
Abstract
Cnidarians are astonishingly diverse in body form and lifestyle, including the presence of a jellyfish stage in medusozoans and its absence in anthozoans. Here, we sequence the genomes of Aurelia aurita (a scyphozoan) and Morbakka virulenta (a cubozoan) to understand the molecular mechanisms responsible for the origin of the jellyfish body plan. We show that the magnitude of genetic differences between the two jellyfish types is equivalent, on average, to the level of genetic differences between humans and sea urchins in the bilaterian lineage. About one-third of Aurelia genes with jellyfish-specific expression have no matches in the genomes of the coral and sea anemone, indicating that the polyp-to-jellyfish transition requires a combination of conserved and novel, medusozoa-specific genes. While no genomic region is specifically associated with the ability to produce a jellyfish stage, the arrangement of genes involved in the development of a nematocyte—a phylum-specific cell type—is highly structured and conserved in cnidarian genomes; thus, it represents a phylotypic gene cluster.
Similar content being viewed by others


Main
The Cnidaria is an ancient phylum considered a sister group to all bilaterian animals1,2. Cnidarian body plans are relatively simple, with two major evolutionary trends: while anthozoans, such as Nematostella or Acropora, possess only planula larva and polyp stages, the alternation of polyp and jellyfish generations is typical for medusozoans (Fig. 1a and Supplementary Fig. 1). A free-swimming jellyfish stage requires specialized cell types and organs for active locomotion, such as striated muscles, statocysts and visual systems of varying complexity, which are absent in corals and sea anemones. Because Anthozoa and Medusozoa are sister groups, two alternative evolutionary scenarios are possible: the jellyfish stage is a medusozoan-specific novelty or the jellyfish stage has been lost in anthozoans3.

a, Typical metagenetic life cycle of medusozoans (Aurelia) compared with the life cycle of anthozoans (Nematostella). b, Adult jellyfish of Aurelia. Scale bar: 1 cm. c, Metamorphosis of Aurelia from the Pacific Ocean (left) and the Atlantic Ocean (right). Scale bar: 1 mm. d, Morbakka polyp. Scale bar: 1 mm. e, Morbakka jellyfish. Scale bar: 10 cm. f, Phylogeny of the sequenced species, based on 133 proteins conserved across 47 taxa. Maximum likelihood with the LG substitution model, rooted with a yeast protein set. All nodes have maximum bootstrap support except for 1 node with 90% support.
From ancient times, jellyfish have attracted human attention for their beautiful symmetries, acute toxicity and economic impact (Supplementary Fig. 2 and Supplementary Note 1). Several cnidarian genomes have been published to date, but until the recent publication by Gold et al.4, none of the species sequenced so far had a jellyfish stage1,2,5,6. To explore molecular mechanisms responsible for the origin of a jellyfish body plan, we sequenced the genomes of Aurelia aurita (a scyphozoan; Fig. 1b,c) and Morbakka virulenta (a cubozoan; Fig. 1d,e) using Illumina technology and libraries with insert sizes in the 600-base pair (bp) to 20-kilobase range (Supplementary Figs. 3 and 4). In Aurelia, which is a complex of several cryptic species, two specimens belonging to genetically different ‘strains’ were used (see Supplementary Note 1.1). One sequenced individual from the Baltic sea (PRJNA494057) is referable to the type species of the genus A. aurita (Linnaeus, 1758), while the other represents a widely used laboratory strain termed ‘Roscoff’, which—contrary to its name—originates from the Pacific Ocean (PRJNA494062). Following the assembly step, the genome of the Baltic sea individual (ABSv1) was selected as the primary Aurelia reference due to its higher quality and continuity (Table 1, Supplementary Fig. 4a,d,e and Supplementary Tables 1 and 3). Our genome assemblies of Aurelia provide valuable data to compare with the recently published genome of an Aurelia jellyfish from California (713-megabase pair (Mbp) assembly; 25,454 scaffolds; N50 = 0.124 Mbp)4, promising insights into speciation processes in this circumglobal genus. The Morbakka specimen (PRJNA494057) was collected in Ondo Fishing Port near Hiroshima (Table 1 and Supplementary Table 1). As a source of comparative data, we also sequenced transcriptomes of 11 cnidarian species representing both medusozoans and anthozoans (Supplementary Fig. 1 and Supplementary Tables 1–3).
Draft genome assemblies of Aurelia and Morbakka comprised 377 and 952 Mbp, respectively, with scaffold N50 values of 1.04 and 2.17 Mbp, thereby allowing long-range synteny analysis between two jellyfish types, as well as comparisons of their genome architecture with those of other cnidarians and bilaterians (Table 1, Supplementary Fig. 4 and Supplementary Tables 3 and 4). Mapping of quality-filtered RNA sequencing (RNA-Seq) data against the genome assemblies resulted in mapping of 94.8 and 98.3% of reads in Aurelia and Morbakka, respectively, indicating that the euchromatic regions containing the majority of expressed genes are included in the assemblies.
In Aurelia, ~45% of the genome consists of interspersed repeats with drastic differences in repeat content between Atlantic and Pacific strains (Supplementary Fig. 5a,b and Supplementary Note 1.5). Among the annotated elements, the most abundant type was the long interspersed nuclear element (LINE)/L2 (~5%), but most of the repeats (22.6 and 34.7%, respectively) seem to be novel and potentially Cnidaria specific. In Morbakka, 40.2% of the genome is occupied by repetitive elements, with LINE/RTE (10.5%) and LINE/Dong-R4 (7.4%) being the most abundant (Supplementary Fig. 5c). In contrast with Aurelia, only 5.7% of Morbakka repeats could not be identified in the repeat databases and are putatively novel (Supplementary Note 1.5).
Using ab initio prediction and RNA-Seq data, we identified 28,625 and 24,278 complete or partial protein-coding genes for Aurelia and Morbakka, respectively, which is comparable to the numbers reported for Nematostella (27,273), Hydra (32,338), Acropora (23,700), Aiptasia (29,269) and Aurelia from California (29,964) (Table 1 and Supplementary Fig. 3; see Supplementary Note 1 for details)1,2,4,5,6. Here, we report the analysis of the gene sets and signalling pathways involved in the development of the jellyfish-specific structures and cell types of two medusozoan species.
Results and discussion
Molecular phylogeny of Cnidaria
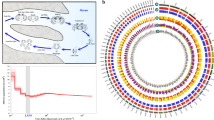
Palaeontological evidence suggests the presence of jellyfish-like organisms in the Early Cambrian7,8, while anthozoans with skeletons—ancestors of extant corals—emerged much later, ~240 million years ago (Ma)5. To address phylogenetic relationships within the Cnidaria, we selected 133 proteins conserved in Aurelia and Morbakka, as well as in 45 genomes and transcriptomes of selected eukaryotes ranging from yeast to higher vertebrates (Fig. 1f, Supplementary Fig. 6 and Supplementary Table 5). In accordance with previously published phylogenetic reconstructions1,3,9, three medusozoan classes—Hydrozoa, Scyphozoa and Cubozoa—grouped together and were separated with a deep split from representative anthozoans. The topology of the relationships among Medusozoa (Fig. 1f and Supplementary Figs. 6–8) is identical to the recently published phylogenetic tree based on a 75-taxon dataset from Kayal and others10. From a broader evolutionary perspective (Supplementary Figs. 7 and 8), the genetic distance between the anthozoans and medusozoans is equivalent to that between Anthozoa and Deuterostomia1,11. Cnidarians are monophyletic, but the magnitude of genetic differences among them is equivalent to that within the whole bilaterian lineage (Supplementary Fig. 8). Consequently, cubozoan and scyphozoan jellyfish, as similar as they might seem at first glance, average roughly the same degree of genetic differences as sea urchins and humans (Supplementary Figs. 7 and 8).
Molecular dating estimated the separation of the major cnidarian clades more than 500 Ma, and each group has undergone a long period of independent evolution (Fig. 2a and Supplementary Fig. 9a). Although precise geological dating might be a matter of debate, ancestors of Hydrozoa, Cubozoa and Scyphozoa separated relatively rapidly, probably coinciding with the emergence of pelagic medusa stages. Interestingly, diversification of species inhabiting the Pacific and Atlantic oceans, such as Tripedalia and Copula, as well as two Aurelia strains, took place during a similar time frame (about ~170–240 Ma) coinciding with the geological period when the Atlantic Ocean itself started to form12. The results of phylogenetic reconstructions and molecular dating strongly corroborate previous reports regarding the high degree of genetic diversity among Aurelia strains worldwide (Fig. 1f and Supplementary Fig. 6)4,13.

a, Separation of the major cnidarian groups occurred >500 Ma. Each group underwent an extended period of independent evolution. Species names are colour coded according to cnidarian classes: Anthozoa (green), Hydrozoa (blue), Cubozoa (orange) and Scyphozoa (red). Horizontal grey bars represent the 95% credibility intervals derived from posterior distributions. b, Aurelia–Nematostella synteny map. Scaffold groups that belong to ancestral Nematostella–human linkage groups (PALs) are marked with coloured boxes: PAL A (red), PAL B (green) and PAL C (blue). c, The most prominent linkage groups between Aurelia and Nematostella correspond to regions where the highest conservation also exists between Nematostella and human genomes. Coloured lines connect the locations of orthologous genes in the scaffolds of Aurelia and Nematostella. Scaffolds are depicted as black vertical lines. Scaffold number is shown at the bottom of each line. d, Aurelia–Morbakka synteny map, indicating that the genome of Morbakka has been strongly reshuffled. Fewer synteny blocks remain compared with the Nematostella–Aurelia pair despite the scaffolds of Morbakka being on average ~4× longer than those of Nematostella.
Genome architecture and ancient linkage groups
Several conserved macrosynteny blocks, which date back to the common ancestor of the Cnidaria and Bilateria, were previously identified in the Nematostella genome1. Our analysis reveals that at least 11 linkage groups are shared by the genomes of Aurelia and Nematostella (Fig. 2b and Supplementary Fig. 10). Among them, four groups of scaffolds directly correspond to three ancient linkage groups (termed putative ancestral linkage groups (PALs)) that are strongly conserved between Nematostella and humans (PALs A, B and C in ref. 1). Aurelia scaffolds 4, 6 and 23 correspond to Nematostella scaffolds 5, 3, 53, 46 and 26, which in turn correspond to the segments of human chromosomes where HoxB, HoxD, HoxC and HoxA clusters are located (Fig. 2c)1. This indicates remarkable conservation of macrosyntenic linkage among Aurelia, Nematostella and humans during a period of more than 500 Myr.
In contrast, the genome of Morbakka exhibited much lower levels of synteny conservation than the Aurelia–Nematostella pair (Fig. 2d and Supplementary Fig. 11). This was surprising because scaffolds in Morbakka assembly were on average ~4× longer than in Nematostella, and theoretically should have contained more orthologues per scaffold pair. Syntenic blocks were also detectable (Fig. 2d), but they contained fewer orthologous pairs per scaffold (Supplementary Figs. 11 and 12). Except for regions containing the Nk2, Otx and minicollagen genes, the overall genomic architecture in Aurelia and Morbakka was very different, suggesting the absence of universal gene clusters in jellyfish with a level of developmental importance analogous to the Hox cluster in Bilateria14. Thus, the arrangement of genes within medusozoan genomes is not directly correlated with their ability to create a jellyfish stage, and the Aurelia genome retained more structural traits from the common ancestor of Cnidaria and Bilateria than that of Morbakka.
Gene sets of Cnidaria and stage-specific genes
Cnidarians are extremely diverse in appearance, physiology and life histories (Fig. 1a–e and Supplementary Fig. 1). As shown in Fig. 3a, differences among cnidarian classes are clearly visualized by a comparison of their gene sets (see Supplementary Note 3.2 and Supplementary Table 6). Based on the number of orthologous genes shared among species, four clusters corresponding to Anthozoa, Hydrozoa, Scyphozoa and Cubozoa are evident. Analysis of gene sets underscores high similarities between the Scyphozoa and Cubozoa, the intermediate position of Hydrozoa, and a large genetic distance between Anthozoa and Medusozoa (Fig. 3a and Supplementary Table 6).

a, Numbers of shared orthologous genes among cnidarian groups (Supplementary Table 6). Based on gene sets, there are four clusters corresponding to Anthozoa, Hydrozoa, Cubozoa and Scyphozoa. Representative bilaterians were used as an outgroup. Abbreviated genus/species names are shown at the bottom. The colours and order correspond to the genus/species names shown on the right-hand side. The colour key shows the number of shared orthologous genes among cnidarian groups. b, Left, expression of polyp- and jellyfish-specific genes in Aurelia. Middle, a total of 1,231 genes are jellyfish-specific, 2,487 are polyp-specific and 24,886 genes do not show stage-specific expression. Right, presence or absence of Aurelia genes with stage-specific expression in the genomes of Acropora and Nematostella. BLASTP cut-off: 1 × 10−4. c, Venn diagram showing the number orthologous genes with jellyfish-specific expression in Aurelia and Morbakka. A list of 13 homeobox genes that are common markers of a jellyfish stage in Scyphozoa and Cubozoa is shown below. d, Scanning electron microscope (SEM) image of a juvenile medusa. e-g, Genes exclusively expressed in striated muscles of Aurelia revealed by in situ hybridization. WD40-repeat protein gene (s1_g264; that is, gene 264 in scaffold 1) (e), putative taxonomically restricted gene with novel repetitive domains (s226_g16) (f) and novel myosin tail protein 4 gene, MTP4 (s206_g6/7) (g). Scale bars: 200 µm. h, Expression of novel and conventional myosins in various stages and tissues of Nemopilema, Aurelia, Tripedalia, Morbakka and Chironex. s88_g59 and so on represent gene IDs in the Aurelia genome (see Supplementary Note 3.1). Numbers following ‘Myh’ or ‘Myo’ represent the classification of medusozoan myosins based on phylogenetic reconstruction and their domain composition. 12 h and 20 h represent the time after metamorphosis induction. ecto-, ectoderm; endo-, endoderm; FPKM, fragments per kilobase of transcript per million mapped reads; j-r-t-mnb, jellyfish without rhopalia, tentacles and manubrium; m-cells, mesoglea cells; meta-, metamorphosis; mnb, manubrium; muscles, ectodermal striated muscles; nseg, non-segmented part; o. arms, oral arms of a jellyfish; seg, segments. Captions for the jellyfish stages and muscle tissues are shown in red text. i, Maximum-likelihood phylogenetic tree of myosin proteins based on the alignment of their Myosin_tail_1 domains. Proteins of Aurelia and Morbakka belong to a distinct clade highlighted in grey. LG substitution model was used. Bootstrap support for all nodes is shown in Supplementary Fig. 14. j, Genomic localization of novel MTPs in the genomes of Morbakka and Aurelia. Scaffolds (S) and the genes within the scaffolds are numbered. A conventional myosin gene is located in Morbakka scaffold 128, adjacent to novel MTP genes. IQ, calmodulin-binding domain; MN, myosin N-terminal domain. k, A possible evolutionary scenario of MTP development from ancestral conventional myosin by gene duplication, followed by subsequent gene family expansions. MTPs of Aurelia and Morbakka seem to be of common origin, but have already acquired structural differences, such as additional repetitive domains in Aurelia.
The major difference between medusozoans and anthozoans is the presence of a jellyfish stage (Fig. 1a). Jellyfish-specific organs and tissues, such as eyes, statocysts and striated swimming muscles, are absent in polyps, and their development requires both structural genes and transcription factors that must be activated only during polyp-to-jellyfish transition and in the adult15,16. To what extent are these genes novel or shared with those present in Anthozoa? What proportion of genes is devoted to the production of stage-specific structures? To answer these questions, we categorized Aurelia genes as polyp- or jellyfish-specific based on RNA-Seq data (see Supplementary Note 3.3). Genes with exclusive stage-specific expression, or with fourfold higher expression in the polyp or jellyfish, were referred to as stage specific15. In Aurelia, 1,231 (4.3%) and 2,487 (8.7%) genes are expressed in a jellyfish- or polyp-specific manner, respectively (Fig. 3b and Supplementary Tables 7 and 8). Thus, approximately 13% of Aurelia genes are potentially involved in the creation of alternative body plans. Next, we checked the genomes of Acropora and Nematostella for genes that exhibited jellyfish- or polyp-specific expression dynamics in Aurelia (BLASTP search with a cut-off of 1 × 10−4). Our analysis revealed that 726 (59%) of jellyfish-specific genes of Aurelia had counterparts in both anthozoans, while 400 (32%) did not have clear anthozoan orthologues (Fig. 3b). Compared with the gene set without stage-specific expression (6,193 genes out of 24,886; 25%), both stage-specific sets are significantly enriched for the genes that are not present in the genomes of Acropora and Nematostella (P < 0.001, χ2 test; see Supplementary Note 3.3). Thus, taxonomically restricted genes seem to represent an important fraction of Aurelia genes with stage-specific expression. Overall, in terms of functional composition, the set of genes with jellyfish-specific expression is enriched for extracellular matrix proteins, ion channels, myosins and homeobox transcription factors, while the polyp stage expresses a large number of proteases, phosphatases and metabolic enzymes, especially those associated with lipid metabolism (see Supplementary Tables 7–10). Similar trends were also observed in the previous Aurelia transcriptomic surveys15,16.
Comparison of gene sets with jellyfish-specific expression between Aurelia and Morbakka yielded unexpected results. Although the total number of genes with jellyfish-specific expression in each species exceeds 1,000, the intersection included only 97 genes (Fig. 3c Supplementary Table 11). In retrospect, this is not particularly surprising, as these two jellyfish species are widely divergent in medusozoan phylogeny (Supplementary Fig. 8). The shared set includes 13 jellyfish-specific homeobox transcription factors and probably represents the ancient complement of regulatory and structural genes retained from an ancestral organism that existed before the divergence of Cubozoa and Scyphozoa (Fig. 3c and Supplementary Table 11).
Stage-specific gene sets, as well as evolutionary processes within Medusozoa, are well exemplified by genes involved in jellyfish propulsion (see Supplementary Note 3.4). Striated swimming muscles, which develop during the polyp-to-medusa transition (Fig. 3d), utilize genes having various degrees of conservation (Fig. 3e–g and Supplementary Fig. 13b–e). For example, WD40-repeat proteins are highly conserved among scyphozoan and cubozoan jellyfishes (Fig. 3e). Others are restricted to Scyphozoa (Fig. 3f), or represent proteins of common origin that have diversified independently in each lineage (Fig. 3g). The latter group includes a novel family of muscle-specific proteins with myosin tail domains (Fig. 3h–j and Supplementary Figs. 13c and 14). These proteins represent a medusozoan-specific invention, which probably emerged from conventional myosins and expanded in the Scyphozoa and Cubozoa (Fig. 3i–k). Proteins with such a domain organization are absent in Anthozoa and are the major developmental markers of jellyfish striated muscles (see Supplementary Note 3.4)15,17,18.
Evolution of Wnt genes and metagenetic life cycles
Unexpectedly large complements of Wnt genes have been reported previously in Nematostella and Hydra19,20. That finding contrasts with the situation in the protostomian lineage, where several groups of Wnt genes exhibit patchy distributions20,21. In Aurelia and Morbakka, the family of Wnt gene ligands is represented by 15 and 14 members, respectively (Fig. 4a,b). Except for Wnt-9 and Wnt-10, all members of the Wnt gene family are present. Medusozoan and anthozoan sequences always belong to distinct clades, reflecting the presence of the corresponding Wnt families in the last common ancestor, as well as a long period of independent evolution (Fig. 4a and Supplementary Fig. 15a).

a, Maximum-likelihood phylogenetic relationships (with the LG substitution model) between Wnt genes from the Anthozoa and Medusozoa. b, Several lineage-specific Wnt gene duplications occurred in Aurelia and Morbakka. a,bWnt gene paralogues. c, Wnt genes are dynamically expressed throughout the life cycle of Aurelia. head, segm and nseg refer to the head, segments and non-segmented part of a strobila. d, Several Wnt genes exhibit tissue-specific expression. A schematic representation of the Aurelia jellyfish is shown with ectoderm in green, muscle in red and the gastrovascular (canal) system in magenta. Filaments, gastric filaments. e–i, SEM images of a polyp and four strobilation stages in Aurelia. Scale bar: 200 µm. j–s, Expression of Wnt-11a (j–n) and Bmp5/8 (o–s) during metamorphosis of Aurelia. Transcripts were detected in the oral side of each developing ephyra anlage. Scale bars: 200 µm in j–l and o–q; 100 µm in in m, n, r and s. t, Ectopic activation of the Wnt cascade during Aurelia metamorphosis with the addition of azakenpaulone (bottom) causes defects in development and separation of ephyra compared with the control (top). u, The addition of azakenpaulone before the induction of metamorphosis completely blocks the development of segments (bottom) compared with the control (top). Scale bars: 1 mm in t and u.
In the course of Aurelia life cycle progression, Wnt genes are differentially regulated (Fig. 4c). Except for Wnt-7b and Wnt-8a, which are predominantly expressed in the polyp stage, there are no other cases of exclusive stage specificity (Fig. 4c,d). Similar complex expression dynamics of Wnt genes is observed during the polyp-to-jellyfish transition in Tripedalia—a cubozoan jellyfish (Supplementary Fig. 15b). Another interesting observation is tissue-specific expression of the majority of Wnt genes in a jellyfish stage. In Aurelia, Wnt-3, Wnt-4b and Wnt-6 are highly upregulated at the bell margin and in the oral arms, while Wnt-8b and Wnt-11b expression is predominantly localized to the endodermal gastrovascular system (Fig. 4d and Supplementary Fig. 15b). This complexity of expression patterns strongly supports the hypothesis that the combinatorial Wnt gene code might be the ancestral mechanism responsible for tissue-layer identity and anterio-posterior polarity in Cnidaria19,22.
The Wnt gene pathway also seems to be important for polyp-to-jellyfish transition in Aurelia (Fig. 4e–i). During strobilation, a polyp is partitioned into multiple ‘segments’ of fixed size, which further develop into small jellyfishes, called ephyra. The oral part of each developing ‘segment’ is marked by ring-shaped Wnt-11a expression in the ectoderm (Fig. 4j–n) and accompanied by endodermal BMP-5/8 expression in the developing gut (Fig. 4o–s). Hyperactivation of the Wnt gene signalling cascade by azakenpaulone treatment causes either a lack of separation between ‘segments’ (Fig. 4t) or a reduction of strobilation to a single, giant jellyfish-like anlage, resembling that in species with monodiscoid strobilation, such as Morbakka or Cassiopea (Fig. 4u and Supplementary Fig. 1e; see Supplementary Note 3.5).
The presence of lineage-specific paralogues and the large number of Wnt genes retained in jellyfish may reflect their complex anatomical organization. It is also important to mention that Wnt genes are more conserved between Cnidaria and Bilateria and among anthozoans and medusozoans than Antennapedia homeobox genes are. Thus, Wnt genes may be more important for body polarity determination and tissue identities of Cnidaria than Hox genes22.
Evolution of Hox genes in Cnidaria
Owing to their phylogenetic position, cnidarians are important for understanding homeobox gene evolution14,23. Anterior Hox genes and ParaHox genes were present in the common ancestor of Cnidaria and Bilateria, but several issues remain to be solved concerning the origin and evolution of the Hox cluster14,24,25. In Aurelia and Morbakka, we identified 79 and 78 genes, respectively, with homeobox domains, and 12 and 13 of these belonged to the Antennapedia class (Supplementary Fig. 16a and Supplementary Table 16).
As in Anthozoa and Hydra, a bilaterian-like Hox cluster is absent in Aurelia and Morbakka1,2,14. Moreover, there is a drastic difference in the genomic organization of homeobox genes between anthozoans and medusozoans. In the vicinity of Hox1, the order of homeobox genes is conserved and their clustering is observed in both Anthozoa and Medusozoa, but neither gene order nor content is conserved between these two cnidarian groups (Fig. 5a). There are also differences in their expression domains across cnidarian lineages. For example, in Aurelia, Hox1 is a specific marker of the subumbrella region where striated muscles are located, while in Clytia, it is a marker of statocysts at the bell rim26. Muscles and statocysts can hardly be considered as anterior or posterior structures. Therefore, our data corroborate previous observations suggesting that the function of Hox genes seems to have diverged considerably within cnidarian groups, as well as between Cnidaria and Bilateria14,26.

a, A conserved Hox cluster is not present in the Cnidaria, and genomic regions where Hox genes are located are considerably different in the Anthozoa and Medusozoa. Syntenic genes are connected with lines, and genes with a homeobox domain are shown as red boxes. Syntenic genes without a homeobox domain are shown as white boxes. Black boxes represent genes without orthologues within genomic segments. b, Mini-clusters of Medusozoa-specific Hox genes are present in Aurelia (scaffold 7) and Morbakka (scaffold 70). Members of these clusters are expressed strictly in polyp (green boxes) or jellyfish stages (red boxes). c, Expression dynamics of Hox genes in polyp and jellyfish stages of Aurelia and Morbakka. Groups of genes with polyp- or jellyfish-specific expression are present. d, Putative ParaHox cluster in Aurelia, and the corresponding genomic arrangement in Morbakka, where the ParaHox cluster is absent. e, Expression of Hox and ParaHox genes in tissues of Aurelia jellyfish. Bell-proxy, slice of the bell without edge part; ecto-up, ectoderm of exumbrella. f, Three ParaHox genes are present in the Medusozoa and are linked in Aurelia, while Cdx has not been identified in the anthozoans.
Although the anthozoan-like cluster around the Hox1 gene is not present in representatives of the Medusozoa (Fig. 5a), they have their own mini-clusters that are potentially of high importance in the context of their life-cycle regulation. In Aurelia (scaffold 7) and Morbakka (scaffold 70), groups of three Hox genes were identified (Fig. 5b). These conserved mini-clusters include genes with exclusive polyp- or jellyfish-specific expression (Fig. 5b). Moreover, several Hox genes in Aurelia and Morbakka exhibit conspicuous stage-specific expression dynamics during life-cycle progression by switching on and off during polyp-to-jellyfish transition (Fig. 5c). Interestingly, their orthologues in Nemopilema and Tripedalia are also stage specific (Supplementary Fig. 16b). Based on these observations, we propose that a subset of medusozoan homeobox genes might function as ‘control switches’ that regulate the transition from a polyp to a jellyfish stage. In the absence of body segmentation, Hox genes in Aurelia and Morbakka may not be responsible for the anterior–posterior specification of tissues and body parts in space, as they are in Bilateria. Instead, they may be involved in temporal regulation that defines alternative body plans (that is, a polyp or a jellyfish).
ParaHox cluster in Aurelia, but not in Morbakka
Unexpectedly, we identified a putative ParaHox cluster in Aurelia (Fig. 5d and Supplementary Fig. 17). It is difficult to conclude whether it is an ancestral feature because the gene order (Cdx > Gsx > Xlox) is different from that in Bilateria27. Expression of Xlox is strictly jellyfish specific in Aurelia, and Gsx and Cdx are expressed in all stages of the life cycle. In Morbakka, no cluster of Xlox, Cdx and Gsc exists, although all three genes are present and none of them is located at the edge of a scaffold with at least three flanking genes at either side (Fig. 5d and Supplementary Fig. 17). Cdx is duplicated in all cubozoan species analysed, with one paralogue retaining expression in both polyp and jellyfish stages, as in Aurelia, while the other is strictly jellyfish specific (Supplementary Fig. 16a). In Aurelia, Xlox and Cdx are strongly expressed in the endoderm, specifically in the gastrovascular system (Fig. 5e). Gsx is upregulated in the oral arms, bell edge, tentacles and striated muscles. Complements of ParaHox genes differ between Medusozoa and Anthozoa (Fig. 5f). Cdx has not been identified in corals, Nematostella and Aiptasia, while Xlox and Gsx are present and are also clustered28. The presence of all three types of ParaHox genes in scyphozoans and cubozoans, including their genomic colocalization in Aurelia, may indicate that some form of a ParaHox cluster existed in the last common ancestor of Cnidaria and Bilateria.
Phylotypic clusters of nematocyte-specific genes
Despite enormous morphological and genetic diversity within Cnidaria, all representatives of the phylum are united by the presence of highly specialized stinging cells (nematocytes), which are utilized for prey capture and defence (Fig. 6a)29. Although cnidarian nematocytes underwent extensive morphological and functional diversification29,30, the central structure of all stinging cells is the nematocyst, which is mainly constructed from several types of minicollagen proteins and is filled with a cocktail of toxins (Fig. 6b–d). In Aurelia polyps, as in Hydra, nematocytes proliferate in the body column (Fig. 6e,f). In a jellyfish stage, they are mostly produced in the epithelium of the bell and in the basal parts of the tentacles at the bell margin, resembling the situation in Clytia (Fig. 6g–j)31. Here, we compared the set of nematocyst-specific proteins of Hydra with the proteomes of medusozoans and anthozoans derived from their genomes and transcriptomes (see Supplementary Note 3.7 and Supplementary Table 17). As shown in Fig. 6k, three groups of nematocyst proteins with variable degrees of conservation are present in Cnidaria (Supplementary Table 17). Group I contains 103 proteins with the highest degree of divergence. Most of them are present only in Hydra species and therefore represent clear examples of taxonomically restricted genes with narrow distributions at the genus level30,32. Group II contains 173 proteins with patchy distributions. In many cases, their copy number varies considerably among the cnidarian classes. Group III contains 58 proteins with the highest degree of conservation. Most of them are enzymes and toxins, such as proteases and phospholipases (Supplementary Table 17).

a, Scanning electron cryomicroscopy image of a firing stinging cell (stenotel) in Hydra. Scale bar: 1 µm. b–d, Visualization of minicollagen distribution in stinging cells of transgenic Hydra. Green fluorescent protein (GFP) fused to cystein-rich domains (CRDs) of minicollagen was expressed under the control of the 5′ region of the minicollagen-6 gene. A reporter construct causes GFP integration into the capsule wall and tubule of all types of nematocytes. Scale bars: 5 µm in b and c; 1 µm in d. e–j, Expression of minicollagen-6 gene in the polyp (e), strobila (f), ephyra (g) and jellyfish of Aurelia (h–j). Scale bars: 200 µm in e and g; 100 µm in in f and j; 1 mm in h and i. k, Conservation of 334 nematocyst proteins in Hydrozoa, Scyphozoa, Cubozoa and Anthozoa (colour coded as in Fig. 2a). Genes were clustered according to their copy number and phylogenetic distribution. Three groups with various levels of conservation are marked as I, II and III. The gradation of colour represents the gene copy number. l, Minicollagen genes are clustered in cnidarian genomes and have a common direction of transcription. Genes with minicollagen CRDs and poly-proline tracks are shown as red boxes, while other genes are shown as black rectangles. Arrows show the direction of transcription. Scaffolds and the genes within the scaffolds are numbered (see Supplementary Note 3.7). Green dots with numbers represent the level of similarity to the Aurelia minicollagen gene (s4_g186) based on a BLASTP search.
Although we failed to detect any bilateria-like clusters involved in anterior–posterior polarity, the situation with nematocyte development was different. Minicollagen genes, which encode the major structural components of nematocysts, are organized into clusters with collinear expression in representatives of Anthozoa, Hydrozoa, Cubozoa and Scyphozoa (Fig. 6l and Supplementary Note 3.7). In all species studied, at least two genomic regions contain groups of minicollagen genes, and there are more of these in Morbakka, Hydra and Aurelia than in Nematostella, which correlates well with the greater complexity of the nematocyte repertoire in the Medusozoa29. Clustering of functionally important genes is a widely used strategy, with the Hox cluster being the most famous example in the Bilateria. It seems that cnidarians also have a phylotypic cluster that is used to generate nematocytes, which are specific to the Cnidaria. Our finding adds to the growing body of data that gene clustering—as observed among fluorescent proteins in Acropora33, photoproteins in Mnemiopsis34 and allorecognition genes in Hydractinia35,36—is an important strategy for establishing phylum- or species-specific functions in early-branching non-bilaterian organisms.
Conclusion
Our study provides a comparative analysis of genome architectures and gene sets among anthozoans, scyphozoans and cubozoans. Scyphozoa and Cubozoa are united by similarities in life style and overall organization, but the genetic differences among them turned out to be considerable. Surprisingly, very few synteny blocks are shared between two types of jellyfish: Aurelia and Morbakka. Comparative analysis of the genome architectures clearly demonstrates that Aurelia is much more similar to Nematostella, which in turn is the most ‘bilaterian-like’ cnidarian sequenced so far1,11. It might be premature to generalize based on just two medusozoan species, but our data suggest that the Scyphozoa have retained more ancestral traits in their genomes than the Cubozoa. The presence of a putative ParaHox cluster in Aurelia and its dispersed state in Morbakka might be an additional hint towards greater genome structural conservation in the scyphozoans.
The set of genes with jellyfish-specific expression is enriched for genes not present in the Anthozoa (see Fig. 3b and Supplementary Tables 7 and 8). In Aurelia, there are 400 genes belonging to this category (32% of all genes with jellyfish-specific expression), and the association between stage specificity and their absence in Nematostella and Acropora is highly significant. Taking into consideration that most of these genes are also lacking in humans, these genes are probably restricted to the Medusozoa. This observation corroborates previous reports about the importance of taxonomically restricted genes for developmental processes and environmental adaptations in the Cnidaria31,37,38,39. At the same time, it is important to mention that 726 genes with jellyfish-specific expression are also present in the Anthozoa (59%). Thus, it is the combination of conserved and putatively novel genes that is important for the functioning of a jellyfish stage.
The magnitude of variation between Aurelia and Morbakka makes it rather difficult to know how the common ancestor of the Medusozoa looked in terms of morphology and genome organization. However, our data provide an interesting perspective on the issue of the ancestral cnidarian body plan before the Anthozoa–Medusozoa split. The jellyfish stage is absent in the Anthozoa, but a polyp stage is typical of both groups and is usually considered to be homologous (Fig. 1a). Interestingly, Aurelia polyps and jellyfish express similar proportions of medusozoa-specific genes (38% in polyps and 32% in jellyfish; see Fig. 3b). This observation implies that in terms of ‘novelty’ (relative to Nematostella and Acropora), these two stages are similar. The anthozoan polyp stage is therefore equally remote genetically from the medusozoan polyp and jellyfish stages. Hence, the old question about which came first, the chicken or the egg (or in this case, the polyp or jellyfish), turns out to be conceptually wrong. Drastic anatomical differences between anthozoan and medusozoan polyps are textbook knowledge dating back to the nineteenth century40,41,42,43. Molecular data strongly support old morphological observations and indicate that anthozoan polyps, medusozoan polyps and a jellyfish stage are equally different from one another. Thus, the only truly conserved stage among the Anthozoa and Medusozoa might be the planula larva, which becomes the best candidate for the cnidarian ancestral body plan.
Cnidarians have proven to be much more diverse in their genomic organization, gene sets and regulation of body plan formation than was previously anticipated. Genetic differences within the phylum are almost equivalent to the variation in the protostomian and deuterostomian clades taken together, with many evolutionary trends of the Bilateria (the development of striated muscles, camera-type eyes and clusters of genes with collinear expression) being independently represented by the cnidarians. The genomes of Aurelia and Morbakka provide an important comparative resource for understanding medusozoan biology, particularly the developmental and evolutionary aspects of their complex life cycles. They also contribute to our understanding of evolutionary processes among cnidarians and in the animal kingdom as a whole.
Methods
Biological materials and sampling
High-molecular-weight DNA for genome sequencing was isolated from spermatozoa of a single Aurelia jellyfish of the Baltic sea strain, collected in the Bay of Kiel (see Supplementary Table 1). Genomic DNA was also extracted from purified mesoglea cells of Roscoff-strain jellyfish because sexually mature male medusae were not available. Jellyfish with a bell diameter of 5–7 cm were dissected, and blocks of mesoglea (pure extracellular matrix with mesoglea cells without any traces of ectodermal cells or gastrovascular system) were digested with Clostridium collagenase (Sigma–Aldrich C0130–100MG; 0.1 mg ml−1 dissolved in filtered sea water), and mesoglea cells were collected by centrifugation (5 min at 500g). After three rounds of washing in filtered sea water, mesoglea cells were pelleted, lysed in DNA extraction buffer (10 mM Tris-HCl, pH 8.0, 75 mM ethylenediaminetetraacetic acid and 1% N-lauroylsarcosine), and DNA was extracted using the standard phenol-chloroform method.
M. virulenta medusae were collected at Ondo Fishing Port, Hiroshima Prefecture, Japan (see Supplementary Table 1). Gonads of three male specimens were dissected and deep frozen. Then, 500 mg of gonad tissue was ground up in liquid nitrogen and mixed with 5 ml of extraction buffer (10 mM Tris-HCl, pH 8.0, 75 mM ethylenediaminetetraacetic acid and 1% N-lauroylsarcosine), and a standard phenol-chloroform DNA extraction procedure was performed.
Genome sequencing, assembly, gene prediction and annotation
High-molecular-weight DNA was quality checked using agarose gel electrophoresis. For paired-end library preparations, it was fragmented by sonication (Covaris M220). Size selection was done by electrophoresis on a BluePippin system (Sage Science). For paired-end libraries, the DNA fraction with a mean fragment size of 600 bp was used. Mate-pair libraries of various fragment sizes ranging from 1–20 kilobases were constructed using a Nextera Mate Pair Sample Prep Kit (Illimina). Following tranposase-mediated fragmentation, DNA fractions of the desired sizes were selected by electrophoresis on a BluePippin system. The resulting paired-end and mate-pair libraries were sequenced on a MiSeq system with 600-cycle chemistry (2 × 300 reads). Raw Illumina reads were quality filtered (Q20; 99% accuracy) to remove low-quality bases using Trimmomatic (version 0.30)44. Raw mate-pair reads were additionally filtered and reverse-complemented with NextClip (version 0.8)45.
Genome assembly was conducted using Newbler version 2.9 software and 37 Gb (~90×) and 34 Gb (~30×) Illumina reads for Aurelia and Morbakka, respectively (see flowchart in Supplementary Fig. 3). Several assemblies with different parameters and sequence data quantities were performed and their results were compared. The best assemblies were selected for scaffolding (see Supplementary Note 1.3), which was performed with SSPACE version 3.0 (ref. 46) and mate-pair reads ranging from 1–20 kilobase pairs. GapCloser version 1.12 was used for filling gaps in the scaffolds. Next, one round of the Haplomerger2 processing pipeline47 was applied to eliminate redundancy in scaffolds and to merge haplotypes. Gene models were predicted using AUGUSTUS version 3.0.2 (ref. 48). RNA-Seq transcripts were mapped to the genome assembly using the PASA version 2.0.1 pipeline49. Resulting transcript models (*.gff3 file) were converted by ‘gff2gbSmallDNA.pl’ into GenBank format and used as a training set for AUGUSTUS (autoAugTrain.pl). Exon and intron hints for gene predictions were generated by mapping raw RNA-Seq reads and complementary DNAs from transcriptome assemblies to the genome sequence with BLAT version 34.
Transcriptome sequencing, assembly and annotation
A total of 11 cnidarian species, representing Anthozoa, Hydrozoa, Scyphozoa and Cubozoa, were used as a source of comparative transcriptomic data (Supplementary Table 2). In all species, messenger RNA was extracted by oligo-dT affinity chromatography. Whole animals at various stages of the life cycle, their tissues, isolated cells or body parts were used (Supplementary Table 2). All sequencing libraries were produced using an Illumina TruSeq Stranded mRNA Sample Prep Kit, quantified by Real-Time PCR (StepOnePlus; Applied Biosystems) and quality controlled using capillary electrophoresis on a Bioanalyzer. Libraries were sequenced on MiSeq and HiSeq 2500 instruments using 600-cycle or 2 × 100–150-cycle chemistry, respectively. In total, 16 reference transcriptomes from 11 species were generated (see Supplementary Tables 1 and 2).
Raw reads were quality filtered (Q20) and trimmed at both ends to remove low-quality regions with Trimmomatic version 0.30. Transcripts were assembled de novo with Trinity (versions r20140717, 2.0.6 and 2.3.2)50. Peptides encoded by transcripts were predicted with ESTScan 3.0.3 (ref. 51) or TransDecoder52. Only peptides with more than 70 amino acids were retained and used in further analyses. The resulting peptides were searched against the non-redundant National Center for Biotechnology Information (NCBI) peptide database and several selected protein sets (human, Nematostella, Acropora and Hydra) using BLASTP. Redundancy in protein sets was removed with CD-HIT version 4.6.1 with a 95% similarity cut-off value53. Protein domains and their coordinates were identified by search with HMMER version 3.1b2 using release 29 of the Pfam-A database (ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam29.0/).
Transcriptome assemblies and raw reads were used as hints for gene model prediction (see flow diagram in Supplementary Fig. 3), and for analysis of gene expression dynamics at various stages of the life cycle and in different tissues (Figs. 3–5). To generate expression tables, quality-filtered reads from all RNA-Seq libraries available for a given species were mapped back to the reference transcripts with Bowtie 2, and transcript abundance was estimated with RSEM version 1.2.5 (ref. 54). Finally, transcript sequences, peptide predictions, BLAST search results and expression values were imported and integrated into a relational database (MySQL 5.6.15). Transcriptomes are accessible via web browser at the Okinawa Institute of Science and Technology (OIST) BLAST server (http://203.181.243.155/aurelia/).
Repeat analysis
Repetitive elements in the draft genome assemblies of Aurelia and Morbakka were identified de novo with RepeatScout version 1.0.5 (ref. 55) and RepeatMasker version 4.0.6 (ref. 56). Repetitive elements were filtered by length and occurrence so that only sequences longer than 50 bp and present more than 10 times in the genome were retained. The resulting sets of repetitive elements were annotated by BLASTN and BLASTX searches against RepeatMasker.lib (35,996 nucleotide sequences) and RepeatPeps.lib (10,544 peptides) bundled with RepeatMasker version 4.0.6. The results of both searches were combined, and BLASTX results were given priority in cases where both BLASTN and BLASTX searches gave hits.
Annotated repeats of Aurelia and Morbakka were added to the OIST BLAST server as combined database ‘Repeats_in_ABSv1_ARSv1_MVIv1_genomes’. They are also stored in the ‘Downloads’ section of the OIST Genome browser (http://marinegenomics.oist.jp/gallery/).
The files ‘AUR21_r04_250316_repeats.fa.gz’ and ‘MOR05_r06_genome_repeats.fa.gz’ include 19,704 (82.1% novel) and 13,698 (49.7% novel) distinct repetitive elements, respectively. Repeat information was also added as ‘Repeat’ tracks to the genome browser of each species.
Molecular phylogeny, macrosynteny analysis and further characterization of the genomes
A full description of the methods and software used can be found in Supplementary Notes 2 and 3.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Datasets associated with this genome project have been registered at NCBI under the BioProject accessions PRJNA494057 (A. aurita Baltic sea strain), PRJNA494062 (A. aurita Roscoff strain) and PRJNA494059 (M. virulenta). Genome assemblies have been deposited at the DNA DataBank of Japan/European Nucleotide Archive/GenBank under accession numbers REGM00000000 (A. aurita Baltic sea), REGL00000000 (A. aurita Roscoff) and RDPX00000000 (M. virulenta). Transcriptome assemblies have been deposited in the NCBI Transcriptome Shotgun Assembly Sequence Database under accession numbers GHAG00000000 (A. aurita Baltic sea), GHAI00000000 (A. aurita Roscoff), GHAS00000000 (A. aurita Kujukushima), GHAK00000000 (A. aurita White sea), GHAF00000000 (M. virulenta), GHAR00000000 (Nemopilema), GHBG00000000 (Copula), GHAQ00000000 (Tripedalia), GHAX00000000 (Chironex), GHBC00000000 (Xenia), GHAW00000000 (Clavularia), GHBA00000000 (Porpita), GHAZ00000000 (Velella) and GHBB00000000 (Physalia). Sequencing reads of the genomes and transcriptomes have been deposited in the NCBI Sequence Read Archive under the study accessions SRR7992476, SRR7992477, SRR7992488, SRR7992489, SRR7992486, SRR7992487, SRR7992484, SRR7992485, SRR7992482, SRR7992483, SRR7992480, SRR7992481, SRR7992474, SRR7992469, SRR7992468, SRR7992475, SRR7992472, SRR7992473, SRR7992470, SRR7992471, SRR7992478 and SRR7992479 (A. aurita Baltic sea), SRR8040393, SRR8040394, SRR8040410, SRR8040411, SRR8040408, SRR8040409, SRR8040406, SRR8040407, SRR8040404, SRR8040405, SRR8040402, SRR8040403, SRR8040391, SRR8040401, SRR8040400, SRR8040399, SRR8040398, SRR8040397, SRR8040392, SRR8040389, SRR8040390, SRR8040387, SRR8040388, SRR8040395 and SRR8040396 (A. aurita Roscoff), SRR7983773, SRR7983772, SRR7983775, SRR7983774, SRR7983769, SRR7983768, SRR7983771 and SRR7983770 (M. virulenta), SRR8089701, SRR8089700, SRR8089699, SRR8089698, SRR8089705, SRR8089704, SRR8089703 and SRR8089702 (A. aurita Kujukushima), SRR8090261, SRR8090262, SRR8090257, SRR8090258, SRR8090263, SRR8090264, SRR8090255, SRR8090256, SRR8090259, SRR8090260, SRR8090265 and SRR8090266 (A. aurita White sea), SRR8101520, SRR8101519, SRR8101522, SRR8101521, SRR8101524, SRR8101523, SRR8101526, SRR8101525 and SRR8101518 (Tripedalia), SRR8101709, SRR8101708 and SRR8101707 (Nemopilema), SRR8115525 (Velella), SRR8115524 (Porpita), SRR8116635 (Physalia) and SRR8116636 (Copula). Genome browsers, genome assemblies, gene models and transcriptomes, together with the annotation files, are available from the Marine Genomics Unit web site (http://marinegenomics.oist.jp/gallery/) and OIST BLAST server (http://203.181.243.155/aurelia/).
Change history
02 May 2019
The version of this article originally published was not open access, but should have been open access. The error has been corrected, and the paper is now open access with a CC-BY license.
References
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007).
Chapman, J. A. et al. The dynamic genome of Hydra. Nature 464, 592–596 (2010).
Collins, A. G. et al. Medusozoan phylogeny and character evolution clarified by new large and small subunit rDNA data and an assessment of the utility of phylogenetic mixture models. Syst. Biol. 55, 97–115 (2006).
Gold, D. A. et al. The genome of the jellyfish Aurelia and the evolution of animal complexity. Nat. Ecol. Evol. 3, 96–104 (2019).
Shinzato, C. et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323 (2011).
Baumgarten, S. et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl Acad. Sci. USA 112, 11893–11898 (2015).
Cartwright, P. et al. Exceptionally preserved jellyfishes from the Middle Cambrian. PLoS ONE 2, e1121 (2007).
Liu, A. G., Matthews, J. J., Menon, L. R., McIlroy, D. & Brasier, M. D. Haootia quadriformis n. gen., n. sp., interpreted as a muscular cnidarian impression from the Late Ediacaran period (~560 Ma). Proc. Biol. Sci. 281, 20141202 (2014).
Zapata, F. et al. Phylogenomic analyses support traditional relationships within Cnidaria. PLoS ONE 10, e0139068 (2015).
Kayal, E. et al. Phylogenomics provides a robust topology of the major cnidarian lineages and insights on the origins of key organismal traits. BMC Evol. Biol. 18, 68 (2018).
Steele, R. E., David, C. N. & Technau, U. A genomic view of 500 million years of cnidarian evolution. Trends Genet. 27, 7–13 (2011).
Seton, M. et al. Global continental and ocean basin reconstructions since 200 Ma. Earth Sci. Rev. 113, 212–270 (2012).
Schroth, W., Jarms, G., Streit, B. & Schierwater, B. Speciation and phylogeography in the cosmopolitan marine moon jelly, Aurelia sp. BMC Evol. Biol. 2, 1 (2002).
Kamm, K., Schierwater, B., Jakob, W., Dellaporta, S. L. & Miller, D. J. Axial patterning and diversification in the Cnidaria predate the Hox system. Curr. Biol. 16, 920–926 (2006).
Fuchs, B. et al. Regulation of polyp-to-jellyfish transition in Aurelia aurita. Curr. Biol. 24, 263–273 (2014).
Brekhman, V., Malik, A., Haas, B., Sher, N. & Lotan, T. Transcriptome profiling of the dynamic life cycle of the scypohozoan jellyfish Aurelia aurita. BMC Genomics. 16, 74 (2015).
Steinmetz, P. R. et al. Independent evolution of striated muscles in cnidarians and bilaterians. Nature 487, 231–234 (2012).
Kraus, J. E., Fredman, D., Wang, W., Khalturin, K. & Technau, U. Adoption of conserved developmental genes in development and origin of the medusa body plan. EvoDevo 6, 23 (2015).
Kusserow, A. et al. Unexpected complexity of the Wnt gene family in a sea anemone. Nature 433, 156–160 (2005).
Lengfeld, T. et al. Multiple Wnts are involved in Hydra organizer formation and regeneration. Dev. Biol. 330, 186–199 (2009).
Miller, D. J., Ball, E. E. & Technau, U. Cnidarians and ancestral genetic complexity in the animal kingdom. Trends Genet. 21, 536–539 (2005).
Guder, C. et al. The Wnt code: cnidarians signal the way. Oncogene 25, 7450–7460 (2006).
Ferrier, D. E. & Holland, P. W. Ancient origin of the Hox gene cluster. Nat. Rev. Genet. 2, 33–38 (2001).
Finnerty, J. R., Pang, K., Burton, P., Paulson, D. & Martindale, M. Q. Origins of bilateral symmetry: Hox and Dpp expression in a sea anemone. Science 304, 1335–1337 (2004).
Chourrout, D. et al. Minimal ProtoHox cluster inferred from bilaterian and cnidarian Hox complements. Nature 442, 684–687 (2006).
Chiori, R. et al. Are Hox genes ancestrally involved in axial patterning? Evidence from the hydrozoan Clytia hemisphaerica (Cnidaria). PLoS ONE 4, e4231 (2009).
Quiquand, M. et al. More constraint on ParaHox than Hox gene families in early metazoan evolution. Dev. Biol. 328, 173–187 (2009).
Ying, H. et al. Comparative genomics reveals the distinct evolutionary trajectories of the robust and complex coral lineages. Genome Biol. 19, 175 (2018).
David, C. N. et al. Evolution of complex structures: minicollagens shape the cnidarian nematocyst. Trends Genet. 24, 431–438 (2009).
Balasubramanian, P. G. et al. Proteome of Hydra nematocyst. J. Biol. Chem. 287, 9672–9681 (2012).
Denker, E., Manuel, M., Leclère, L., Le Guyader, H. & Rabet, N. Ordered progression of nematogenesis from stem cells through differentiation stages in the tentacle bulb of Clytia hemisphaerica (Hydrozoa, Cnidaria). Dev. Biol. 315, 99–113 (2008).
Khalturin, K., Hemmrich, G., Fraune, S., Augustin, R. & Bosch, T. C. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 25, 404–413 (2009).
Shinzato, C., Shoguchi, E., Tanaka, M. & Satoh, N. Fluorescent protein candidate genes in the coral Acropora digitifera genome. Zoolog. Sci. 29, 260–264 (2012).
Schnitzler, C. E. et al. Genomic organization, evolution, and expression of photoprotein and opsin genes in Mnemiopsis leidyi: a new view of ctenophore photocytes. BMC Biol. 10, 107 (2012).
Nicotra, M. L. et al. A hypervariable invertebrate allodeterminant. Curr. Biol. 19, 583–589 (2009).
Rosa, S. F. et al. Hydractinia allodeterminant alr1 resides in an immunoglobulin superfamily-like gene complex. Curr. Biol. 20, 1122–1127 (2010).
Forêt, S. et al. New tricks with old genes: the genetic bases of novel cnidarian traits. Trends Genet. 26, 154–158 (2010).
Fraune, S. et al. In an early branching metazoan, bacterial colonization of the embryo is controlled by maternal antimicrobial peptides. Proc. Natl Acad. Sci. USA 107, 18067–18072 (2010).
Franzenburg, S. et al. Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl Acad. Sci. USA 110, E3730–E3738 (2013).
Haeckel, E. Die Gastraea-Theorie, die Phylogenetische Classification des Thierreiches und die Homologie der Keimblätter (Jena Z. Naturwiss, 1873).
Faurot, L. Etudes sur l’Anatomie, l’Histologie et le Développement des Actinies (typ. A. Hennuyer, 1895).
Technau, U. & Steele, R. E. Evolutionary crossroads in developmental biology: Cnidaria. Development 138, 1447–1458 (2011).
Steinmetz, P. R. H., Aman, A., Kraus, J. E. M. & Technau, U. Gut-like ectodermal tissue in a sea anemone challenges germ layer homology. Nat. Ecol. Evol. 1, 1535–1542 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Leggett, R. M., Clavijo, B. J., Clissold, L., Clark, M. D. & Caccamo, M. NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568 (2014).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Huang, S., Kang, M. & Xu, A. HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579 (2017).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Iseli, C., Jongeneel, C. V. & Bucher, P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 138–148 (1999).
Haas, B. & Papanicolaou, A. TransDecoder (find coding regions within transcripts) (2018); http://transdecoder.github.io
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0 (Institute for Systems Biology); http://www.repeatmasker.org
Acknowledgements
We thank the following individuals for essential contributions: H. Akiyama (Kujukushima Aquarium) for furnishing the Aurelia jellyfish samples; T. Shaposhnikova and G. Jarms for providing Aurelia polyp cultures; J. Wittlieb (CAU) for performing Hydra embryonic microinjections; S. Gorb (CAU) for providing valuable advice and help with scanning electron cryomicroscopy; K. Hisata and H. Miyagi for creating the genome browsers; S. D. Aird (OIST) for editing the manuscript; and I. Rudsky and I. Tikhomirov for offering valuable advice and discussion. K.K. was supported by grants from the Japan Society for the Promotion of Science (JSPS 17K07420) and Russian Foundation for Basic Research (РФФИ 13-04-01795). S.T. was supported by the grant from the Japan Society for the Promotion of Science (JP18K14791).
Author information
Authors and Affiliations
Contributions
K.K. and N.S. designed the project. K.K., C.S., M.K., M.H., M.F., R.K., M.K., H.G., M.T. and S.T. performed the experiments, prepared the transcriptomic and genomic libraries, and performed sequencing and analysis of the data. K.K. assembled genomes and transcriptomes of Aurelia and Morbakka, with support from C.S. and R.K. F.A-E. performed confocal laser scanning microscopy and scanning electron microscopy, and analysed the microscopic data. K.K. and N.S. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–17 and Supplementary Notes 1–3
Supplementary Data 1
Supplementary Tables 1–17
Supplementary Data 2
Sequences of cnidarian Wnt proteins used for phylogenetic reconstruction shown in Fig. 4a and Supplementary Fig. 15a
Supplementary Data 3
Phylogenetic tree of cnidarian Wnt proteins shown in Fig. 4a and Supplementary Fig. 15a
Supplementary Data 4
Sequences of cnidarian Homeobox proteins used for phylogenetic reconstruction shown in Supplementary Fig. 16a
Supplementary Data 5
Phylogenetic tree of cnidarian Homeobox proteins shown in Supplementary Fig. 16a
Supplementary Data 6
Phylogenetic relationships of 46 taxa representing the Cnidaria, Bilateria, Placozoa, Sponges and Ctenophores inferred by the maximum-likelihood approach (shown in Supplementary Fig. 8).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khalturin, K., Shinzato, C., Khalturina, M. et al. Medusozoan genomes inform the evolution of the jellyfish body plan. Nat Ecol Evol 3, 811–822 (2019). https://doi.org/10.1038/s41559-019-0853-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-019-0853-y
This article is cited by
-
Evolution of glial cells: a non-bilaterian perspective
Neural Development (2024)
-
The origin and evolution of Wnt signalling
Nature Reviews Genetics (2024)
-
Genomic and single-cell analyses reveal genetic signatures of swimming pattern and diapause strategy in jellyfish
Nature Communications (2024)
-
Role of cell proliferation in strobilation of moon jellyfish Aurelia coerulea
Fisheries Science (2024)
-
Highly conserved and extremely evolvable: BMP signalling in secondary axis patterning of Cnidaria and Bilateria
Development Genes and Evolution (2024)
