV-associated neurological complications in humans.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Change history
08 January 2018
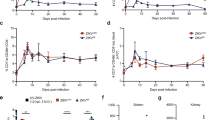
In the version of this Letter originally published, technical problems led to errors in Figs. 3e and 4f. In Fig. 3e, the bottom right graph had an incorrect title of ‘CD8+ T cells’; it should have instead been ‘CD4+ T cells’. In Fig. 4f, the bottom panels had an incorrect title of ‘CD8 competent Ifnar–/–7 d.p.i. cerebral cortex’; it should have instead been ‘CD8 depleted Ifnar–/– 7 d.p.i. cerebral cortex’. These errors have now been corrected in all versions of the Letter.
References
Rasmussen, S. A., Jamieson, D. J., Honein, M. A. & Petersen, L. R. Zika virus and birth defects—reviewing the evidence for causality. New Engl. J. Med. 374, 1981–1987 (2016).
Cao-Lormeau, V. M. et al. Guillain–Barre syndrome outbreak associated with Zika virus infection in French Polynesia: a case–control study. Lancet 387, 1531–1539 (2016).
Oehler, E. et al. Zika virus infection complicated by Guillain–Barre syndrome—case report, French Polynesia, December 2013. Euro. Surveill. 19, 20720 (2014).
Mecharles, S. et al. Acute myelitis due to Zika virus infection. Lancet 387, 1481 (2016).
Carteaux, G. et al. Zika virus associated with meningoencephalitis. New Engl. J. Med. 374, 1595–1596 (2016).
Soares, C. N. et al. Fatal encephalitis associated with Zika virus infection in an adult. J. Clin. Virol. 83, 63–65 (2016).
Grant, A. et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890 (2016).
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Dowall, S. D. et al. A susceptible mouse model for Zika virus infection. PLoS Negl. Trop. Dis. 10, e0004658 (2016).
Rossi, S. L. et al. Characterization of a novel murine model to study Zika virus. Am. J. Trop. Med. Hyg. 94, 1362–1369 (2016).
Elong Ngono, A. et al. Mapping and role of the CD8+ T cell response during primary Zika virus infection in mice. Cell Host Microbe 21, 35–46 (2017).
Iwasaki, A. Immune regulation of antibody access to neuronal tissues. Trends Mol. Med. 23, 227–245 (2017).
Daniels, B. P. et al. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J. Clin. Invest. 127, 843–856 (2017).
Manangeeswaran, M., Ireland, D. D. & Verthelyi, D. Zika (PRVABC59) infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57BL/6 mice. PLoS Pathog. 12, e1006004 (2016).
Winkler, C. W. et al. Adaptive immune responses to Zika virus are important for controlling virus infection and preventing infection in brain and testes. J. Immunol. 198, 3526–3535 (2017).
Cormier, S. A. et al. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J. Virol. 88, 9350–9360 (2014).
Shaw, A. C., Goldstein, D. R. & Montgomery, R. R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13, 875–887 (2013).
Sousa, A. Q. et al. Postmortem findings for 7 neonates with congenital Zika virus infection. Emerg. Infect. Dis. 23, 1164–1167 (2017).
Retallack, H. et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl Acad. Sci. USA 113, 14408–14413 (2016).
Dirlikov, E. et al. Guillain–Barre syndrome during ongoing Zika virus transmission—Puerto Rico, January 1–July 31, 2016. Morb. Mortal. Wkly Rep. 65, 910–914 (2016).
Yockey, L. J. et al. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166, 1247–1256.e4 (2016).
Van den Pol, A. N., Mao, G., Yang, Y., Ornaghi, S. & Davis, J. N. Zika virus targeting in the developing brain. J. Neurosci. 37, 2161–2175 (2017).
Iijima, N. & Iwasaki, A. Access of protective antiviral antibody to neuronal tissues requires CD4 T-cell help. Nature 533, 552–556 (2016).
Acknowledgements
The authors thank A.N. van den Pol for providing anti-ZIKV rat serum, and H. Dong and Y. Kumamoto for animal care and technical assistance, respectively. This study was in part supported by the National Institutes of Health (1R21AI131284 to A.I., T32GM007205 to L.J.Y. and 4T32AI007019-41 to K.A.J.). A.I. is an investigator of the Howard Hughes Medical Institute. K.A.J. is a recipient of the Burroughs Wellcome Postdoctoral Enrichment Program.
Author information
Authors and Affiliations
Contributions
K.A.J. and A.I. planned the project, designed experiments, analysed and interpreted data and wrote the manuscript. K.A.J., P.W.W., S.L. and L.J.Y. designed and performed experiments. A.J.H. assisted with histopathological analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A correction to this article is available online at https://doi.org/10.1038/s41564-017-0101-7.
Electronic supplementary material
Supplementary Information
Supplementary Figures 1–7.
Rights and permissions
About this article

Cite this article
Jurado, K.A., Yockey, L.J., Wong, P.W. et al. Antiviral CD8 T cells induce Zika-virus-associated paralysis in mice. Nat Microbiol 3, 141–147 (2018). https://doi.org/10.1038/s41564-017-0060-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-017-0060-z
This article is cited by
-
Bystander activated CD8+ T cells mediate neuropathology during viral infection via antigen-independent cytotoxicity
Nature Communications (2024)
-
In human astrocytes neurotropic flaviviruses increase autophagy, yet their replication is autophagy-independent
Cellular and Molecular Life Sciences (2022)
-
Microglia are involved in phagocytosis and extracellular digestion during Zika virus encephalitis in young adult immunodeficient mice
Journal of Neuroinflammation (2021)
-
IL-22 hinders antiviral T cell responses and exacerbates ZIKV encephalitis in immunocompetent neonatal mice
Journal of Neuroinflammation (2020)
-
Inhibition of Na+/K+ ATPase blocks Zika virus infection in mice
Communications Biology (2020)
