
Abstract
Bats are the longest-lived mammals, given their body size. However, the underlying molecular mechanisms of their extended healthspans are poorly understood. To address this question we carried out an eight-year longitudinal study of ageing in long-lived bats (Myotis myotis). We deep-sequenced ~1.7 trillion base pairs of RNA from 150 blood samples collected from known aged bats to ascertain the age-related transcriptomic shifts and potential microRNA-directed regulation that occurred. We also compared ageing transcriptomic profiles between bats and other mammals by analysis of 298 longitudinal RNA sequencing datasets. Bats did not show the same transcriptomic changes with age as commonly observed in humans and other mammals, but rather exhibited a unique, age-related gene expression pattern associated with DNA repair, autophagy, immunity and tumour suppression that may drive their extended healthspans. We show that bats have naturally evolved transcriptomic signatures that are known to extend lifespan in model organisms, and identify novel genes not yet implicated in healthy ageing. We further show that bats’ longevity profiles are partially regulated by microRNA, thus providing novel regulatory targets and pathways for future ageing intervention studies. These results further disentangle the ageing process by highlighting which ageing pathways contribute most to healthy ageing in mammals.
Similar content being viewed by others

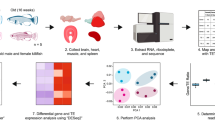

Main
Ageing is the leading risk factor for major life-threatening conditions such as cancer, neurodegeneration and cardiovascular disorders1. Despite centuries of study, the complexity of the ageing process has hampered our understanding of what drives ageing, with multiple theories of how and why we age but little consensus2,3. Given that the predicted increase in human lifespan (50% increase in people aged >60 years, 340% increase in people aged >80 years by 2050)4 is not associated with a similar increase in healthspan, we urgently need to understand ageing to relieve its maladies5. Most ageing studies have been carried out in laboratory model species, as these are easier to manipulate and house and have shorter lifespans6,7. Although substantial progress has been made in extending lifespan and healthspan in these short-lived model organisms, there is limited evidence that these approaches will be effective in more long-lived species such as humans5. An alternative approach is to explore ageing in species that are even more ‘ageing-resistant’ than humans and have naturally evolved longer healthspans6,7,8,9.
By far the most successful mammals in this regard are bats. Bats account for 20% of all living mammals, have evolved self-powered flight and have exceptional longevity10. Nineteen species of mammals live longer than humans, given their body size, of which 18 are bats6. Specifically, some wild individuals of the ~7-g Brandt’s bat (Myotis brandtii) can live >41 years, over ten times longer than expected for their body size (humans live only five times longer than expected), showing little signs of ageing6,7. Logistically it is difficult to study bats in an ageing context, as most are only found in the wild and not easily maintained in captivity11. Initial longitudinal ageing studies suggest that the longest-lived genus (Myotis) maintains the length of their telomeres with age without developing cancer12 and do not show an increased level of mitochondrial damage as expected given their metabolic rate13. This is potentially due to adaptations in their DNA repair and maintenance mechanisms13. However, to date, longitudinal ageing studies in bats have focused on only one ageing pathway or process at a time—for example, telomeres12, mitochondria13, microbiome14 or metabolome11. As ageing is an inherently complex process, a better approach is to study the interplay of multiple biological pathways to uncover the molecular mechanisms of healthy ageing evolved in bats. Comparative, population-level transcriptomics provides an alternative means that can potentially capture a full repertoire of RNA species and reveal subtle age-related transcriptional variation across individuals.
Recent transcriptome studies have catalogued signatures of ageing in model organisms and reveal a remarkable conservation of age-related pathways15,16,17. These molecular signatures illustrate a combination of degenerative phenotypes, notably reflected by low-grade, chronic inflammation, dysregulation of metabolic activities and insufficient cellular maintenance16,18. By contrast, new evidence has shown that long-lived mammals, including bats, may harbour species-specific transcriptional signatures that are beneficial for the maintenance of cellular functions into old age19,20,21,22. Here, focusing on a unique, longitudinal capture–mark–recapture study of a wild population of long-lived Myotis myotis bats (maximum lifespan, 37.1 years)12, we performed a systems-level comparative analysis (Fig. 1a) to ascertain the age-related transcriptional changes and microRNA (miRNA)-directed regulation that may underlie bats’ exceptional longevity.

a, Schematic drawing of the data analyses. b, Evaluation of the transcriptome complexity. Cumulative percentage of total transcriptome output was contributed by transcripts that were sorted from most to least based on expression values (n = 48,749). Lines represent average values across samples of the same age cohort, and the more lightly shaded adjacent regions reflect 95% confidence intervals. The intersection between the cumulative lines and dashed line indicates the overall contribution of the top 100 highly expressed transcripts across cohorts. c, Evaluation of gene expression variation. Residual variance represents the contribution from uncharacterized variables. d, Pairwise differential gene expression analyses across age cohorts. The values in each square represesnt the number of differentially expressed genes. Red (upper triangular matrix) indicates up-regulation while blue (lower triangular matrix) indicates down-regulation. GO, gene ontology.
Results and discussion
Overview of M. myotis ageing transcriptomes
Using a non-lethal sampling process developed to maximize transcript representation from bat blood (>60% of all protein-coding genes represented)23, we deep-sequenced ~1.7 trillion base pairs of RNA from 100 bat blood samples (69.6 ± 9.0 s.d. million reads per sample) using Illumina RNA-sequencing (RNA-Seq). These blood samples (~50–200 μl) were collected from 70 individual bats ranging in age from 0 to >7 years (for example, first caught as an adult 6 years before subsequent recapture) at five colonies in Brittany, France (Supplementary Tables 1–3 and Supplementary Fig. 1). The majority of the raw reads (98.5%) showed high quality (>Q30). On average, 77% of clean reads were successfully mapped to the reference genome (Myotis lucifugus), 64.2% of which had unique mapping coordinates. Of all mapped reads, 88.1% were concordantly aligned. Only those reads that were uniquely and concordantly mapped were used for downstream analyses. To gain an overview of the bat blood transcriptome, we developed a comprehensive pipeline (see Methods) to assemble and annotate full-length transcripts from 100 RNA-Seq samples, which resulted in 31,460 protein-coding transcripts (corresponding to 12,263 protein-coding genes), 10,775 long non-coding RNAs (lncRNAs) and 6,514 miscellaneous RNAs (miscRNAs) (Supplementary Fig. 2). More details regarding samples, transcriptome assembly and annotation are described in Methods and Supplementary information.
Similar to humans24, the M. myotis blood transcriptome was dominated by a few highly abundant transcripts, with ~75% of the overall transcriptional output derived from the top 100 most highly expressed transcripts (Fig. 1b). Interestingly, these dominant transcripts were enriched in several cellular maintenance bioprocesses, such as autophagy and DNA repair (Fig. 1b). This high level of cellular maintenance expression profile was not observed in human or mouse blood, based on a comparative cross-sectional analysis (Supplementary Fig. 3; see also Methods). From a global perspective, transcript correlation analyses showed high concordance across samples (average Spearman’s rank correlation coefficient = 0.89, Supplementary Fig. 4), and gene expression variation analyses indicated that ‘age’ accounted for only 8.7% of total variance (Fig. 1c), suggesting that the age-related transcriptomic changes in M. myotis are subtle. This percentage of age-related variance is comparable to that of humans (7.8) but lower than that of short-lived mice (12.8; see Methods). Consistent with the long-lived naked mole rat20, only a small proportion of genes showed differential expression (false discovery rate (FDR) < 0.05) with increasing age (adults aged 1 to >7 years) (Fig. 1d), suggesting that maintenance of transcriptomic profiles may be crucial for longevity in mammals.
A transcriptomic signature of ageing in bats
Next, we identified 6,692 age-associated genes that have been tagged at least once as differentially expressed between any two age cohorts (Supplementary Table 4). We performed weighted gene co-expression network analyses (WGCNA) on this dataset and identified seven gene expression modules that exhibited distinct ageing patterns in M. myotis (Fig. 2a–c, Supplementary Table 5 and Supplementary information). M. myotis bats showed canonical age-related transcriptomic signatures shared with other mammals16, such as age-related declines in the expression of genes associated with the adaptive immune response and mitochondrial activity (M4 and M5; Fig. 2b). In contrast, M. myotis showed an up-regulation of pathways involved in DNA damage signalling and DNA repair with age (M1–M3; Fig. 2a). These pathways were interconnected, suggesting an interplay between DNA damage sensing, signalling, repair and replication, a network that could maintain genomic stability in M. myotis (Fig. 2a). In particular, we conducted a network analysis using all the genes (n = 107) enriched in DNA repair (GO: 0006281) that exhibited a positive correlation with age in M. myotis. Based on various sources of evidence (see Methods), the majority of genes (95 out of 107) were functionally related (Fig. 3a). Remarkably, we found that a number of genes directly involved in several mammalian DNA repair pathways (for example, nucleotide excision repair, mismatch repair, base excision repair and double-strand break repair) were strongly enriched and connected in the network (Fig. 3b). This suggests that M. myotis bats may have evolved an elaborate DNA repair machinery to maintain genome stability. This age-related increase in DNA maintenance could underlie the low level of cancer incidence reported in bats12. An alternative explanation is that the enhancement of DNA repair pathways could be a response to cumulative DNA damage over time. However, our earlier longitudinal studies suggest that bats do not experience increased levels of DNA damage with age12,13 as compared to other mammals. Further estimation of the level of DNA damage experienced with ageing by bats is required to test these hypotheses.

a–c, Gene expression modules showed positive (M1–M3; a), negative (M4, M5; b) and no correlation (M6, M7; c) with age. The networks indicate the enriched gene ontology terms and their connectivity in the modules under a–c, respectively. In each module, n indicates the number of genes in the module. The expression regression line was generated using the Loess curve-fitting method, and P values indicate the significance of Spearman’s rank correlation coefficient between the eigengene and age. The eigengene is a central gene whose expression pattern can represent the whole module. SSUrRNA, small subunit ribosomal RNA.

a, The gene product network. The network included 107 genes (nodes) enriched in DNA repair that exhibited a positive correlation with age in M. myotis. The thickness of edges indicates the strength of data support (see Methods). Coloured nodes indicate their engagement in different DNA repair pathways. b, The numbers of genes enriched in different mammalian DNA repair pathways. KEGG, Kyoto Encyclopedia of Genes and Genomes.
In our previous study we speculated that telomeres, which did not significantly shorten with age, may be maintained through the mechanism of alternative lengthening of telomeres (ALT) in M. myotis12. This hypothesis is strongly supported by up-regulation of large proportions of genes directly involved in ALT (for example, MRE11A, PCNA, BLM, DNA2, MND1 and WRN) and inactivation of TERT over age, as observed in this study (Supplementary Data 1). An age-related increase in the expression of genes involved in cell cycle arrest was also observed in M. myotis. Induction of cell cycle control may represent a universal tumour-suppressant mechanism in mammals, but one that results in cellular senescence25,26. Interestingly, M. myotis exhibited enhanced autophagy activity with advancing age (Fig. 2a), which may potentially eliminate cumulative damaged organelles and protein aggregates and thereby promote cell survival.
We did not detect up-regulation of any enriched gene ontology terms associated with chronic inflammatory responses with advancing age (M6 and M7; Fig. 2c). This is unusual given that increased inflammation is a typical hallmark of ageing in mammals1,27. However, this supports the hypothesis that bats have evolved unique immune systems that enable them to dampen the constant sterile inflammation they experience from their high metabolic rates25,26. This may in turn limit inflammation-driven ageing and also underlie their apparent tolerance of pathogens (for example, those responsible for Ebola, rabies and severe acute respiratory syndrome)28,29.
Comparison of longitudinal age-related signatures across mammals
To ascertain whether these age-related transcriptomic changes are unique to bats, we conducted comparative transcriptomic analyses between M. myotis and three other mammals, Homo sapiens (human, aged ~25–75 years, n = 147), Mus musculus (mouse, aged ~0.2–2.5 years, n = 25) and Canis lupus (wolf, aged ~1–9 years, n = 26) (see Methods). When we focused on the top 200 genes that exhibited the strongest correlation with age, each species displayed unique age-correlated pathways (Fig. 4a). These top age-correlated genes found in M. myotis are mainly enriched in the maintenance of genome stability (for example, DZIP3 and PLK4) and anti-cancer activity (for example, WDR12 and WRAP53) (Fig. 4b and Supplementary Data 2). The majority of these have not been associated directly with ageing in humans or model species (Fig. 4b). However, many of these genes do interact upstream or downstream with known human ageing-associated genes, as curated in the GenAge database (Supplementary Table 6). Although age-related immunosenescence and reduced mitochondrial activity did occur in the blood transcriptome of all four mammal species, the pathways associated with genome maintenance and cell cycle regulation had higher positive correlations with age in bats (Fig. 4c and Supplementary Data 3). These pathways were down-regulated in the other three mammals (Fig. 4c). Despite being of similar size to a bat, mice uniquely exhibited increased nutrient sensing and immuno-inflammatory gene expression profiles during ageing, which have been shown to accelerate the ageing process16. Uniquely seen in bats, our results indicate that increased genome maintenance and cell cycle regulation may represent an evolutionary adaptation that enables bats to achieve exceptional longevity.

a, Distribution of Spearman’s correlation coefficient between gene expression and age across the four species. Gene ontology terms were enriched for the top 100 genes (both upper and lower). b, Top 20 genes that exhibited the strongest positive and negative correlation with age in M. myotis. c, Comparison of the pathway expression pattern with age across the four species. Within each species, the median z-scores of all genes under each of enriched age-associated gene ontology terms are used to represent their overall expression pattern with age. The z-scores were converted from Spearman’s correlation coefficients. The values following the gene ontology terms indicate the number of genes enriched.
Longevity signatures evolved naturally in long-lived bats
Research on the biology of ageing has discovered a wealth of genes that can be transcriptionally modified to prevent age-related diseases and promote longevity in model organisms30. We therefore investigated 207 human ageing-associated genes that are curated in the GenAge database31 (see Methods) and compared their correlation with age across humans, mice, bats and wolves to ascertain whether bats have naturally evolved known ‘life-extension’ expression profiles. A large body of these candidate genes showed contrasting expression changes in M. myotis compared with the other mammals (Supplementary Data 4). In total, 23 genes had the opposite direction of expression with age in M. myotis compared to each of the other three mammals (Fig. 5a), suggesting a role in driving bat longevity. Intriguingly, the expression changes of certain candidate genes occurring naturally in M. myotis during ageing were observed to extend lifespan in model organisms when modulated. For example, PTEN (Spearman’s rs = 0.395, P = 0.002; Fig. 5b) is a well-established tumour suppressor involved in DNA damage repair and cell cycle arrest32. Transgenic mice carrying an extra PTEN dosage displayed enhanced protection from cancer and presented a modest (16%) extension of lifespan33. Also, overexpression of SIRT1, GCLM and BUB1B (Fig. 5b) was verified as prolonging mean or maximum lifespan in fruit flies and mice via in vivo functional assays34,35,36.

a, Of 207 human ageing-associated genes, those that exhibited the opposite direction of expression changes with age in bat compared to human, mouse and wolf are shown. b, Examples of gene expression patterns during ageing in M. myotis that were observed to extend lifespan in model species. Spearman’s correlation coefficients with age are presented for all four species.
Conversely, repression of IKKB and MYC, as seen in bats, increased longevity and enhanced healthspan in mice37,38. In particular, MYC has been shown to be evolving under divergent selection in bat lineages12. MYC is a highly conserved proto-oncogene that is critically engaged in many essential cellular processes39. Despite its importance in maintaining cell function, its overexpression has been documented in a variety of cancers40. In humans, the product of MYC directly activates telomerase by inducing transcription of telomerase reverse transcriptase (TERT)41, whose overexpression is detected in ~90% of malignant cancer types40. This co-expression probably occurred also in M. myotis, as supported by the high expression correlation between MYC and TERT (Spearman’s rs = 0.305, P = 0.017) according to our analysis. Being under selective pressure exclusively along the bat ancestral branch, down-regulation of MYC, coupled with its synergistic effect on TERT inactivation, may underlie the long lifespan and low cancer incidence observed in M. myotis, despite its small size and high metabolic rate.
These results suggest that study of the ageing transcriptome in wild, long-lived bats can uncover molecular mechanisms that underlie their increased healthspans, and that the novel candidates identified (for example, TGIF2, WRAP53 and IPP2; Fig. 4b and Supplementary Data 2) are suitable targets for future ageing intervention studies.
MiRNA regulation of longevity signatures in bats
To ascertain the molecular mechanisms that may underlie these transcriptional signatures of longevity, we sequenced 50 blood miRNomes from the same population (Supplementary Table 7 and Supplementary information); 98% of the miRNA sequences were obtained from the same blood samples used for messenger RNA-Seq (Supplementary Table 1). Principle component analysis (PCA) of the miRNA expression profile (Fig. 6a) revealed a striking similarity to that of the mRNA transcriptome (Supplementary Fig. 5), with a noticeable shift between juvenile and adult. However, expression analyses demonstrated that a major change also occurred between 4- and 5-year-old cohorts, with most up-regulated miRNAs acting as tumour suppressors (for example, miR-146a/b, miR-30a) and down-regulated miRNAs promoting cell cycle or carcinogenesis (for example, miR-18a, miR-29c; Fig. 6b and Supplementary Fig. 6). miR-146a, which plays a suppressive role in prostate and lung cancer by inhibiting cell proliferation and migration42, was up-regulated 2.27-fold (P < 0.001) while miR-18a, used as a sensitive screening biomarker in a wide variety of human cancers43, was down-regulated 2.08-fold (P < 0.001). Although currently no direct evidence of lifespan extension was observed in model organisms by genetic modification of any of these differentially expressed miRNAs, some (for example, miR-146a and miR-18a) did show enhanced protection from various cancer types and improved health in humans42,43. No miRNAs were differentially expressed amongst the older age cohorts (5, 6 and 7+ years; Supplementary Fig. 6), suggesting that this anti-cancer miRNA expression profile is maintained into old age in M. myotis.

a, PCA based on 117 miRNA expression data. b, Cluster analysis of 117 miRNAs. The miRNAs in red indicate their differential expression between 4 and 5 years of age (red, up-regulation; black, down-regulation). c, The regulatory network between miRNAs and their top targeted biological pathways. The connectivity indicates that certain miRNAs regulate genes that are grouped under particular gene ontology terms. The top ten miRNAs with the highest numbers of targets are highlighted. d, Examples of predicted miRNA–mRNA pairs that were negatively correlated. These mRNAs are mainly involved in DNA repair and cell cycle regulation. Both miRNA and mRNA expression values were normalized using the TMM method and were further log2-transformed. P values indicate the significance of negative correlation. The grey shading reflects the 95% confidence interval. ROS, reactive oxygen species.
Next, we established a gene regulatory network to gauge the impact of miRNA on gene expression. The miRNA–mRNA interactions were determined by in silico miRNA target prediction44 and were further validated by confirming a negative correlation between expression of the predicted miRNA–mRNA pair (see Methods and Supplementary information). Collectively, 10,108 miRNA–mRNA interactions were established between 117 known miRNAs and 7,051 mRNAs, only 9.45% of which (955 out of 10,108) exhibited significant negative correlation (FDR < 0.1). The enrichment analysis illustrated that miRNA mediated a wide spectrum of bioprocesses, encompassing nutrient sensing, immunity and cellular maintenance pathways (Fig. 6c and Supplementary Fig. 7). In particular, we noticed that a large number of genes associated with cell cycle regulation and DNA repair were mediated by miRNA (Fig. 6d and Supplementary Table 8). For example, the up-regulation of EP300, a gene involved in DNA repair, correlated with the down-regulation of miR-148b; on the other hand, SHC1 and MTOR, genes that induce cell growth and proliferation, appeared to be progressively suppressed by increased expression of miR-30a and miR-330 with advancing age. These results are suggestive of the prominent roles of miRNA in the regulation of ageing pathways that may underpin the extreme longevity observed in M. myotis. As these candidates are suggested on the basis of in silico analyses, future functional assays are necessary to confirm their roles in bat longevity.
This unique longitudinal study of ageing in a wild population of long-lived bats revealed that M. myotis did not show the same transcriptomic changes with age commonly observed in other mammals. Their unique ageing-transcriptomic shifts suggest that the regulation of genes associated with DNA repair, autophagy, immunity and tumour suppression underlies their extraordinary longevity. Our results show that bats have naturally evolved transcriptomic signatures that are known to extend lifespan in model organisms, and also identify potential targets for future ageing intervention studies. The regulatory network uncovered suggests that these pathways are partially mediated by miRNA and provide a potential molecular mechanism that underlies their longevity-associated transcriptomic signature. Using cutting-edge molecular techniques coupled with longitudinal field studies, we used the diversity within nature to identify key targets and regions that may regulate and control extraordinary ageing in mammals. These results will drive a better understanding of the ageing process and can provide molecular targets for future ageing intervention studies, highlighting a new role for longitudinal field studies of non-model organisms.
Methods
Sampling
Bat capture and blood sampling were implemented in accordance with the permits and ethical guidelines issued by ‘Arrêté’ by the Préfet du Morbihan and the University College Dublin ethics committee. The sampling procedures are exhaustively described in ref. 23. Briefly, female M. myotis bats, aged 0 to 7+ years (for example, first marked with transponders as adult 6 years before subsequent recapture), were captured in Brittany, France. These individuals have been marked with unique transponders since 2010, and the initial ages of bats when first captured were determined by examining the epiphyseal cartilage in their finger bones. Individuals were recorded as juvenile (0 years old) if the epiphyseal plates in their finger bones were open; otherwise, they were recorded as adult (1+ years old, true age unknown). Each year, individuals recaptured were identified by their transponder number and new captures (juvenile and adult) were transponded. For each individual, a volume of ~50–200 μl of blood, depending on the weight of the individual, was collected from the uropatagial vein using a sterile needle (26 gauge). Blood samples were pipetted into cryotubes (2 ml, Nalgene labware) and immediately flash-frozen in liquid nitrogen. Haemostatic gel (Bloxang, Bausch & Lomb) was applied to the puncture to prevent further bleeding. The bats were rapidly released after being offered water and food. Blood samples were maintained at −150 °C for long-term storage before RNA extraction.
RNA extraction
Total RNA was extracted from whole blood using a RNAzol BD kit (No. RB192, Molecular Research Center, Inc.), following the manufacturer’s instructions with minor modifications. RNA extraction is described in ref. 23. The quantity and quality of RNA were assessed using a NanoDrop Spectrophotometer (Thermo Scientific) and a Bioanalyzer 2100 (Aligent Technologies), respectively. All samples that met the criteria of having >2 μg of total RNA and an RNA Integrity Number score >8.0 were chosen for mRNA-Seq Illumina sequencing library preparation. For small RNA library preparation, 3 μg of total RNA per sample was required.
Library preparation and Illumina sequencing
For both RNA-Seq and small RNA library preparation, all qualified RNA samples were initially purified using a Turbo DNA-free kit (No. AM1907, Ambion) to remove residual DNA. The Globin-Zero Gold rRNA Removal kit (Epicentre Illumina) was further employed to deplete unwanted rRNA and globin mRNA. Nevertheless, this kit was specifically designed for human and mouse, and performed less effectively on other species. A large amount of ribosomal RNA (mostly 7 s rRNA) still remained after depletion based on the MiSeq result (Fasteris SA). Therefore, an additional step of 7 s rRNA depletion was carried out (Fasteris SA). The probes specifically designed for 7 s rRNA removal are 5′-TCCTTAGGCAACCTGGTGG-3′, 5′-GGGAGGTCACCATATTGATG-3′ and 5′-GGCAACCTGGTGGCCCCCCGCTCCCGGGAGG-3′. To assess the efficiency of the 7 s rRNA probes, we constructed two libraries with and without 7 s rRNA depletion from the same sample, HWJ-8. The MiSeq titration run demonstrated that the percentage of short reads that mapped to 7 s rRNA reduced from 31.65 to 17.78 after depletion (Supplementary Fig. 8), implying that the 7 s rRNA depletion step performed effectively.
RNA-Seq libraries were prepared using the TruSeq stranded mRNA kit (Illumina), following the manufacturer’s protocols. Sequencing was performed on Illumina HiSeq 2500 platforms, with the sequencing depth to a minimum of 50 million and 125-bp paired-end reads per sample. Small RNA libraries were constructed using the TruSeq small RNA kit (Illumina) and were further sequenced on Illumina HiSeq 2500 platforms, to generate a minimum of 8 million 50-bp single-end reads per sample.
Summary of sequenced samples
We deep-sequenced 100 transcriptomes and 50 miRNomes from 150 blood samples which were collected from 71 female M. myotis bats (Supplementary Table 1). These individuals were caught in five colonies in Brittany between 2013 and 2016 (Supplementary Table 2). Among these individuals, two were caught four times in consecutive years, two were caught three times and 20 were captured twice, with the remainder being caught only once. Of all samples, 49 had the transcriptome and corresponding miRNome sequenced simultaneously (Supplementary Table 1). The statistics derived from these samples are summarized in Supplementary Tables 3 and 7.
Individual transcriptome assembly
The quality control pipeline is extensively described in ref. 23. To enable a rapid genome-mapping process required for the referenced-based assembly, the identical quality control reads, which consume large amounts of computational resources but make little contribution to the complexity of transcriptome assembly, were removed using FastUniq45, leaving only one pair represented. These adaptor-free, high-quality and non-redundant paired-end reads were further used as inputs for assembly. We employed both de novo and reference-based methods to assemble individual transcriptomes, and the optimal strategy was selected on the basis of the assembly quality. For the reference-based method, since the M. myotis genome is not yet available, the M. lucifugus genome was used as a reference as this is the phylogenetically closest, well-assembled genome available. However, due to the genetic divergence (~30 million years) between these two species46, we carried out a series of tests to determine the optimal number of mismatches per alignment for the genome-mapping step. In brief, the filtered RNA-Seq data were aligned to the M. lucifugus genome (MyoLuc 2.0) using Tophat2 (v.2.1.0)47, with different numbers of mismatches (~2–10). For all RNA-Seq samples, the mapping rates were categorized into different groups based on the number of mismatches, and Student’s t-tests (all data followed normal distribution, Shapiro–Wilk test, P > 0.05) were conducted between groups N2 and N3, groups N3 and N4 and so on, up to groups N9 and N10. Group N6 (six mismatches per alignment) was selected as the optimal mismatch number, because there were no significant differences observed between groups N6 and N7 based on the mapping rates (P > 0.05) (Supplementary Fig. 9). To exclude any ambiguous mappings, we retained only the unique and concordant alignments using Samtools48. The resulting BAM file was further used to reconstruct transcripts by Cufflinks (v.2.2.1)49, with the parameters –GTF–guide and –F 0.1. –GTF–guide allows the assembly of unannotated regions in the genome, while –F 0.1 removes dubious isoforms with extremely low expression. The final assembly was determined by keeping those transcripts with fragment per kilobase of transcript per million (FPKM) mapped reads higher than zero. De novo assembly was carried out using Trinity (v.2.1.1)50 in the strand-specific and paired-end mode with the default parameters. For each sample, the quality of the reference-based and de novo assemblies was assessed using CEGMA (v.2.5)51. On average, the completeness of the reference-based assembly was 15% higher than that of the de novo assembly (Supplementary Fig. 10). Given their better quality, the reference-based assemblies were used to construct the ‘super’ transcriptome reference.
‘Super’ transcriptome assembly and annotation
The reference-based assemblies from all 100 samples were merged using Cuffmerge (v.2.2.1)49. The parameter —min-isoform-fraction 0.5 was used to discard the unreliable isoforms poorly supported by low sequencing coverage. The redundancy of the merged assembly was removed using CD-HIT52 with a sequence identity threshold of 95% (-c 0.95), and FrameDP53 was employed to correct the misassembled transcripts with unexpected ‘indels’ or stop codons.
Functional characterization of the super transcriptome assembly was carried out using the following pipeline. Briefly, all transcripts were categorized into three groups: protein-coding RNA, lncRNA and miscRNA. To annotate the protein-coding transcripts, the open reading frame (ORF) of each transcript was predicted using FrameDP53. The transcripts with potential ORFs were queried against the Uniprot and Nr databases using BLASTX54, with an E-value <10−6. The transcripts, with the effective hits which shared at least 60% similarity and 70% sequence coverage with the entries, were accordingly annotated as protein-coding transcripts. The ORF potential transcripts with no effective hits in either database were regarded as miscRNA. lncRNA were predicted from the transcripts with no obvious ORFs defined by FrameDP53. Firstly, these transcripts were compared to the Pfam database55 to search for conserved domains using BLASTX54 with an E-value <10−6. Those transcripts that contained or overlapped with any conserved motifs were excluded. In addition, those transcripts with coding potential scores >0.3, as evaluated by CPAT56, were also discarded. The threshold of the coding potential score is suggested in ref. 56. The final set of lncRNAs was determined by removing those filtered transcripts with lengths shorter than 200 bp. The remainder of the uncharacterized transcripts were considered as miscRNA.
Transcript expression analyses
For each RNA-Seq sample, adaptor-free, high-quality reads (without redundancy removal) were used to quantify the reference transcripts using Salmon (v.0.9)57 with the parameters –ISF and –F 31, indicating the library type (I, first-forward; S, strand-specific; F, paired-end) and k-mer length, respectively. Before expression analysis, RNA-Seq samples with a CEGMA index of the reference-based assembly <0.85, were removed. A transcript was considered expressed if it had a cumulative raw read count >100 across all samples. This led to a matrix containing expression data of 48,749 transcripts across 88 samples.
To examine the complexity of the bat blood transcriptome, the contribution of each transcript to total transcriptional output was measured in each sample. To achieve this, all transcript per million (TPM) values from each sample were sorted from most to least and their relative fractions were calculated by dividing them by the sum of all TPM values respectively. A functional enrichment analysis of the top 100 highly expressed transcripts was performed using Metascape58. We also explored the global transcriptomic similarity across 88 samples. Based on the expression data of 48,749 transcripts, pairwise Spearman’s rank correlation coefficient tests were carried out using the R package cor (v.3.0). Before the analysis, TPM values were log2-transformed (log2 (TPM + 1)). The average linkage method was applied to all correlation tests.
Gene expression variance analyses
In this study, we focused largely on gene-level analysis and investigated only 31,460 protein-coding transcripts, as most lncRNAs and miscRNAs are currently functionally unknown. To obtain gene expression estimates, raw expression counts of the transcripts which had the same BLAST hits in the Uniprot or Nr databases were accordingly aggregated to the gene-level counts using the Bioconductor R package tximport59. Thus, 31,460 protein-coding transcripts corresponded to 12,263 protein-coding genes. All downstream analyses were conducted based on these 12,263 genes.
For the age cohort analyses, samples with unambiguous ages (0, 1, 2, 3, 4, 5, 6) were employed, together with 7+ regarded as the oldest age cohort (Supplementary Fig. 1a). Gene expression counts (Supplementary Data 5) were trimmed mean of M-values (TMM)-normalized, log2-transformed and further converted to z-scores. A linear mixed model (LMM) was used to evaluate the contribution of potential variables (see below) to gene expression variation. Normalized gene counts (n = 12,263) were considered as dependent variables, whereas age was considered as an explanatory variable together with other variables including the individual, colony, year of capture and sequencing batch effect. With the exception of age being modelled as a fixed effect, all other variables were modelled as random effects. The LMM was implemented using the Bioconductor R package variancePartition60 with the following formula:
Gene co-expression network analyses
To identify gene expression changes with age, pairwise differential gene expression analyses were performed using the R packages DESeq2 (ref. 61) and EdgeR62. For both methods, FDR < 0.05 and an absolute value of log2 (fold change) > 0.5 were used to define differentially expressed genes. To reduce the rate of false-negatives and obtain a wide range of age-related candidates for pattern analysis, we maximized the number of differentially expressed genes from both methods. All network analyses were implemented in R. Gene co-expression analysis was performed using the R package WGCNA (v.1.63)63. Genes that exhibited no differential expression between any pairs of age cohorts were excluded from this analysis because these non-varying genes usually represent noise for pattern detection. The differentially expressed genes from pairwise differential expression analyses were clustered into different modules, and the correlation between each module and age was calculated (Supplementary information). After Benjamini–Hochberg correction, modules with P < 0.05 were considered to have significant correlation with age. For each module, a functional enrichment analysis was carried out using Metascape58. In particular, we established a gene product network encompassing 107 genes that were enriched in DNA repair (Gene Ontology: 0006281) using STRING64. These genes were clustered in modules that exhibited a positive correlation with age in M. myotis. The interaction was predicted based on different sources of evidence, including curated databases, functional experiments and gene neighbourhood, co-occurrence and co-expression. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis was performed using STRING64.
Comparative transcriptomic analyses across mammals
To ascertain how the longevity-associated genes and pathways observed in M. myotis changed with age in other mammals, we took advantage of existing ageing blood transcriptome datasets available from three independent cohort studies on other mammals and compared their ageing transcriptomic signatures to those of bats. These comparative datasets included human (H. sapiens, n = 147, age ~25–75 years)24, mouse (Mus musculus, n = 25, ~0.2–2.5 years)15 and wolf (Canis lupus, n = 26, ~1–9 years)65. For human and mouse, raw gene expression counts were obtained from refs. 15 and 24, respectively, while for wolf we analysed the raw RNA-Seq data to generate gene expression estimates using the pipeline established for M. myotis, as described above. For human and mouse, gene ontology term enrichment analyses of the top 100 highly expressed genes were performed using Metascape55, and the percentage of age-related gene expression variance was estimated using the same method as for M. myotis.
To gain a global view of transcriptomic shifts during ageing, for each species PCA was performed based on expression data of all expressed genes using the R package prcomp (Supplementary Fig. 5). For each species, gene expression data were normalized using TMM and further transformed to z-score. To compare age-related gene expression patterns across species, for each gene we measured the Spearman’s rank correlation coefficient between its expression and age across all four mammals. Since the age cohorts in human, mouse and wolf datasets started from adult, samples from juveniles (0 years old) were excluded from correlation analyses in M. myotis. We plotted the distribution of Spearman’s correlation coefficient for each species and investigated the top 200 genes (100 positive and 100 negative) that exhibited the strongest correlation with age (Fig. 4a). Functional enrichment analyses of these top genes were performed using Metascape58.
To investigate the expression pattern at the pathway level, within each species we employed the median z-scores of all genes under each enriched gene ontology term to represent their overall pattern with age. The z-scores were transformed from Spearman’s correlation coefficient within a species. These enriched gene ontology terms were selected on the basis of their distinct expression patterns with age in M. myotis (Fig. 2a–c) and were associated with ageing processes. The full list of gene ontology terms that were used for comparison is given in Supplementary Data 3.
In addition, we also ascertained the expression levels of 307 genes curated in the GenAge database (Build 19) that are highly associated with human ageing16, across these four mammals. Only 207 genes were commonly expressed, so their correlation with age was investigated (Supplementary Data 4). For the top 20 most age-correlated genes (Fig. 4b), we further investigated their functional association with 207 human ageing-associated genes using the STRING database64 (Supplementary Table 8).
miRNA expression analyses
Additionally, we also sequenced 50 small RNA libraries from M. myotis whole-blood samples, 49 of which originated from the same samples that had corresponding transcriptomes previously sequenced (Supplementary Table 1). For each library, miRNA profiling and quantification were analysed using the miRDeep2 pipeline66. The detailed steps and parameters used are described fully in ref. 19. By searching the miRBase (v.32)67, mature miRNA candidates were categorized into the ‘known’ and ‘novel’ groups. Here we focused only on the ‘known’ miRNAs, as these are well documented and experimentally validated. The novel miRNAs were usually expressed at a lower level, thus having limited impact on gene regulation. For miRNA expression analysis, conserved miRNAs across all libraries were extracted as described in ref. 19. Only miRNAs that were presented in at least 80% of all samples were used in the expression analysis.
The samples from individuals 0, 1, 2, 3, 4, 5, 6 and 7+ years of age were used for age cohort analysis. Quantile normalization was applied to the raw miRNA expression counts (Supplementary Data 6). The normalized values were further log2-transformed and converted to z-scores. PCA was performed using prcomp (v.3.0) in R68. Pairwise differential miRNA expression analysis was conducted using DESeq2 (ref. 61), and differentially expressed miRNAs were defined according to the criteria stated above.
miRNA–mRNA expression correlation analyses
miRNA–mRNA interaction was investigated to elucidate the mechanisms of miRNA-directed regulation. First, 3′-UTR sequences were initially extracted from the assembled protein-coding transcripts (n = 31,460) using ExUTR44. miRNA targets were predicted using miRanda69, with the parameters -strict and -en -20. For each miRNA–mRNA pair predicted above, Spearman’s rank correlation coefficient was calculated. To achieve this, samples that had both transcriptome and miRNome sequenced were selected. The raw expression counts were quantile-normalized and log2-transformed. For each pair, the correlation coefficient was computed using cor (v.3.0) in R68. Negatively correlated miRNA–mRNA pairs were determined according to the threshold of FDR < 0.1. Functional enrichment analysis of miRNA-mediated mRNA was performed using Metascape58. In particular, we investigated those genes that were involved in cell cycle regulation and DNA repair pathways and that are simultaneously regulated by miRNA in M. myotis.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw data used in this study have been deposited in the National Center for Biotechnology Information’s BioProject under the accession PRJNA503704. The additional data supporting the conclusions in this paper can be available in the Supplementary Data 1–6.
Code availability
The custom scripts have been deposited in GitHub (https://github.com/UCDBatLab/Longitudinal_myoMyo_transcriptome).
References
Lopez-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Gladyshev, V. N. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15, 594–602 (2016).
Kirkwood, T. B. Understanding the odd science of aging. Cell 120, 437–447 (2005).
Ageing and Health: Fact Sheet 404 (WHO, 2015).
de Magalhaes, J. P. The scientific quest for lasting youth: prospects for curing aging. Rejuvenation Res. 17, 458–467 (2014).
Austad, S. N. Methuselah’s Zoo: how nature provides us with clues for extending human health span. J. Comp. Pathol. 142 (Suppl. 1), S10–S21 (2010).
Munshi-South, J. & Wilkinson, G. S. Bats and birds: exceptional longevity despite high metabolic rates. Ageing Res. Rev. 9, 12–19 (2010).
Seluanov, A., Gladyshev, V. N., Vijg, J. & Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer 18, 433–441 (2018).
Tian, X., Seluanov, A. & Gorbunova, V. Molecular mechanisms determining lifespan in short- and long-lived species. Trends Endocrin. Met. 28, 722–734 (2017).
Teeling, E. C. et al. Bat biology, genomes, and the bat1k project: to generate chromosome-level genomes for all living bat species. Annu. Rev. Anim. Biosci. 6, 23–46 (2018).
Ball, H. C., Levari-Shariati, S., Cooper, L. N. & Aliani, M. Comparative metabolomics of aging in a long-lived bat: insights into the physiology of extreme longevity. PloS One 13, e0196154 (2018).
Foley, N. M. et al. Growing old, yet staying young: the role of telomeres in bats’ exceptional longevity. Sci. Adv. 4, eaao0926 (2018).
Jebb, D. et al. Population level mitogenomics of long-lived bats reveals dynamic heteroplasmy and challenges the free radical theory of ageing. Sci. Rep. 8, 13634 (2018).
Hughes, G. M., Leech, J., Puechmaille, S. J., Lopez, J. V. & Teeling, E. C. Is there a link between aging and microbiome diversity in exceptional mammalian longevity? PeerJ 6, e4174 (2018).
Aramillo Irizar, P. et al. Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nat. Commun. 9, 327 (2018).
de Magalhaes, J. P., Curado, J. & Church, G. M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 25, 875–881 (2009).
Fushan, A. A. et al. Gene expression defines natural changes in mammalian lifespan. Aging Cell 14, 352–365 (2015).
Peters, M. J. et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 6, 8570 (2015).
Huang, Z., Jebb, D. & Teeling, E. C. Blood miRNomes and transcriptomes reveal novel longevity mechanisms in the long-lived bat, Myotis myotis. BMC Genomics 17, 906 (2016).
Kim, E. B. et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011).
Seim, I. et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 4, 2212 (2013).
Li, Y. & de Magalhaes, J. P. Accelerated protein evolution analysis reveals genes and pathways associated with the evolution of mammalian longevity. Age 35, 301–314 (2013).
Huang, Z. et al. A nonlethal sampling method to obtain, generate and assemble whole blood transcriptomes from small, wild mammals. Mol. Ecol. Resour. 16, 150–162 (2016).
Mele, M. et al. Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665 (2015).
Blagosklonny, M. V. Cell cycle arrest is not senescence. Aging 3, 94 (2011).
de Magalhaes, J. P. & Passos, J. F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 170, 2–9 (2018).
Franceschi, C., Garagnani, P., Vitale, G., Capri, M. & Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrin. Met. 28, 199–212 (2017).
Kacprzyk, J. et al. A potent anti-inflammatory response in bat macrophages may be linked to extended longevity and viral tolerance. Acta Chiropt. 19, 219–228 (2017).
Wang, L. F., Walker, P. J. & Poon, L. L. Mass extinctions, biodiversity and mitochondrial function: are bats ‘special’ as reservoirs for emerging viruses? Curr. Opin. Virol. 1, 649–657 (2011).
Gems, D. & Partridge, L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu. Rev. Physiol. 75, 621–644 (2013).
Tacutu, R. et al. Human Ageing Genomic Resources: new and updated databases. Nucleic Acids Res. 46, D1083–D1090 (2018).
Ortega-Molina, A. & Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 24, 184–189 (2013).
Ortega-Molina, A. et al. PTEN positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 15, 382–394 (2012).
Baker, D. J. et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol. 15, 96–102 (2013).
Orr, W. C. et al. Overexpression of glutamate-cysteine ligase extends life span in Drosophila melanogaster. J. Biol. Chem. 280, 37331–37338 (2005).
Satoh, A. et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430 (2013).
Hofmann, J. W. et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell 160, 477–488 (2015).
Zhang, G. et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497, 211–216 (2013).
Henriksson, M. & Luscher, B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Adv. Cancer Res. 68, 109–182 (1996).
Jafri, M. A., Ansari, S. A., Alqahtani, M. H. & Shay, J. W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 8, 69 (2016).
Wu, K. J. et al. Direct activation of TERT transcription by c-MYC. Nat. Genet. 21, 220–224 (1999).
Sun, Q. et al. miR-146a functions as a tumor suppressor in prostate cancer by targeting Rac1. Prostate 74, 1613–1621 (2014).
Komatsu, S. et al. Circulating miR-18a: a sensitive cancer screening biomarker in human cancer. Vivo 28, 293–297 (2014).
Huang, Z. & Teeling, E. C. ExUTR: a novel pipeline for large-scale prediction of 3′-UTR sequences from NGS data. BMC Genomics 18, 847 (2017).
Xu, H. et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PloS One 7, e52249 (2012).
Ruedi, M. et al. Molecular phylogenetic reconstructions identify East Asia as the cradle for the evolution of the cosmopolitan genus Myotis (Mammalia, Chiroptera). Mol. Phylogenet. Evol. 69, 437–449 (2013).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Gouzy, J., Carrere, S. & Schiex, T. FrameDP: sensitive peptide detection on noisy matured sequences. Bioinformatics 25, 670–671 (2009).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Wang, L. et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Tripathi, S. et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17, 483 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 (2017).
Charruau, P. et al. Pervasive effects of aging on gene expression in wild wolves. Mol. Biol. Evol. 33, 1967–1978 (2016).
Friedlander, M. R., Mackowiak, S. D., Li, N., Chen, W. & Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52 (2012).
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A. & Enright, A. J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144 (2006).
Team RC. R: A language and environment for statistical computing (2013).
Enright, A. J. et al. MicroRNA targets in Drosophila. Genome Biol. 5, R1 (2003).
Acknowledgements
We acknowledge and thank the members of Bretagne Vivante and local volunteers and students from University College Dublin for their extensive help in sample collection, and the various owners/local authorities for allowing access to their sites. We would also like to thank M. Bekaert, M. Clarke, G. Hughes and J. Kacprzyk for helpful discussions of the analyses. We acknowledge the Irish Centre for High-End Computing for the provision of computational facilities and support. This project was funded by a European Research Council Research Grant (No. ERC-2012-StG311000 to E.C.T.), a UCD Wellcome Institutional Strategic Support Fund, financed jointly by University College Dublin and SFI-HRB-Wellcome Biomedical Research Partnership (No. 204844/Z/16/Z to E.C.T.), an Irish Research Council Consolidator Laureate Award (to E.C.T.) and a China Scholarship Council studentship (under the UCD-CSC funding programme to Z.H.). The French fieldwork was supported by a Contrat Nature Grant awarded to Bretagne Vivante.
Author information
Authors and Affiliations
Contributions
E.C.T and Z.H. devised the study. M. myotis samples were collected by F.T., S.J.P., E.C.T., E.J.P., N.M.F., D.J., C.V.W. and Z.H. RNA extraction was performed by C.V.W. and Z.H. All data analyses were performed by Z.H. Z.H. is responsible for the Figures presented throughout. The manuscript was written by E.C.T. and Z.H. with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information
Supplementary Methods, Supplementary Figs. 1–14 and Supplementary Tables 1–9
Supplementary Data 1
Spearman’s correlation coefficients between gene expression and age (n = 12,263).
Supplementary Data 2
Top 20 genes that exhibited the strongest correlation (both positive and negative) with age in M. myotis.
Supplementary Data 3
The full list of GO term expression pattern with age across 4 species.
Supplementary Data 4
Spearman’s correlation coefficients between expression of 207 human aging-associated genes and age.
Supplementary Data 5
Raw gene expression counts (n = 12,263).
Supplementary Data 6
Raw mature miRNA expression counts (n = 117).
Rights and permissions
About this article

Cite this article
Huang, Z., Whelan, C.V., Foley, N.M. et al. Longitudinal comparative transcriptomics reveals unique mechanisms underlying extended healthspan in bats. Nat Ecol Evol 3, 1110–1120 (2019). https://doi.org/10.1038/s41559-019-0913-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-019-0913-3
This article is cited by
-
Experimental evidence for cancer resistance in a bat species
Nature Communications (2024)
-
Correlated evolution of social organization and lifespan in mammals
Nature Communications (2023)
-
Considerations for reproducible omics in aging research
Nature Aging (2023)
-
The Mystery of Cancer Resistance: A Revelation Within Nature
Journal of Molecular Evolution (2023)
-
Next Generation Sequencing Revolutionizes Organismal Biology Research in Bats
Journal of Molecular Evolution (2023)
