
Abstract
Butterflies are a diverse and charismatic insect group that are thought to have evolved with plants and dispersed throughout the world in response to key geological events. However, these hypotheses have not been extensively tested because a comprehensive phylogenetic framework and datasets for butterfly larval hosts and global distributions are lacking. We sequenced 391 genes from nearly 2,300 butterfly species, sampled from 90 countries and 28 specimen collections, to reconstruct a new phylogenomic tree of butterflies representing 92% of all genera. Our phylogeny has strong support for nearly all nodes and demonstrates that at least 36 butterfly tribes require reclassification. Divergence time analyses imply an origin ~100 million years ago for butterflies and indicate that all but one family were present before the K/Pg extinction event. We aggregated larval host datasets and global distribution records and found that butterflies are likely to have first fed on Fabaceae and originated in what is now the Americas. Soon after the Cretaceous Thermal Maximum, butterflies crossed Beringia and diversified in the Palaeotropics. Our results also reveal that most butterfly species are specialists that feed on only one larval host plant family. However, generalist butterflies that consume two or more plant families usually feed on closely related plants.
Similar content being viewed by others

Main
Butterflies have long captivated naturalists, scientists and the public, and they have played a central part in studies of speciation, community ecology, plant–insect interactions, mimicry, genetics and conservation. Despite being the most intensely studied insect group, the evolutionary history and drivers of butterfly diversification remain poorly understood1,2. Butterflies are thought to have diversified in relation to multiple abiotic and biotic factors, including adaptations to novel climates and species interactions, with caterpillar–host interactions and geographic history playing a major role3. However, these hypotheses have not been tested because a robust phylogenetic framework at the taxonomic scale that would be needed to examine their evolution has not been available. Furthermore, host plant and distribution data have largely been scattered across literature, museum collections, and local databases, limiting our ability to conduct broad, comparative macroevolutionary studies.
We sequenced 391 genes from nearly 2,300 butterfly species to reconstruct a new phylogenomic tree of butterflies representing 92% of all genera (Fig. 1 and Supplementary Fig. 1), assembled a comprehensive host association dataset and aggregated global distribution records. Using our tree, we inferred the evolutionary timing, patterns of host use, and biogeographic history of butterflies. We addressed three long-standing questions related to butterfly evolution: (1) did butterflies originate in the northern (Laurasia) or southern (Gondwana) hemisphere4; (2) what plants did the ancestor of butterflies feed on5; and (3) are host repertoires (that is, diets) of butterfly species and clades constrained by host phylogeny6,7?

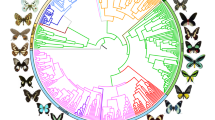
Time-calibrated tree of 2,244 butterfly species based on 391 loci and 150 amino acid partitions. Branches show distinct changes in diversification (circles) as estimated by clade-specific models. Letters at nodes refer to clades with significant rate shifts (see section 6 of Supplementary Results). Coloured lines in the outer ring beside tips indicate association with one of the 13 host modules (see section 17 of Extended Online Methods). Black lines in the host association ring indicate species without data, and asterisks denote non-monophyletic subfamilies. Supplementary Fig. 1 shows this tree with visible species names and ages for all nodes.
Results and discussion
To elucidate patterns of global butterfly diversification in space and time, we used targeted exon capture8 to assemble a dataset of 391 gene regions (161,166 nucleotides and 53,722 amino acids) from 2,244 butterfly species (Supplementary Table 1). The majority (1,914 specimens) of butterflies sampled were newly sequenced for this study, representing all families, subfamilies and tribes, and 92% of recognized genera, from 90 countries. These were obtained from 28 specimen collections across the world (see section 2 of the Extended Online Methods). Phylogenomic trees were inferred with nucleotides or translated amino acids with nine different subsets and partitioning schemes. Our trees were highly congruent, with strong support for the monophyly of all families and nearly all subfamilies with branch support metrics (SH-aLRT, ultrafast bootstrap) and multispecies coalescent species tree analyses (Supplementary Table 2). We also conducted four-cluster likelihood mapping to identify potentially conflicting signals in our datasets (Supplementary Table 3). Our results strongly support the need for revision of the classification of at least 36 butterfly tribes (27% of total) as currently circumscribed (Supplementary Table 2).
We conducted 24 dating analyses using different fossil and secondary calibration schemes along with sensitivity analyses to assess the impact of analytical and sampling bias. Across analyses, our results revealed largely congruent timing of butterfly divergence events (Supplementary Table 4). Butterflies originated from nocturnal, herbivorous moth ancestors around 101.4 million years ago (Ma) (102.5–100.0 Ma), providing evidence for a mid-Cretaceous origin of butterflies2,9.
To determine the geographic origin of butterflies, we used our dated tree (Fig. 1) to conduct a global biogeographic analysis with 15,764 newly aggregated country-level distribution records (Supplementary Table 5). Modelling with three different area categorizations, models of range evolution and parameters (adjacency matrices, time slices, etc.) consistently recovered butterflies as originating in the Americas, in what is present-day western North America or Central America (Fig. 2 and Supplementary Tables 6 and 7). All extant butterfly families excluding the Neotropical Hedylidae diversified ~10–30 Ma after the Cretaceous Thermal Maximum, ~90 Ma, when the global climate cooled by nearly 5 °C (ref. 10) (Figs. 1 and 2). During the Cretaceous, butterflies dispersed out of the Neotropics at a much higher rate than that of any other dispersal route (Supplementary Fig. 2). As new butterfly lineages became established in other bioregions, interbioregion dispersals became more frequent, particularly out of the present-day Indo-Australian Archipelago (Supplementary Figs. 3 and 4). Beginning around 60 Ma, the Neotropics served as an important bioregion with high in situ butterfly speciation (Supplementary Fig. 5), and many lineages dispersed out of this region to other areas (Supplementary Fig. 6). The relative rate of dispersal out of the Neotropics remained high during the early Cenozoic, although not as much as it was during the Cretaceous (Supplementary Figs. 2 and 3). Over the course of evolution, butterfly speciation was substantially higher in the tropics than in temperate zones (Supplementary Data 1). More dispersal events originated in the tropics (Supplementary Fig. 6), as evidenced by relative mean out-of-tropics dispersal rates from the temperate Eastern Palaearctic, and from the Neotropics to the Nearctic (Fig. 3). This pattern differs from that seen in mammals, which are thought to have dispersed primarily in the opposite direction during the Pliocene11,12,13. Our estimates of within-area dispersal rates (Supplementary Figs. 7 and 8) indicate that some butterflies, including swallowtails (Papilionidae), contradicted the general trend and dispersed into the Neotropics at high rates, corroborating previous findings14. Most dispersal events between the Neotropics and the Nearctic took place after the Eocene/Oligocene boundary, ~33.9 Ma (Supplementary Fig. 4), congruent with a previous biogeographic study15. Two lineages dispersed from the Eastern Palaearctic around 17 Ma, and these appear to be the first colonizers of Europe: ancestors of the Nymphalini subclade including Aglais, Nymphalis and Polygonia, and a clade of chequered skippers (Carcharodini; Supplementary Table 7). Butterflies were present on what are now all modern continental landmasses by the late Eocene (Supplementary Table 8).

Bioregion shading indicates the number of butterfly lineages that were associated with that bioregion during that time period, as determined by BioGeoBEARS ancestral state reconstruction. Each map corresponds to a 15-Ma interval of butterfly evolution. Results are based on data from this study.

Numbers beside each arrow are average rates from 1,000 simulations using biogeographic stochastic mapping in BioGeoBEARS. These numbers were divided by 100 for ease of comparison (raw values can be found in Supplementary Data 5). E., Eastern; W., Western.
To understand the evolution of larval host plant use, we compiled 31,456 butterfly host records from 186 books, published papers, and public and private databases (Supplementary Table 9). We found that butterfly origin and diversification lagged far behind the origin of angiosperms16,17,18, corroborating previous studies8,19. We used a recently developed network approach to create host plant modules to infer the associations of butterflies and plants6,20. Butterfly host plants include more than 80 orders and ~300 families21, rendering standard ancestral state reconstruction intractable. Our analyses provide support for Fabaceae as the larval host plant of the most recent common ancestor of butterflies (Supplementary Tables 10 and 11 and Supplementary Fig. 9), a widely accepted hypothesis5 that has lacked empirical support. The crown age of the most recent common ancestor of Fabaceae is thought to be ~98 Ma (refs. 16,18), largely coincident with the origin of butterflies.
Although most butterflies in our dataset are herbivores as larvae, a small number also feed on detritus, lichens or other insects (Supplementary Table 9). The oldest associations in the entirely entomophagous Miletinae (Lycaenidae) appear to originate by 58.4 Ma (58.9–57.1 Ma), a date that largely corresponds with an earlier estimation of the origin of this subfamily22 (Supplementary Tables 4 and 12). Lycaenidae, with caterpillars that are ancestrally symbiotic with ants8,23, date back to 64.5 Ma (65.4–63.7 Ma) (Supplementary Fig. 10), long after the origin of ants (139–158 Ma)24. Together with plants, ants appear to have provided a template for diversification of Lycaenidae and some members of its sister clade, Riodinidae. Our host database provides an important resource for future studies on butterfly feeding patterns.
We examined host plant specificity on the butterfly phylogeny (Fig. 1) and found that more than two-thirds of extant butterfly species feed on a single plant family (67.7%), whereas less than a third (32.3%) are generalists feeding on two or more (Supplementary Table 13), a pattern largely in agreement with ecological studies25. Butterflies feeding on grass and legumes (Poaceae and Fabaceae) are often host specific; the majority do not feed on plants from other families (Supplementary Table 9). These two plant families are geographically widespread and abundant in almost every ecosystem26,27, and most grasses and legumes lack potent defensive chemicals that restrict insect feeding28. These plant traits may have allowed butterflies to remain associated with these plant families for millions of years. We also found that 94.2% of generalists feed on plant families that are significantly closely related compared with a randomly sampled null distribution, suggesting that ‘generalists’, although capable of feeding on different host families, still consume closely related plants. This finding supports the pattern proposed by Ehrlich and Raven29 in which related butterflies feed on related plants.
Our study provides a robust baseline for future studies of this model insect lineage. The consistency of results obtained using different approaches for each of our analyses suggests that our conclusions are robust. Our data support the hypothesis that butterflies originated in the Americas in the late Cretaceous, 100 million years after the origin of angiosperms, and that they first fed on legumes. Butterflies dispersed from the Americas to the Eastern Palaearctic across Beringia ~75 Ma before diversifying in the Palaeotropics. Although our analyses point to a Nearctic origin, evidence for a North American versus a Central American origin is not strong, and we therefore tentatively conclude that a Laurasian origin is likely. Larval host plants played an important part in the evolution of butterflies, and some groups became host specific whereas others retained a wide host breadth. The molecular, host plant and geographic data provided here serve as a baseline for future comparative analyses of butterflies.
Methods
Taxon sampling and sequence acquisition
A total of 2,248 butterfly specimens representing 2,244 species in 1,644 genera were included for the molecular component of this study, along with ten outgroups from other lepidopteran superfamilies (Supplementary Table 1). The ingroup included genera from all families, subfamilies and tribes of butterflies according to the current classification. We aimed to include at least one species from every valid genus and sequenced the type species of each genus whenever possible. We obtained 92% of all described valid butterfly genera when the initial dataset was assembled (July 2019).
We obtained marker loci for phylogenetic analysis by (1) anchored hybrid enrichment exon capture of DNA extracts and subsequent Illumina sequencing30 or (2) bioinformatically removing these sequences from published genomes and transcriptomes. We used the BUTTERFLY1.0 probe set8 and selected a 391-locus subset that was captured reliably in at least 60% of samples. We chose this approach because it has been proven to resolve relationships of many different butterfly groups31,32,33,34. The BUTTERFLY1.0 probe set includes 13 genes (12 nuclear genes and the COI mitochondrial gene) that have been widely used in butterfly phylogenetics9,35, also termed ‘legacy genes’36, and additional protein-coding genes that may be used to address broad questions pertaining to butterfly biology, such as vision, host use and olfaction8.
Specimens were collected in 90 countries over a 70-year period by over 300 people and deposited in one of the 28 specimen collections from which we obtained tissue samples (Supplementary Table 1). We successfully captured and sequenced DNA from decades-old museum specimens37, which enabled us to include taxa that are rare or live in areas where collecting fresh material is difficult. The oldest sample was a pinned specimen collected on 22 April 1946: Dira clytus (Nymphalidae) (LEP79391). Images of 460 representative voucher specimens are shown in Supplementary Data 2, and specimen repositories are listed in Supplementary Table 1. All voucher specimens, at minimum, had their wings and genitalia retained for identification and future research.
We obtained sequence data from 343 published genomes and transcriptomes. Ten of these were outgroups representing nine moth families that are closely related to butterflies according to published studies on lepidopteran phylogeny9,38,39,40,41,42.
We extracted DNA from 1,915 specimens that were (1) stored in ethanol and frozen; (2) dried and stored in glassine envelopes under ambient conditions (papered); or (3) dried, spread and pinned in a museum collection. Locus assembly and sequence clean-up followed the pipeline of Breinholt et al.42. Published sequences comprised (1) genome assemblies, (2) genomic reads, and (3) paired or (4) single-end transcriptomes. Three sequence datasets were created for this study: a nucleotide dataset with all codon positions (nt123); a nucleotide dataset that excludes all synonymous changes (degen), created using the Perl script Degen1 v.1.4 (refs. 43,44); and an amino acid (aa) dataset translated from the nt123 dataset (Supplementary Data 3).
Phylogenetic analysis and dating
Maximum likelihood (ML) tree inference was conducted on all three datasets (nt123, degen and aa) in IQ-TREE 2.0 (ref. 45); parameter settings for each analysis can be found in Supplementary Table 14. Branch support was calculated with 1,000 ultrafast bootstrap replicates (UFBS; ‘-B 1000’ command)46,47 and Shimodaira–Hasegawa approximate likelihood ratio tests (SH-aLRT; ‘-alrt 1000’ command)48. Quartet sampling was performed on the degen359 and aa154 trees with the highest likelihood score. Four-cluster likelihood mapping analyses49 were performed on the degen and aa datasets to assess the placement of particular butterfly clades that have been the subject of previous phylogenetic studies. We applied this approach in addition to standard branch support metrics, because the latter can be subject to inflated estimates49.
We obtained divergence time estimates using a penalized-likelihood based approach implemented in treePL50. We implemented three different methods for calibrating trees and assessed similarities among results. Method 1 involved dating with secondary calibrations only. We used the 95% credibility intervals of Lepidoptera ages from Fig. S12 of Kawahara et al.38 to assign minimum and maximum ages to 27 ingroup and six outgroup nodes in our tree. Method 2 involved dating with fossils and one secondary root calibration. In this approach, we followed the guidelines of Parham et al.51 by calibrating nodes with 11 butterfly fossils that could be assigned to the geological age of a butterfly lineage with confidence as verified by de Jong52. None of the outgroup nodes could be calibrated because reliable fossils associated with our non-butterfly Lepidoptera were too young to influence deeper node ages representing multisuperfamily clades. Consequently, preliminary treePL analyses yielded highly dubious age estimates for deep nodes on the tree, hundreds of millions of years older than expected based on the literature. We therefore added a single secondary calibration to the root of the tree. Although combining secondary and fossil calibrations in a single analysis can create redundancy that negatively affects the resulting age estimates53, the limited fossil record of Lepidoptera made it a necessity to obtain comparable results derived primarily from fossils. We ran two versions of this method, each with a different root calibration. Method 2A used a maximum-age estimate of 139.4 Ma, based on the angiosperm age estimate of Smith and Brown17. Method 2B used a more conservative maximum-age estimate of 251 Ma, based on the older end of the credibility interval for the age of angiosperms in Foster et al.54. Both calibrations were used under the assumption that butterflies diverged from their moth ancestors after their most frequently used host plants, angiosperms, were already present55,56. Method 3 involved secondary calibrations and six fossils. In this approach, we combined the 33 secondary calibrations from Method 1 with six fossil calibrations, including some of the fossils used in Method 2. Fossils previously used to calibrate trees of Kawahara et al.38 were excluded from this analysis to avoid circularity and redundancy with secondary calibrations. Whenever possible, redundant fossil calibrations from Method 2 were replaced with calibrations from unrelated fossils that could be associated with a different node in the same clade.
Diversification rate analyses
We performed a Bayesian analysis of macroevolutionary mixtures using the program BAMM v.1.10.4 (ref. 57) to detect shifts in diversification rates between clades. Reversible-jump Markov chain Monte Carlo was run for 50 million generations and sampled every 50,000 generations. Priors were estimated with the R package BAMMtools v.2.1.6 (ref. 58) using the command ‘setBAMMpriors’. The tree was trimmed in Mesquite v.3.6 (ref. 59) to remove all outgroups. Six analyses were performed using different priors for expected numbers of shifts (5, 10, 20, 30, 40 and 50 shifts).
We conducted a series of analyses in HiSSE (Hidden State Speciation and Extinction) and a BiSSE-like (Binary State Speciation and Extinction) implementation of HiSSE60 in the R package hisse61 to evaluate whether there is a correlation between butterfly and plant diversification. We pruned outgroups from the aa154 dated tree (Strategy A) and compared 20 HiSSE models and BiSSE-like implementations of HiSSE. The BiSSE equivalent of HiSSE tests whether there are different diversification rates associated with the two host plant use states. Other models were built in the HiSSE framework to test alternative combinations of the presence or absence of hidden state and host plant use associations while also considering different transition rate matrices, net turnover rates, τi (speciation plus extinction: λi + μi) and extinction fractions, εi (extinction divided by speciation: μi/λi) (Supplementary Table 15). We tested whether diversification rates were linked to feeding (A) as a larval specialist or generalist (Supplementary Table 16); (B) on Poales (Supplementary Table 17) in Papilionoidea, Hesperiidae and Nymphalidae; (C) on Fabales (Supplementary Table 18) in Papilionoidea and Nymphalidae; (D) on Brassicales (Supplementary Table 19) in Papilionoidea and Pieridae; (E) on Fagales (Supplementary Table 20); (F) on the Poaceae module (Supplementary Table 21); (G) on the Fabaceae module (Supplementary Table 22); and (H) on Fabaceae in Eudaminae (Supplementary Tables 22 and 23). We compared these different models of HiSSE and BiSSE-like implementations to account for hidden states to alleviate concerns that SSE models can lead to a high incidence of false positive results62.
Fraction files of clade-based taxonomic diversity estimates were created for all HiSSE runs to account for taxonomic sampling bias (Supplementary Table 24). We set the total number of extant butterfly species as 19,500, which is an ~8% increase compared with the butterfly species richness estimate of van Nieukerken et al.63. We added this diversity correction based on many recent new butterfly species descriptions (for example, by Cong et al.64) and morphospecies that we are aware of that have not yet been formally described. We estimated the total number of generalist and specialist species by calculating the percentage of generalists and specialists in our dataset at the family level. We standardized the proportion of species richness in that family compared to all butterflies, based on diversity estimates of van Nieukerken et al.63. For example, 78.61% of all sampled Hesperiidae that had host data were specialists, and Hesperiidae comprise 21.91% of all butterfly species richness; thus, we estimated Hesperiidae specialists as 19,500 × 0.2191 × 0.7861 = 3,359 species. Applying these calculations for all families yielded totals of 12,969 specialist species and 6,531 generalist species (Supplementary Table 25); these numbers were used to estimate fractions of generalists and specialists in our dataset.
Calculating the fraction of species sampled within each host plant module proved more challenging. To estimate the true butterfly species richness for each module, we used unpublished estimates of species richness for all butterfly genera by G.L. and assumed that if a species was known to belong to a module, so would some of its congeners. These calculations were revised because some genera had large host ranges with species assigned to multiple modules. For example, the three species of Vanessa with host records in our dataset were assigned to three different modules. As there is an estimated total of 24 Vanessa species, we calculated that approximately 24/3 = 8 Vanessa species belonged in each of those modules. Calculations for all genera in all modules, and the resulting estimates of module totals and fractions sampled, are provided in Supplementary Table 26.
Biogeography
To reconstruct the biogeographic history of butterflies, we aggregated global distribution data from multiple sources to create a butterfly checklist for each country. Data sources included: (1) the Lepidoptera and Other Life Forms Database (http://ftp.funet.fi/index/Tree_of_life/insecta/lepidoptera); (2) WikiSpecies (https://species.wikimedia.org); and (3) the type locality of each species or subspecies in our list of valid butterfly names, which was obtained from 1, above. This initial global checklist was vetted using published country checklists and the ButterflyNet Trait Database65. Trait data from ca. 100 comprehensive and country-specific field guides have been entered into this database, allowing us to generate species lists to cross-validate checklists assembled66.
We designated 14 biogeographic regions across the globe (Supplementary Fig. 11 and Supplementary Table 27), determined which of these regions were occupied by each species in our tree and developed a 14-state character matrix. Six countries (Canada, China, Indonesia, Mexico, Russia, US) spanned two or three bioregions, which required manual evaluation of whether species in these countries were found in one or more of the adjoining bioregions. US and Canadian species were assigned to East and/or West Nearctic bioregions based on the palaeogeographic history of North America (that is, whether the species were east or west of the continental divide) with reference to locality records from Butterflies and Moths of North America (https://www.butterfliesandmoths.org). Russian species were assigned to Eastern and/or Western Palaearctic bioregions based on locality records assembled by the Lepidoptera and Other Life Forms Database67. Some countries did not have complete distribution lists and were thus evaluated manually by coauthors. Chinese species were assigned to Eastern Palaearctic and Oriental bioregions by H.W. Indonesian species were assigned to Oriental, Wallacean and Australian bioregions by D.J.L. and D.P. Mexican species were assigned to East Nearctic, West Nearctic and Central American bioregions by J.I.M.
The majority of butterfly species are distributed in fewer than five bioregions. Some species are more widespread, but we found that this was often due to recent anthropogenic introductions. Consequently, a final round of data cleaning was performed in which records of species found in at least five bioregions were manually verified and edited to accurately reflect true native species’ ranges. Cleaned bioregion and tropicality data were converted to character matrices to be used for subsequent distribution analyses (Supplementary Tables 28 and 29).
We estimated the ancestral area of origin and geographic range evolution for butterflies using two approaches: the ML approach of the DECX model68 as implemented in the C++ version69,70 (https://github.com/champost/DECX); and the program BioGeoBEARS v.1.1.2 (ref. 71). DECX uses a time-calibrated tree, the modern distribution of each species for a set of geographic areas and a time-stratified geographic model that is represented by connectivity matrices for specified time intervals spanning the evolutionary history of clade of interest72.
We also ran BioGeoBEARS with seven and eight areas to estimate immigration and emigration rates (Supplementary Figs. 12 and 13 and Supplementary Table 27). BioGeoBEARS could not be run with 14 states owing to the complexity of our dataset (2,248 tree tips). The seven and eight bioregions largely corresponded to the biogeographic realms defined by Udvardy73. We implemented both the Dispersal Extinction Cladogenesis (DEC)68,74 and the Likelihood equivalent of the Dispersal-Vicariance approach (DIVALIKE)75 models and different adjacency matrices (Supplementary Data 4). Both approaches gave largely consistent results, regardless of the model and parameters used (Supplementary Tables 6 and 30).
We performed biogeographic stochastic mapping to examine in situ speciation, immigration and emigration between the seven bioregions in BioGeoBEARS. We followed Li et al.76 and ran 1,000 simulations with the DEC model, and calculated relative mean dispersal rates between all permutations of bioregions (Fig. 3 and Supplementary Data 5). These mean dispersal rates represent dispersal of butterfly lineages throughout the entire evolutionary history of Papilionoidea and thus cannot reveal changes in rates over time. To look at historical biogeography of butterflies during different epochs, rates along all possible interbioregion colonization rates were calculated at specific time intervals of 5 million years (Supplementary Table 31). These relative rates were averaged to represent relevant geological time periods (Supplementary Figs. 2–4).
Larval host plant analyses
Larval host records were compiled from nine sources: (1) the Database of the World’s Lepidopteran Hostplants (HOSTS)21, which summarizes data from ~270 other sources; (2) the Lepidoptera and Other Life Forms Database (http://ftp.funet.fi/index/Tree_of_life/insecta/lepidoptera/); (3) 40 years of food plant rearing records from Costa Rica by D.H.J., W.H., and colleagues (http://janzen.sas.upenn.edu/); (4) the ButterflyNet Trait Database65, which includes host plant records from 109 butterfly field guides and other resources; (5) a comprehensive database for host records for all butterflies in Japan77; (6) a set of papers documenting the hosts of butterflies in India78,79,80,81,82,83,84; (7) a database of hosts and ant symbionts of larval Lycaenidae and Riodinidae compiled from 85 literature sources by N.E.P. and members of her laboratory; (8) a database of butterfly host records from Ecuador based on field observations and literature records compiled by K.R.W.; and (9) 88 papers from the primary literature or relevant websites (Supplementary Table 9 and Supplementary Data 6). Whenever possible, we retained the following information for each host record, if available: (1) the taxon and taxonomic authority of butterfly to the lowest available taxonomic level (family, subfamily, tribe, genus, species or subspecies); (2) the taxon and taxonomic authority of host to the lowest available taxonomic level (family, genus, species, subspecies or variety); (3) plant part eaten; (4) record certainty (novel plant accepted in captivity, oviposition record with no observation of herbivory, etc.); (5) geographic location of observation; and (6) relevant information on all non-plant hosts. The extensive data recorded in the host (food plant) database of D.H.J., W.H., and colleagues were simplified to retain the fields of butterfly genus and specific epithet, as well as plant family, genus and specific epithet, together with an indication of whether the plant was introduced to Costa Rica. This database contains many records of informal, non-ICZN-compliant names of butterfly cryptic species. Rather than discarding the large number of records that would not be compatible with any other data source, we regarded these as the nominal species (for example, Battus polydamas instead of Battus polydamasDHJ01). The number of records for each butterfly species × plant species interaction was recorded.
We examined relationships between individual butterfly species and host families that are consumed by their larvae. For these analyses, we chose the rank of plant family because it has been adopted as the standard taxonomic rank for examining host use evolution6,85. For each plant-feeding butterfly species in our tree, we quantified host plant richness and phylogenetic distance using six different metrics implemented in the R package picante v.1.8.2 (ref. 86). To calculate these metrics, we used the calibrated tree of seed plants from Smith and Brown17.
As the number of host groups in our dataset was too large for an ancestral state reconstruction (approximately 200 of the 300 known host plant families21 plus host insects), we first reduced the number of host groups by using a network analysis. The Beckett algorithm87, as implemented in the function ‘computeModules’ from the package bipartite88 in R v.3.6.2 (ref. 89), assigns plants and butterflies to modules and computes the modularity index, Q. By maximizing Q, the algorithm finds groups of butterflies and hosts that interact more with each other than with other taxa in the network. Thus, hosts that are assigned to the same module tend to be used by the same butterflies. We found 13 modules for butterfly host associations in our module analysis (Supplementary Tables 32 and 33). We then conducted three larval host ancestral state reconstruction analyses using stochastic character mapping with SIMMAP in phytools v.0.7-70 (refs. 90,91) using the ‘make.simmap’ command. We reconstructed the ancestral state of (A) generalist versus specialist feeding (two states, Supplementary Data 7); (B) plant, lichen, Hemiptera or Hymenoptera as a food source (four states, Supplementary Data 8); and (C) plant module (13 states, Supplementary Data 9).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All supplementary data archives are available on Figshare (https://doi.org/10.6084/m9.figshare.21774899). Genomic data for all newly sequenced specimens in this study have been uploaded to GenBank as part of BioProject PRJNA714105. Individual BioSample accession numbers for each specimen are provided in Supplementary Table 1.
Code availability
All new code developed to aid with the analyses in this study has been uploaded to GitHub and made publicly available. GitHub URLs for specific scripts are provided in the Methods and Extended Online Methods sections.
References
Chazot, N. et al. Priors and posteriors in Bayesian timing of divergence analyses: the age of butterflies revisited. Syst. Biol. 68, 797–813 (2019).
Allio, R. et al. Whole genome shotgun phylogenomics resolves the pattern and timing of swallowtail butterfly evolution. Syst. Biol. 69, 38–60 (2020).
Boggs, C. L., Watt, W. B. & Ehrlich, P. R. Butterflies: Ecology and Evolution Taking Flight (University of Chicago Press, 2003).
Braby, M. F., Trueman, J. W. H. & Eastwood, R. When and where did troidine butterflies (Lepidoptera: Papilionidae) evolve? Phylogenetic and biogeographic evidence suggests an origin in remnant Gondwana in the Late Cretaceous. Invertebr. Syst. 19, 113–143 (2005).
Janz, N. & Nylin, S. Butterflies and plants: a phylogenetic study. Evolution 52, 486–502 (1998).
Braga, M. P., Landis, M. J., Nylin, S., Janz, N. & Ronquist, F. Bayesian inference of ancestral host–parasite interactions under a phylogenetic model of host repertoire evolution. Syst. Biol. 69, 1149–1162 (2020).
Braga, M. P., Janz, N., Nylin, S., Ronquist, F. & Landis, M. J. Phylogenetic reconstruction of ancestral ecological networks through time for pierid butterflies and their host plants. Ecol. Lett. 24, 2134–2145 (2020).
Espeland, M. et al. A comprehensive and dated phylogenomic analysis of butterflies. Curr. Biol. 28, 770–778.e5 (2018).
Wahlberg, N., Wheat, C. W. & Peña, C. Timing and patterns in the taxonomic diversification of Lepidoptera (butterflies and moths). PLoS ONE 8, e80875 (2013).
Linnert, C. et al. Evidence for global cooling in the Late Cretaceous. Nat. Commun. 5, 4194 (2014).
Domingo, L., Tomassini, R. L., Montalvo, C. I., Sanz-Pérez, D. & Alberdi, M. T. The Great American Biotic Interchange revisited: a new perspective from the stable isotope record of Argentine Pampas fossil mammals. Sci. Rep. 10, 1608 (2020).
Carrillo, J. D. et al. Disproportionate extinction of South American mammals drove the asymmetry of the Great American Biotic Interchange. Proc. Natl Acad. Sci. USA 117, 26281–26287 (2020).
Rolland, J., Condamine, F. L., Beeravolu, C. R., Jiguet, F. & Morlon, H. Dispersal is a major driver of the latitudinal diversity gradient of Carnivora. Glob. Ecol. Biogeogr. 24, 1059–1071 (2015).
Condamine, F. L., Silva-Brandão, K. L., Kergoat, G. J. & Sperling, F. A. H. Biogeographic and diversification patterns of neotropical Troidini butterflies (Papilionidae) support a museum model of diversity dynamics for Amazonia. BMC Evol. Biol. 12, 82 (2012).
Chazot, N. et al. Conserved ancestral tropical niche but different continental histories explain the latitudinal diversity gradient in brush-footed butterflies. Nat. Commun. 12, 5717 (2021).
Magallón, S., Gómez-Acevedo, S., Sánchez-Reyes, L. L. & Hernández-Hernández, T. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. N. Phytol. 207, 437–453 (2015).
Smith, S. A. & Brown, J. W. Constructing a broadly inclusive seed plant phylogeny. Am. J. Bot. 105, 302–314 (2018).
Li, H.-T. et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5, 461–470 (2019).
Heikkilä, M., Kaila, L., Mutanen, M., Peña, C. & Wahlberg, N. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. R. Soc. B Biol. Sci. 279, 1093–1099 (2011).
Braga, M. P., Guimarães, P. R., Wheat, C. W., Nylin, S. & Janz, N. Unifying host-associated diversification processes using butterfly–plant networks. Nat. Commun. 9, 5155 (2018).
Robinson, G. S., Ackery, P. R., Kitching, I. J., Beccaloni, G. W. & Hernández, L. M. HOSTS – A Database of the World’s Lepidopteran Hostplants (accessed 1 August 2020); http://www.nhm.ac.uk/our-science/data/hostplants/
Kaliszewska, Z. A. et al. When caterpillars attack: biogeography and life history evolution of the Miletinae (Lepidoptera: Lycaenidae). Evolution 69, 571–588 (2015).
Pierce, N. E. et al. The ecology and evolution of ant association in the Lycaenidae (Lepidoptera). Annu. Rev. Entomol. 47, 733–771 (2002).
Moreau, C. S. & Bell, C. D. Testing the museum versus cradle tropical biological diversity hypothesis: phylogeny, diversification, and ancestral biogeographical range evolution of the ants. Evolution 67, 2240–2257 (2013).
Forister, M. L. et al. The global distribution of diet breadth in insect herbivores. Proc. Natl Acad. Sci. USA 112, 442 (2015).
Kelly, C. K. & Southwood, T. R. E. Species richness and resource availability: a phylogenetic analysis of insects associated with trees. Proc. Natl Acad. Sci. USA 96, 8013–8016 (1999).
Kennedy, C. E. J. & Southwood, T. R. E. The number of species of insects associated with British trees: a re-analysis. J. Anim. Ecol. 53, 455–478 (1984).
Rutz, A. et al. The LOTUS initiative for open knowledge management in natural products research. eLife 11, e70780 (2022).
Ehrlich, P. R. & Raven, P. H. Butterflies and plants: a study of coevolution. Evolution 18, 586–608 (1965).
Lemmon, A. R., Emme, S. A. & Lemmon, E. M. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst. Biol. 61, 727–744 (2012).
Valencia-Montoya, W. A. et al. Evolutionary trade-offs between male secondary sexual traits revealed by a phylogeny of the hyperdiverse tribe Eumaeini (Lepidoptera: Lycaenidae). Proc. R. Soc. B Biol. Sci. 288, 1–10 (2021).
Toussaint, E. F. A. et al. Anchored phylogenomics illuminates the skipper butterfly tree of life. BMC Evol. Biol. 18, 101 (2018).
Espeland, M. et al. Four hundred shades of brown: higher level phylogeny of the problematic Euptychiina (Lepidoptera, Nymphalidae, Satyrinae) based on hybrid enrichment data. Mol. Phylogenet. Evol. 131, 116–124 (2019).
Carvalho, A. P. S. et al. Diversification is correlated with temperature in white and sulfur butterflies. Preprint at bioRxiv https://doi.org/10.1101/2022.09.22.509088 (2022).
Kawahara, A. Y. et al. Phylogenetics of moth-like butterflies (Papilionoidea: Hedylidae) based on a new 13-locus target capture probe set. Mol. Phylogenet. Evol. 127, 600–605 (2018).
Toussaint, E. F. A. et al. Afrotropics on the wing: phylogenomics and historical biogeography of awl and policeman skippers. Syst. Entomol. 46, 172–185 (2021).
Nunes, R. et al. Predictors of sequence capture in a large-scale anchored phylogenomics project. Front. Ecol. Evol. 10, 943361 (2022).
Kawahara, A. Y. et al. Phylogenomics reveals the evolutionary timing and pattern of butterflies and moths. Proc. Natl Acad. Sci. USA 116, 22657–22663 (2019).
Regier, J. C. et al. A large-scale, higher-level, molecular phylogenetic study of the insect order Lepidoptera (moths and butterflies). PLoS ONE 8, 1–23 (2013).
Mayer, C. et al. Adding leaves to the Lepidoptera tree: capturing hundreds of nuclear genes from old museum specimens. Syst. Entomol. 46, 649–671 (2021).
Rota, J. et al. The unresolved phylogenomic tree of butterflies and moths (Lepidoptera): assessing the potential causes and consequences. Syst. Entomol. 47, 531–550 (2022).
Breinholt, J. W. et al. Resolving relationships among the megadiverse butterflies and moths with a novel pipeline for anchored phylogenomics. Syst. Biol. 67, 78–93 (2018).
Regier, J. C. et al. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 463, 1079–1083 (2010).
Zwick, A. Degeneracy coding web service. GitHub https://github.com/carlosp420/degenerate-dna (2010).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Strimmer, K. & von Haeseler, A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl Acad. Sci. USA 94, 6815 (1997).
Smith, S. A. & O’Meara, B. C. TreePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 28, 2689–2690 (2012).
Parham, J. F. et al. Best practices for justifying fossil calibrations. Syst. Biol. 61, 346–359 (2012).
de Jong, R. Fossil butterflies, calibration points and the molecular clock (Lepidoptera: Papilionoidea). Zootaxa 4270, 1–63 (2017).
Graur, D. & Martin, W. Reading the entrails of chickens: molecular timescales of evolution and the illusion of precision. Trends Genet. 20, 80–86 (2004).
Foster, C. S. et al. Evaluating the impact of genomic data and priors on Bayesian estimates of the angiosperm evolutionary timescale. Syst. Biol. 66, 338–351 (2017).
Shields, O. Fossil butterflies and the evolution of Lepidoptera. J. Res. Lepid. 15, 132–143 (1976).
Wahlberg, N. That awkward age for butterflies: insights from the age of the butterfly subfamily Nymphalinae (Lepidoptera: Nymphalidae). Syst. Biol. 55, 703–714 (2006).
Rabosky, D. L. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE 9, e89543–e89543 (2014).
Rabosky, D. L. et al. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5, 701–707 (2014).
Maddison, W. P. & Maddison, D. R. Mesquite: a modular system for evolutionary analysis. Mesquite https://www.mesquiteproject.org (2018).
Beaulieu, J. M. & O’Meara, B. C. Detecting hidden diversification shifts in models of trait-dependent speciation and extinction. Syst. Biol. 65, 583–601 (2016).
Beaulieu, J., O’Meara, B., Caetano, D., Boyko, J. & Vasconcelos, T. Package ‘hisse’. CRAN https://CRAN.R-project.org/package=hisse (2021).
Rabosky, D. L. & Goldberg, E. E. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst. Biol. 64, 340–355 (2015).
van Nieukerken, E. J. et al. Order Lepidoptera Linnaeus, 1758. In: Zhang, Z.-Q. (Ed.) Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. Zootaxa 3148, 212 (2011).
Cong, Q., Zhang, J., Shen, J. & Grishin, N. V. Fifty new genera of Hesperiidae (Lepidoptera). Insecta Mundi 2019, 0731 (2019).
Shirey, V. et al. LepTraits 1.0 A globally comprehensive dataset of butterfly traits. Sci. Data. 9, 382 (2022).
Pinkert, S., Barve, V., Guralnick, R. P. & Jetz, W. Global geographical and latitudinal variation in butterfly species richness captured through a comprehensive country‐level occurrence database. Glob. Ecol. Biogeogr. 31, 830–839 (2022).
Savela, M. Lepidoptera and some other life forms. FUNET https://www.funet.fi/pub/sci/bio/life/intro.html (2021).
Ree, R. H. & Smith, S. A. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol. 57, 4–14 (2008).
Smith, S. A. Taking into account phylogenetic and divergence-time uncertainty in a parametric biogeographical analysis of the northern hemisphere plant clade Caprifolieae. J. Biogeogr. 36, 2324–2337 (2009).
Beeravolu, C. R. & Condamine, F. L. An extended maximum likelihood inference of geographic range evolution by dispersal, local extinction and cladogenesis. Preprint at bioRxiv https://doi.org/10.1101/038695 (2016).
Matzke, N. J. BioGeoBEARS: biogeography with Bayesian (and likelihood) evolutionary analysis in R scripts (GitHub, 2018).
Allio, R. et al. Genome-wide macroevolutionary signatures of key innovations in butterflies colonizing new host plants. Nat. Commun. 12, 354 (2021).
Udvardy, M. D. F. A classification of the biogeographical provinces of the world. Morges (Switzerland): International Union of Conservation of Nature and Natural Resources. IUCN Occasional Paper no. 18 (IUCN, 1975).
Ree, R. H. Detecting the historical signature of key innovations using stochastic models of character evolution and cladogenesis. Evolution 59, 257–265 (2005).
Ronquist, F. Dispersal-vicariance analysis: a new approach to the quantification of historical biogeography. Syst. Biol. 46, 195–203 (1997).
Li, X. et al. A diversification relay race from Caribbean-Mesoamerica to the Andes: historical biogeography of Xylophanes hawkmoths. Proc. R. Soc. B Biol. Sci. 289, 20212435 (2022).
Saito, M. U., Jinbo, U., Yago, M., Kurashima, O. & Ito, M. Larval host records of butterflies in Japan. Ecol. Res. 31, 491–491 (2016).
Veenakumari, K., Mohanraj, P. & Sreekumar, P. Host plant utilization by butterfly larvae in the Andaman and Nicobar Islands (Indian Ocean). J. Insect Conserv. 1, 235–246 (1997).
Kunte, K. Additions to known larval host plants of Indian butterflies. J. Bombay Nat. Hist. Soc. 103, 119–122 (2006).
Kalesh, S. & Prakash, S. K. Additions to larval host plants of butterflies of the Western Ghats, Kerala, Southern India (Rhopalocera, Lepidoptera). Part 1. J. Bombay Nat. Hist. Soc. 104, 235–238 (2007).
Kalesh, S. & Prakash, S. K. Additions to larval host plants of butterflies of the Western Ghats, Kerala, Southern India (Rhopalocera, Lepidoptera). Part 2. J. Bombay Nat. Hist. Soc. 112, 111–113 (2015).
Naik, D. & Mustak, M. S. Additions to larval host plants of Indian butterflies (Lepidoptera). J. Bombay Nat. Hist. Soc. 112, 181–183 (2015).
Karmakar, T. et al. Early stages and larval host plants of some northeastern Indian butterflies. J. Threat. Taxa 10, 11780–11799 (2018).
Nitin, R. et al. Larval host plants of the butterflies of the Western Ghats, India. J. Threat. Taxa 10, 11495–11550 (2018).
Edger, P. P. et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl Acad. Sci. USA 112, 8362 (2015).
Kembel, S. W. et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464 (2010).
Beckett, S. J. Improved community detection in weighted bipartite networks. R. Soc. Open Sci. 3, 140536 (2016).
Dormann, C. F., Gruber, B. & Fründ, J. Introducing the bipartite package: analysing ecological networks. R News 8, 8–11 (2008).
R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2009).
Revell, L. J. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012).
Wahlberg, N. et al. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proc. R. Soc. B Biol. Sci. 272, 1577–1586 (2005).
Acknowledgements
We thank M. Kuhn and E. Mavrodiev for assembling trait data. S. Epstein, T. Girard-Ang, P. Pezzi and L. Xiao assisted with laboratory work. J. Barber, M. Brownlee, N. Chazot, S. Cinel, J. Daniels, R. Godfrey, A. Gomez, H. Gough, C. Hamilton, G. Hill, P. Houlihan, C. Huang, J. Miller, K. Miner, C. Mitter, K. Mitter, A. Renevier-Faure, J. Rubin, M. Scallion, R. Singh, Y. Sondhi and L. Wu provided specimens or helped to improve the manuscript. C. Wheat and N. Wahlberg kindly provided input when the project began. RAPiD Genomics (Gainesville, FL, USA) conducted sequencing. K. Meusemann (1KITE) provided four-cluster likelihood mapping scripts. T. Barve, K. Casarella, A. Clark, C. Couch, H. Dansby, R. Merritt, L. Nguyen and X. Zheng helped create figures. Maps in Fig. 3 and in all supplementary figures were created with mapchart.net; these maps are licensed under a Creative Commons Attribution-ShareAlike 4.0 International License (https://creativecommons.org/licenses/by-sa/4.0/). High-performance clusters at Brigham Young University, Harvard University, Smithsonian Institution, University of Florida and Zoological Research Museum Alexander Koenig provided computational support. Funding came from the US National Science Foundation (NSF) GoLife ‘ButterflyNet’ collaborative grant (DEB-1541500, 1541557, 1541560) to A.Y.K., R.P.G., D.J.L. and N.E.P. Specimen collection and preservation was funded by NSF DBI-1349345, 1601369, DEB-1557007 and IOS-1920895 (A.Y.K.), NSF DEB-1120380 (D.J.L.), grants 9285-13 and WW-227R-17 from the National Geographic Society (D.J.L.), NSF DBI-1256742 (A.Y.K. and K.R.W.), NSF DEB-0639861 (K.R.W.) and NSF SES-0750480, DEB-0447244 and DEB-9615760 (N.E.P.). M.E. was supported by the Research Council of Norway (no. 204308) and the Hintelmann Scientific Award for Zoological Systematics. F.L.C. was supported by the European Research Council under the European Union’s Horizon 2020 research and innovation programme (project GAIA, no. 851188). M.P.B. was supported by the Swedish Research Council (IPG no. 2020‐06422). R.V. was supported by the Spanish Ministry of Science and Innovation grant PID2019-107078GB-I00/AEI/10.13039/501100011033. G.T. was supported by the Spanish Ministry of Science and Innovation (grants PID2020-117739GA-I00/AEI/10.13039/501100011033 and RYC2018-025335-I). V.L. was supported by the Russian Science Foundation (grant 19-14-00202) and by the Ministry of Science and Higher Education of the Russian Federation (grant 075-15-2021-1069). M.Y. was supported by MEXT KAKENHI no. 19916010 and JSPS KAKENHI grants 13010131, 23570111, 26440207, 17K07528 and 21H02215. A.B.B.M., H.P.R. and N.O.M. were supported by CNPQ grants proc 563332/2010-7 and 304273/2014-7. We are thankful for the continuous support from the Museum of Comparative Zoology for fieldwork (Putnam Expedition Grants), and providing funds to support Open Access (Wetmore-Colles Fund).
Author information
Authors and Affiliations
Contributions
A.P.S.C., A.Y.K., C.E., C.S., D.M.P., E.A.E., E.F.A.T., F.L.C., H.L.O., J.W.B., M.E., M.P.B., P.B.F., R.A.S. and X.L. contributed analysis. A.Y.K., D.J.L., L.R., J.W.B., M.E., N.E.P. and R.P.G. conceptualized the study. A.Y.K., D.J.L., M.A.J., N.E.P., R.P.G. and W.J. acquired funding. A.Y.K., C.S., D.J.L., D.M.P., E.A.E., E.A.L., F.L.C., H.W., J.P.W.H., J.W.B., L.R., M.A.K.N., M.E., M.G.A.I., N.E.P., R.P.G., V.B., V.S. and W.A.V.-M. performed data assembly. A.Y.K., D.J.L. and N.E.P. were responsible for project administration. A.B.B.M., A.P.S.C., A.Y.K., D.J.L., E.F.A.T., G.T., H.P.R., J.I.M., J.W.B., M.E., M.Y., N.E.P., R.A.S., R.V., P.V., Y.-F.H. and Z.F.F. performed sampling. C.S., J.W.B. and M.E. were responsible for sequence workflow. A.D.W., A.Markee, A.P.S.C., A.Y.K., D.J.L., D.P., H.P.R., J.I.M., J.V.L., K.M.D., M.E., M.Y., N.O.M., R.A.S., R.M., S.C.M., T.D., Y.-L.N. and Z.F.F. identified and prepared specimens. A.Y.K., D.J.L., J.W.B., M.E., N.E.P. and R.P.G. provided supervision. D.J.L., G.L., R.P.G. and V.B. were responsible for taxonomy and curation. A.Markee, A.P.S.C., A.Y.K., D.J.L., F.L.C. and X.L. performed visualizations. A.P.S.C., A.Y.K., D.J.L. and D.M.P. wrote the original draft of the article. A.B.B.M., A.D.W., A.H., A.Markee, A.Monastyrskii, A.P.S.C., A.S., A.Y.K., C.A.M., C.E., C.J.M., C.S., D.H.J., D.J.L., D.J.M., D.M.P., D.P., D.S., E.A.E., E.A.L., E.D., E.F.A.T., F.L.C., G.L., G.N., G.T., H.A.B., H.L.O., H.P.R., H.W., I.J.K., J.A.T.B., J.D.H., J.I.M., J.P.W.H., J.V.L., J.W.B., K.A.-P., K.K., K.M., K.M.D., K.R.W., L.C., L.R., M.A.J., M.A.K.N., M.E., M.F.B., M.G.A.I., M.J.L., M.P.B., M.S., M.Y., N.B.H., N.E.P., N.O.M., N.T.H., P.A.C.B., P.B.F., P.S.P., P.V., R.A.R.C., R.A.S., R.E., R.J.G., R.L.H.S., R.M., R.P.G., R.V., S.C.C., S.C.M., S.Sáfián, S.Schröder, T.B.L., T.D., V.B., V.L., V.S., W.-P.C., W.A.V.-M., W.H., W.J., X.L., Y.-F.H., Y.-L.N., Y.I. and Z.F.F. reviewed and edited the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Ecology & Evolution thanks Nicolas Chazot and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Extended Online Methods, Supplementary Results and Figs. 1–26.
Supplementary Tables
Supplementary Tables 1–52.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kawahara, A.Y., Storer, C., Carvalho, A.P.S. et al. A global phylogeny of butterflies reveals their evolutionary history, ancestral hosts and biogeographic origins. Nat Ecol Evol 7, 903–913 (2023). https://doi.org/10.1038/s41559-023-02041-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-023-02041-9
This article is cited by
-
COI Barcodes combined with multilocus data for representative Aporia taxa shed light on speciation in the high altitude Irano-Turanian mountain plateaus (Lepidoptera: Pieridae)
BMC Ecology and Evolution (2024)
-
The angiosperm radiation played a dual role in the diversification of insects and insect pollinators
Nature Communications (2024)
-
A trans-oceanic flight of over 4,200 km by painted lady butterflies
Nature Communications (2024)
-
Global biogeographic regions for ants have complex relationships with those for plants and tetrapods
Nature Communications (2024)
-
Eversible tentacle organs in caterpillar–ant communication: do they signal partner quality in polyommatinae lycaenid butterflies?
Insectes Sociaux (2024)
