
Abstract
Jellyfish (medusae) are a distinctive life-cycle stage of medusozoan cnidarians. They are major marine predators, with integrated neurosensory, muscular and organ systems. The genetic foundations of this complex form are largely unknown. We report the draft genome of the hydrozoan jellyfish Clytia hemisphaerica and use multiple transcriptomes to determine gene use across life-cycle stages. Medusa, planula larva and polyp are each characterized by distinct transcriptome signatures reflecting abrupt life-cycle transitions and all deploy a mixture of phylogenetically old and new genes. Medusa-specific transcription factors, including many with bilaterian orthologues, associate with diverse neurosensory structures. Compared to Clytia, the polyp-only hydrozoan Hydra has lost many of the medusa-expressed transcription factors, despite similar overall rates of gene content evolution and sequence evolution. Absence of expression and gene loss among Clytia orthologues of genes patterning the anthozoan aboral pole, secondary axis and endomesoderm support simplification of planulae and polyps in Hydrozoa, including loss of bilateral symmetry. Consequently, although the polyp and planula are generally considered the ancestral cnidarian forms, in Clytia the medusa maximally deploys the ancestral cnidarian–bilaterian transcription factor gene complement.
Similar content being viewed by others


Main
In most cnidarians a ciliated, worm-like planula larva settles to produce a polyp. In Anthozoa (corals and anemones), the polyp is the sexually reproductive form but, in the medusozoan branch of Cnidaria, polyps generally produce sexually reproductive jellyfish by a process of strobilation or budding. Jellyfish (medusae) are gelatinous, pelagic, radially symmetric forms found only in Medusozoa. They show complex physiology and behaviour as shown by neural integration of well-defined reproductive organs, digestive systems, locomotory striated muscles and sensory structures. Medusae in many species show some nervous system condensation, notably the nerve rings running around the bell margin1. Some have considered the medusa the ancestral state of cnidarians, with anthozoans having lost this stage (for example, see ref. 2). Under this scenario, the polyp stage was acquired later during medusozoan evolution. Anthozoa would then have evolved from within Medusozoa and so would have lost the medusa stage. However, recent molecular phylogenies support Anthozoa and Medusozoa as sister groups, favouring a benthic, polyp-like adult cnidarian ancestor and an acquisition of the medusa stage in the common branch of Medusozoa3,4. Candidate gene expression studies have shown parallels between medusa and polyp development5 and transcriptome comparisons between species with and without medusae have extended candidate gene lists6,7 but, in general, the genetic foundations of complex medusa evolution within the cnidarian lineage are not well understood.
There are four classes of Medusozoa: Cubozoa (box jellyfish), Scyphozoa (so-called ‘true’ jellyfish), Staurozoa (‘stalked jellyfish’) and Hydrozoa3,8. Life cycles in different medusozoan lineages have undergone frequent modifications, including loss of polyp, planula and medusa stages. Hydra, the classical model of animal regeneration, is a hydrozoan characterized by the loss of the planula and medusa stages from the life-cycle. Compared to anthozoan genomes9,10,11, the Hydra genome is highly diverged and dynamic; it may therefore be atypical of Medusozoa and even Hydrozoa12. Here we report on the genome of Clytia hemisphaerica, a hydrozoan with a typical medusozoan life-cycle, including planula, polyp and medusa stages (Fig. 1). Clytia is easy to maintain and manipulate and amenable to gene function analysis13, allowing mechanistic insight into cellular and developmental processes8,14,15. We analyse transcriptomes from all life-cycle forms, illuminating the evolution of the planula, polyp and medusa and demonstrate how the gene-complement of the cnidarian–bilaterian ancestor provided the foundation of anatomical complexity in the medusa.

a–c, In the Clytia life-cycle (a), the planula larva (b) develops from a fertilized egg and metamorphoses into a primary polyp (c). d–f, The polyp then extends asexually forming a colony composed of feeding polyps (gastrozooids) attached through a common stolon (d) and gonozooids (e) that release swimming medusae (f). Scale bars: b,c,e, 100 μm; d,f, 1 mm. Abbreviations in panel a correspond to the messenger RNA libraries in Supplementary Table 6: EG, early gastrula; P1/P2/P3, planula at 24 h/48 h/72 h after fertilization; PoPr, primary polyp; St, stolon; GO, gonozooid; PH, gastrozooid/polyp head; BMF, baby female medusae 1 day old; MMF, mature female medusa; M, mature male medusa. Credit: a, adapted from ref. 87 Hokkaido University, Japan; b,f, reproduced from ref. 14, Elsevier.
Results
Characteristics of the Clytia genome
We sequenced the Clytia hemisphaerica genome using a whole genome shotgun approach (see Methods; Supplementary Table 1 and Supplementary Fig. 1), giving an assembly with overall length of 445 megabases (Mb). Staining of DNA in prophase oocytes shows the genome is packaged into 15 chromosome pairs (Supplementary Fig. 2). We predicted gene models by aligning expressed sequence reads (RNA-Seq) to the genome. We used sequences derived from a comprehensive set of stages and tissues as well as deeply sequenced mixed-stage libraries (see Methods and Supplementary Table 1). This gave 26,727 genes and 69,083 transcripts. Benchmarking Universal Single-Copy Ortholog (BUSCO) analysis of the presence of universal single copy orthologues indicates a genome coverage of 86% (total ‘complete’ sequences, with 90% for protein set coverage; Supplementary Table 1)16. Using RNA-Seq data we could confirm the trans-spliced-leader sequences previously identified using expressed sequence tags (ref. 17). We did not identify additional ones. The genome GC content is 35%, which is higher than Hydra (29%, ref. 12) but lower than the anthozoan Nematostella (39%, ref. 9).
Reads mapped to the genome suggested a polymorphism frequency of ~0.9%. This is probably an underestimate of heterozygosity in wild populations, as genomic DNA and mRNA for transcriptomes was derived from self-crossed laboratory-reared Clytia Z strains (Methods). The complete mitochondrial genome showed the same gene order as the Hydroidolina ancestor18 (see Supplementary Fig. 1).
The repeat content of ~39%, probably an underestimate given the difficulty of assembling these regions, revealed a rich landscape of uncharacterized interspersed elements in Clytia. While ~97% of the total repeat content could not be classified (~38% of genome length, see Methods), MetaSINE was found to be the most abundant classifiable repeat, with over 5,000 copies19. Many of the most abundant repeats were short (<500 nucleotides), flanked by short inverted repeats and may represent new or divergent MITE (miniature inverted-repeat transposable element) families. For example, 17,035 copies or fragments of the most abundant repeat were detected, with the first and last 16 nucleotides of 242 nucleotides forming an inverted repeat and the element as a whole having no detectable sequence similarity to sequences from other species at protein or nucleic acid levels. In contrast, using the same methods, the most abundant element in the Hydra genome was a LINE (long interspersed nuclear element) present in ~30,000 copies or fragments and >4 kilobases (kb) in length.
Patterns of gene gain and loss
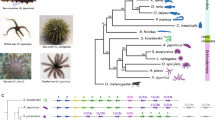
We identified groups of orthologues for a selection of animals with completely sequenced genomes and unicellular eukaryotic outgroups (see Methods). Orthologous group presence or absence was used to infer a Bayesian phylogeny that recapitulated the widely accepted major groupings of bilaterian animals (Fig. 2). Cnidarians were the sister group of Bilateria and within Cnidaria we recovered the expected monophyletic relationships: corals, anemones, anthozoans and hydrozoans. The hydrozoan branch lengths were the longest within the cnidarians, implying elevated rates of gene gain and loss in their lineage, although branches leading to several other species were noticeably longer, including the ecdysozoan models Caenorhabditis and Drosophila, the ascidian Ciona, as well as the ctenophore Mnemiopsis. Clytia and Hydra branch lengths were similar, suggesting that genome evolution has proceeded at comparable rates in these two hydrozoan lineages and that Hydra is not exceptional within this clade. This gene content-based phylogeny positioned sponges (represented by Amphimedon), not ctenophores (represented by Mnemiopsis), as the sister group of all other animals20,21,22, although this relationship has weak support, the lowest of any node in our tree.

The tree was rooted with Saccharomyces cerevisiae. All nodes have a posterior probability of 1 unless indicated. A binary restriction site model and discrete gamma distribution with four site rate categories was used in conjunction with an ascertainment bias correction. See Methods for details.
Among many examples of gene gain in Clytia, we could identify new multigene families and also instances of horizontal gene transfers (HGT), as illustrated by a UDP-glucose 6-dehydrogenase-like (UGDH) gene (Supplementary Fig. 3). UGDH is required for the biosynthesis of various proteoglycans and so to regulate signalling pathways during metazoan embryonic development23. Unexpectedly, the Clytia genome contains two UGDH-like genes, including one acquired in Hydrozoa by HGT from a giant virus of the Mimiviridae family and expressed specifically during Clytia medusa formation. Interestingly, this UGDH-like xenolog, found in most available hydrozoan transcriptomes (including a close relative of Hydra, Ectopleura larynx), was lost in the Hydra lineage and replaced by another UGDH-like acquired through HGT from bacteria (Supplementary Fig. 3)12. Which reactions these enzymes catalyse and their roles during medusa formation remain to be determined. We also detected numerous gene duplications in the hydrozoan lineage, illustrated by the 39 innexin gap junction genes (Supplementary Fig. 4), 14 green fluorescent protein (GFP) and 18 clytin photoprotein genes (Supplementary Fig. 5) found in the Clytia genome. The four GFPs and three clytin sequences previously reported in Clytia are thus transcribed from several recently duplicated genes, probably facilitating the levels of protein production needed to achieve the high cytoplasmic concentrations required for energy transfer between clytins and GFPs24.
Numerous probable gene losses in the hydrozoan lineage (that is, genes absent in Clytia and Hydra but present in Anthozoa and Bilateria) were confirmed by alignment-based phylogenetic analyses. These include at least five Fox family members (FoxAB, FoxE, FoxG, FoxM, FoxQ1) and several homeobox-containing transcription factors involved in nervous system development in Bilateria (Gbx, Mnx, Rax, Ro, Dbx, Pax3-7/PaxD)25,26. Also absent were regulators of the anthozoan directive axis (the axis orthogonal to the oral/aboral axis, possibly related to the bilaterian dorsal/ventral axis; refs. 27,28), including HOX2 (represented in Nematostella by Anthox7/HoxC, Anthox8/HoxD), Gbx, Netrin and its receptor UNC-5 and chordin. (The ‘chordin-like’ gene described in Hydra (ref. 29), is not orthologous to bilaterian and Nematostella chordin30). Comparisons between the two available hydrozoan genomes revealed a much higher number of lost transcription factors in the Hydra lineage (for example, CnoxA, Cdx, DRGX, Ems, Emx, Eve, FezF, FoxD, FoxL2, several FoxQ2 paralogs, Hand, Hmx, Islet, Nkx6, Msxlx, PaxE, Pdx/Xlox, Pknox, POU class 2/3, Six1/2, Twist, Tbx2/3, Tbx4/5, TLX) than in the Clytia lineage. Remarkably, all the conserved homeodomain-containing transcription factors found in the Hydra genome are also present in Clytia while more than 20 of those present in Clytia are missing in Hydra (Supplementary Table 2). We identified seven Clytia transcription factors specifically expressed in the medusa (Cdx, CnoxA, DRGX, FoxL2, Pdx/Xlox, Six1/2, TLX, see below) lost in Hydra but still present in the transcriptome of one of its closest relatives possessing a medusoid stage, Ectopleura larynx (Supplementary Fig. 6 and Supplementary Table 5). These Hydra gene losses thus probably relate to the loss of the medusa stage.
Gene order disruption in the hydrozoan lineage
We tested conservation of gene order between Clytia, Hydra, Nematostella and Branchiostoma floridae, a bilaterian showing a particularly slow rate of loss of syntenic blocks31, by identifying conserved adjacent pairs of orthologues (see Methods) shared between two genomes. Clytia shares most genes in adjacent pairs with Hydra (340), including myc2 and its target CAD32. Fewer pairs were conserved between Clytia and either Nematostella (36) or Branchiostoma (16). Although Nematostella, Hydra and Clytia, as cnidarians, are equally distant phylogenetically from Branchiostoma, the number of genes in adjacent pairs in Clytia/Branchiostoma (16) or Hydra/Branchiostoma (13) is considerably smaller than in Nematostella/Branchiostoma (110). Similar trends emerged from analyses limited to orthologues identified in all four species (Ch/Hv 51; Ch/Nv 8; Ch/Bf 4; Nv/Bf 20), so our conclusions are not biased by an inability to detect more divergent orthologues. Such conservation of adjacent gene pairs possibly relates to coordinated transcription or enhancers being embedded in adjacent genes33. In contrast, even though Clytia and Hydra genomes contain orthologues of most of the Wnt, Fox, NK, ParaHox or Hox anthozoan family members, none of them is found in clusters as described in both Nematostella and bilaterians28,34,35,36,37,38,39 (Supplementary Table 2), reinforcing the idea of rapid evolution of genome organization in the common branch of Clytia and Hydra. Although a few homeobox, Wnt and Fox are found on the same scaffold in Clytia or Hydra, further analysis suggests these pairs are not conserved, as the clustered genes were found to be either recent duplicates or the orthologues in the second species were lost or do not cluster (Hox9-14c and parahox-like CnoxA cluster in Clytia only, Lhx2/9 and Lmx LIM genes cluster in Hydra only; Supplementary Table 2).
Elevated stage-specific gene expression in medusae and polyps
Hydrozoan life cycles are characterized by abrupt morphological transitions: metamorphosis from the planula to polyp; and budding of the compex medusa from gonozooid polyps. To address global trends in differential gene use across the life-cycle we produced a comprehensive replicated transcriptome dataset from 11 samples (Fig. 1a). Principal component analysis (PCA) of the most variably expressed genes across these transcriptomes confirmed sample reproducibility and revealed clear clustering of the three distinct hydrozoan life-cycle stages: (1) the gastrula and planula samples, (2) the polyp and stolon samples and (3) the medusa samples (Fig. 3a). Genes with highest loadings in the first principal component included proteases, as might be expected between feeding adult stages and non-feeding larvae (Supplementary data). Transcriptomes from gonozooids, which are specialized polyp structures containing developing medusae, were intermediate between the polyp and medusa ones. Inter-sample distances on the basis of all genes presented a similar picture to the PCA (Fig. 3b). The main Clytia life-cycle phases thus have qualitatively distinct overall profiles of gene expression, with a distance-based dendrogram showing the polyp and medusa transcriptomes closer to each other than either is to the planula stage.

a, PCA of all libraries from staged mRNA samples. b, Distance-based clustering of all libraries. AU (approximately unbiased, gray) and BP (bootstrap probability, black) values are indicated. Library names are as described in Fig. 1 and Methods. c, Gene expression levels can be partitioned into ‘on’ and ‘off’ categories. Red and blue lines show fitted log-normal distributions and the black line their sum. The blue lines represents genes that we describe as ‘on’, and the red ‘off’. Grey bars correspond to the empirically observed distribution of expression levels. Two example libraries are shown here, P1 and PH. All sample distributions are shown in Supplementary Fig. 7. d and e, Numbers of Clytia genes specific to a single stage (vertices) and genes expressed in two out of three stages (edges). Circle area is proportional to the number of genes. In panel d, top numbers correspond to the total of genes ‘on and up’ in a given stage and ‘off’ in the other stages. Lower bracketed numbers were obtained without the ‘up’ filter. In panel e, numbers of stage-specific transcription factors (TFs) (‘on and up’ filtering) are indicated. Bracketed numbers correspond to homeodomain-containing transcription factors.
By fitting the log-transformed expression data for each library to the sum of two Gaussian distributions40 (Fig. 3c and Supplementary Fig. 7; see Methods), we estimated the number of genes that were ‘on’ in a given library (for example, P1, PH or BMF) and hence stage (planula, polyp or medusa). By these criteria, polyp and medusa stages expressed more genes than embryo and planula stages, with most distinct genes being ‘on’ in the primary polyp library (19,801 genes) and fewest in the early gastrula (13,489 genes).
The majority of predicted genes, 84% (22,472/26,727) were classified as ‘on’ in at least one of our sampled libraries (see Methods; note that our gene prediction protocol includes data from deep sequencing of other mixed libraries) and 41% (10,874/26,727) are expressed in all libraries. We combined results from libraries of the same life-cycle stages (see Methods) and found 335 genes specifically ‘on’ in the planula, 1,534 in the polyp and 808 in the medusa, with 1,932, 284 and 981 genes specifically ‘off’ at these stages respectively (Fig. 3d). We further filtered these data by requiring that genes also show statistically significant expression differences between stages defined as ‘specifically on’ and other stages, allowing a rigorous treatment of the variance between biological replicates (see Methods). This test reduced these lists, but the results showed the same overall trends in numbers of genes unique to stages (Fig. 3d). We conclude that the two adult stages in the Clytia life-cycle show greater complexity of gene expression than the planula larva.
To determine whether the medusa stage was enriched in genes found only in the medusozoan clade, as might plausibly be expected of an evolutionary novelty, we combined these lists of stage-specific genes with a phylogenetic classification of gene age (see Methods; Supplementary Fig. 8). All three main life-cycle stages (planula, polyp and medusa) were enriched in Clytia-specific sequences, indicating that phylogenetically ‘new’ genes are more likely than ‘old’ genes to show stage-specific expression but are not associated with any one life-cycle phase. In general, genes that evolved after the cnidarian/bilaterian split were more likely to be expressed specifically in adult (polyp/medusa) stages.
Stage-specific transcription factors
To address the nature of the molecular differences between stages, we assessed enrichment of gene ontology terms in stage-specific genes relative to the genome as a whole. Planula larvae were found to be significantly enriched in G-protein coupled receptor signalling components, while polyp and medusa were enriched in cell–cell and cell–matrix adhesion class molecules (see Supplementary Table 3). Medusa-specific genes were unique in being significantly enriched in the ‘nucleic acid binding transcription factor activity’ term.
Confirming the strong qualitative distinction in gene expression profiles between planula, polyp and medusa (see Fig. 3a,b) clustering of transcription factor expression profiles recovers the three major life-cycle stages (Fig. 4a). The majority of transcription factors (Supplementary Table 4) specific to a particular stage were specific to the medusa (34, of which 11 are plausibly sex-specific; Fig. 3e and Supplementary Table 5). Twelve were polyp-specific (for example, Vsx, two Hmx orthologues) and a total of 62 transcription factors were expressed at polyp and/or medusa stages but not at the planula stage (12.3% of the total transcription factors). Only three transcription factors showed expression specific to the planula. This pattern is even more striking in the case of the 72 total homeodomain-containing transcription factors: 27.7% are expressed at polyp and/or medusa stages but not at the planula stage, while no homeodomain-containing transcription factors were identified as planula specific.

a, Clustering of libraries by transcription factor expression distances (total number of transcription factors, 505; AU/BP values shown). b, Heatmap showing major classes of transcription factor differential expression. Gene names are taken from orthology assignments of this work. In cases where assignment was not clear, names preceded by * are taken from best human Blast hits. Those followed by ** are genes for which an assignment in Clytia existed in Genbank before this study (for example, Cdx, DRGX). Trailing numbers are unique gene identifiers from this project. Expression units are rlog values from DESeq2 with the row means subtracted. Library names are as described in Fig. 1 and Methods. c, In situ hybridization of medusa-enriched transcription factors; whole female medusa (left, scale bar: 500 μm) and tentacle bulb (right, scale bar: 50 μm). These show a variety of distributions in relation to prominent medusa organs (m, manubrium; g, gonad), and notably with the nerve rings that run around the bell rim and the associated tentacle bulbs (nr, nerve ring; b, bulb). d, Oral and lateral view schematics of Clytia medusa tentacle bulbs indicating transcription factor expression territories.
Among transcription factors expressed strongly in the medusa but poorly at planula stages, we noted a large number with known involvement in neural patterning during bilaterian development (Medusa only: TCF15/Paraxis, Pdx/Xlox, Cdx, TLX, Six1/2, DRGX, FoxQ2 paralogs; Polyp and Medusa: Six3/6, FoxD, FoxQ2 paralogs, FezF, Otx paralogs, Hmx, Tbx4/5, Dmbx, Nkx2a, Nkx6, Neurogenin1/2/3; Fig. 4b). We detected expression of these transcription factors in distinct cell populations of the manubrium, gonads, nerve rings and tentacle bulbs (Fig. 4c,d), structures known to mediate and coordinate feeding, spawning and swimming in response to environmental stimuli1,41,42. The variety of patterns shows an unanticipated degree of molecular and cellular complexity. We propose that, in Clytia, expression of conserved transcription factors in the medusa is associated with diverse cell types, notably with the neural and neurosensory functions of a complex nervous system, with continuous expression of certain transcription factors in post-mitotic neurons being necessary to maintain neuronal identity43. Members of the Sox, PRDL and Achaete scute (bHLH subfamily) orthology groups, commonly associated with neurogenesis44,45 are detectable across all life-cycle stages in Clytia, so our results are unlikely to be simply due to a higher production of nerve cells in the medusa.
Anthozoan larvae and bilaterian embryos express a common set of transcription factors at their respective aboral/anterior ends, including Six3/6, FezF, FoxD, Otx, Rax, FoxQ2 and Irx (refs. 46,47). In the Clytia planula, whose anterior/aboral structures are relatively simple, most orthologues of this transcription factor set are not expressed (Six3/6, FezF, FoxD, Otx orthologues; Figs. 4b and 5b), while another, Rax, was not found in the genome. A FoxQ2 gene (CheFoxQ2a) is expressed aborally in Clytia planulae48 but is not the orthologue of Nematostella aboral and Platynereis apical FoxQ2 (refs. 46,47), which are instead orthologous to CheFoxQ2b, a Clytia polyp–medusa specific gene (Figs. 4b, 5b and Supplementary Fig. 6.2; ref. 48). Irx is the only member of this conserved set of anterior/aboral transcription factors likely to be aborally expressed in Clytia planulae49.

a, Clytia hemisphaerica shows the typical hydrozoan tri-phasic life-cycle, comprising a planula larval stage (bottom), a colonial polyp stage (middle) and a sexually reproducing medusa form (top). Both planula and medusa stages have been lost in the Hydra lineage. Hydrozoan planulae and polyps are morphologically simpler than those of Anthozoa (for example, Nematostella). The comparison of Clytia and Hydra genomes with that of Nematostella shows that the hydrozoan lineage underwent important genome reorganization (see Discussion). b, The planula larva of Nematostella (top) presents a well-defined endoderm and ectoderm and bears an aboral apical organ. The eight internal mesenteries and the pharynx manifest the directive polarity axis, orthogonal to the oral–aboral one. A number of studies have identified a set of conserved transcription factors responsible for setting up the polarity axes and patterning the body. The Clytia planula (bottom) is radially symmetric and has a simpler morphology, with an ectodermal layer surrounding a mass of endodermal cells. Although the oral pole shares a set of developmental regulators with the planula of Nematostella (Bra, Gsc, FoxA, FoxB), the aboral pole appears to be highly divergent: expression of these known aboral transcription factors could not be detected, with the exception of CheFoxQ2a which does not belong to the NvFoxQ2a orthology group. Most Nematostella directive axis regulators have been lost in Hydrozoa. Coloured bars represent expression domains.
The metamorphosis in Clytia from planula to polyp is drastic and the endoderm and oral ectoderm of the morphologically simple Clytia planulae50 do not show continuity with the polyp mouth and digestive structures. In contrast, Nematostella planulae contain developing mesenteries, mouth and pharyngeal structures51, anticipating gradual development into a feeding polyp. Correspondingly, endoderm and mesoderm patterning genes expressed in many bilaterian larvae and Nematostella planulae (Cdx, Pdx/Xlox, Nkx2, Nkx6, Twist, TCF15/Paraxis, Six1/2, Hand)52,53 are not expressed in Clytia planulae. In contrast, despite different gastrulation mechanisms in anthozoans and hydrozoans, orthologues of transcription factors associated with gastrulation and endoderm formation in Nematostella54, including FoxA, FoxB, Brachyury, Snail and Gsc, are also expressed in oral-derived cells at gastrula and planula stages in Clytia49, as well as at polyp and medusa stage.
Discussion
Three lines of evidence suggest that the Clytia genome has undergone a period of rapid evolution since the divergence of Hydrozoa from their common ancestor with Anthozoa (Fig. 5a). First, rates of amino acid substitution appear to be elevated in hydrozoan relative to anthozoan cnidarians55. Second, orthologous gene content analysis shows that the hydrozoans Clytia and Hydra have the longest branches within Cnidaria, with elevated rates of gene gain and loss (Fig. 2). Third, analysis of adjacent gene pairs shows more conservation between Anthozoa and Bilateria than between Hydrozoa and Bilateria.
Gene expression analysis and lost developmental genes point to secondarily simplified planula and polyp structures in Clytia. The planula larva, in particular, shows an absence of key apical (aboral/anterior; Fig. 5b) and endomesoderm patterning genes considered ancestral on the basis of shared expression patterns in Anthozoa and bilaterian larvae46,47,53. Similarly, several genes with roles in patterning the directive axis of the anthozoan planula27,30,51,56,57,58 are lost from the Clytia and Hydra genomes (Chordin, Hox2, Gbx, Netrin), providing support for loss of bilaterality in medusozoans30. Much of the directive axis-patterning gene expression lost in Clytia planulae (Fig. 5b) is, in Nematostella, probably involved in differentiating structures (mesenteries) that are maintained in the adult polyp, supporting the idea that the simple state of the Clytia polyp is secondary. Although bilateral symmetry is observed in a few disparate hydrozoan clades, its sporadic presence suggests convergence59. It will be instructive to test whether, in these cases, bilaterality is under the control of different developmental mechanisms than those reported for Nematostella27,30,51,56,57,58.
The medusa stage, as well as being morphologically complex, expresses a notable number of transcription factors that are conserved between cnidarians and bilaterians. These genes are expressed either specifically in the medusa (for example, DRGX, Twist and Pdx), or in both polyp and medusa but not planula stages (for example, Six3/6, Otx and FoxD), with medusa expression patterns suggesting roles in establishment or maintenance of neural cell-type identity. Hydra has lost the medusa from its life-cycle and has lost orthologues of most transcription factors that in Clytia are expressed specifically in the medusa, further supporting the notion that these genes are regulating the identity of cells now restricted to the medusa.
We propose then that, in part, the rapid molecular evolution we observe at the genome scale in Hydrozoa is connected as much to the simplified planula and polyp as to the more obvious novelty of the medusa. Genomic and transcriptomic studies of the other medusozoan lineages, such as the scyphozoan Aurelia60, whose polyps are less simple than those of Clytia, will show if the expansion of cell type and morphological complexity in the medusa phase has similarly been offset by reduction of key developmental gene use in planula and polyp stages.
Methods
Animals and extraction of genomic DNA
A three-times self-crossed strain (Z4C)2 (male) was used for genomic DNA extraction, aiming to reduce polymorphisms. The first wild-type Z-strain colony was established using jellyfish sampled in the bay of Villefranche-sur-Mer (France). Sex in Clytia is influenced by temperature61 and some young polyp colonies can produce both male and female medusae. Male and female medusae from colony Z were crossed to make colony Z2. Two further rounds of self-crossing produced (Z4C)2 (see Supplementary Fig. 1 for relationships between colonies). For in situ hybridization (and other histological staining) we used a female colony Z4B, a male colony Z10 (offspring of (Z4C)2 × Z4B) as well as embryos produced by crossing Z10 and Z4B strains. (Z4C)2, Z4B and Z10 are maintained as vegetatively growing polyp colonies. For chromosome number determination we performed confocal (Leica SP5) microscopy of isolated fully grown oocytes, in which the duplicated and paired chromosomes are strongly condensed even before meiotic maturation. We stained oocytes with Hoechst dye 33258 and anti-tubulin antibody YL1/2 after fixation in 4% formaldehyde in HEM buffer15 or after fixation in methanol at 20 °C.
For genomic DNA extraction mature (Z4C)2 medusae were cultured in artificial sea water (RedSea Salt, 37‰ salinity) then in Millipore-filtered artificial sea water containing penicillin and streptomycin for 3 to 4 d. They were starved for at least 24 h. Medusae were snap-frozen in liquid nitrogen, ground with mortar and pestle into powder then transferred into a 50 ml Falcon tubes (roughly 50–100 jellyfish/tube). About 20 ml of DNA extraction buffer (200 mM Tris-HCl pH 8.0 and 20 mM EDTA, 0.5 mg ml-1 proteinase K and 0.1% SDS) were added and incubated at 50 °C for 3 h until the solution became uniform and less viscous. An equal volume of phenol was added, vortexed for 1 min, centrifuged for 30 min at 8,000g, then supernatant was transferred to a new tube. This extraction process was repeated using chloroform. X1/10 volume of 5 M NaCl then 2.5 volumes of ethanol were added to the supernatant before centrifugation for 30 min at 8,000g. The DNA precipitate was rinsed with 70% ethanol, dried and dissolved into distilled water. A total 210 µg of DNA was obtained from 270 male medusae.
Genome sequencing and assembly
Libraries for Illumina and 454 sequencing were prepared by standard methods (full details in Supplementary Methods).
Sequence files were error-corrected using Musket62 and assembled using SOAPdenovo263 with a large k-mer size of 91 in an effort to separate haplotypes at this stage. We subsequently used Haplomerger2 to collapse haplotypes to a single more contiguous assembly64.
We performed further genomic scaffolding using a de novo transcriptome assembly. We assembled all RNA-Seq libraries (see below) with Trinity (r20140717) using ‘normalize’ and ‘trimmomatic’ flags, with other parameters as defaults65. Further scaffolding was done using L_RNA_Scaffolder with these transcript sequences66. Within the work reported here, this transcriptome was used as the basis for additional genomic scaffolding and the spliced-leader sequence analysis but not for further analyses.
RNA extraction and transcriptome sequencing
RNA samples were prepared from Z4B female and Z10 male medusae and polyps, as well as embryos generated by crossing these medusae. Animals were starved for at least 24 h before extraction and kept in Millipore-filtered artificial sea water containing penicillin and streptomycin. Then they were put in the lysis buffer (Ambion, RNAqueous MicroKit), vortexed, immediately frozen in liquid nitrogen and stored at –80 °C until RNA preparation.
Total RNA was prepared from each sample using the RNAqueous Microkit or RNAqueous (Ambion). Treatment with DNase I (Q1 DNAse, Promega) for 20 min at 37 °C (2 units per sample) was followed by purification using the RNeasy minElute Cleanup kit (Qiagen). See Supplementary Table 6 for total RNA (evaluated using Nanodrop). RNA quality of all samples was checked using the Agilent 2100 Bioanalyzer. The samples used to generate the expression data presented in Fig. 3a are described in Supplementary Tables 6–9. For the ‘mix’ sample, purification of mRNA and construction of a non-directional complementary DNA library were performed by GATC Biotech, and sequencing was performed on a HiSeq 2500 sequencing system (paired-end 100 cycles). For the other samples, purification of mRNA and construction of a non-directional cDNA library were performed by USC Genomics Center using the Kapa RNA library prep kit and sequencing was performed using either HiSeq 2500 (single-read 50 cycles) or NextSeq (single-read 75 cycles).
Gene prediction and transcript prediction
Genes were predicted from transcriptome data. Using tophat2, we mapped single-end RNA-Seq reads from libraries of early gastrula; 1-, 2- and 3-day-old planula; stolon, polyp head, gonozooid, baby medusa, mature medusa, male medusa (this study); growing oocyte and fully grown oocyte to the genomic sequence15,67. In addition we mapped a mixed library made from the above samples but sequenced with 100 base pair (bp) paired-end reads and a further mature medusa library (100 bp paired-end). Genes were then predicted from these mappings using cufflinks and cuffmerge68. Proteins were predicted from these structures using Transdecoder, with Pfam hit retention69,70 and the protein encoded with the most exons taken as a representative for gene-level analyses.
Where genes are reported as lost, we performed additional tblastn searches of representative sequences from other species directly against the Clytia genome sequence to confirm absence from our data71.
Spliced-leader sequences
A list of spliced-leader sequences (short RNA leader sequences added to the 5’ ends of messenger RNAs by trans-splicing) was previously identified in Clytia using expressed sequence tag data17. Spliced-leader sequences were searched in the Trinity transcriptome assembly (see above) following the same method as previously17. Common sequences of at least 12 nucleotides present at the 5’ end of at least three transcripts were selected and aligned manually to establish a list of putative spliced-leader sequences.
Repetitive elements
A library of de novo identified repetitive elements was created using RepeatScout v.1.0572. Elements were classified using blastn searches against RepBase (20170127), nhmmer searches against Dfam, and hits in the Clytia genome identified using RepeatMasker73.
Protein data sets
We constructed a database of metazoan protein-coding genes from complete genomes, including the major bilaterian phyla, all non-bilaterian animal phyla (including six cnidarian species) and unicellular eukaryotic outgroups. For most species, we used annotation from NCBI and selected one representative protein per gene, to facilitate subsequent analyses (Supplementary Table 10). We used the proteins as the basis for an OMA analysis to identify orthologous groups, v.2.1.1, using default parameters74. We converted the OMA gene OrthologousMatrix.txt file into Nexus format with datatype = restriction and used it as the basis for a MrBayes analysis (v.3.2.6 25/11/2015), using corrections for genes present in fewer than two taxa ‘lset coding = noabsencesites|nosingletonpresence’ and a discrete gamma distribution with four site categories ‘lset rates = gamma’, as described in ref. 20. We performed four MrBayes runs and assessed convergence of chains in each run as an average standard deviation of split frequencies <0.01 (three out of four runs, with all four runs showing the same main topology). The resulting tree was then used in a subsequent OMA run to produce hierarchical orthologous groups (HOGs). These HOGs were used as the basis for the phylogenetic classification of Clytia genes into one category out of eukaryotic, holozoan, metazoan, planulozoan, cnidarian or hydrozoan, on the basis of the broadest possible ranking of the constituent proteins. Genes were presumed to have evolved in the most recent common ancestor of extant leaves and leaves under this node where the gene was not present were presumed to be losses, with the minimum number of losses inferred to explain the observed presence and absence. Clytia-specific genes were identified as those whose encoded proteins had no phmmer hits to the set of proteins used in the OMA analysis.
Where specific genes are named in the text, orthology assignments were taken from classical phylogenetic analysis (or in a few cases pre-existing sequence database names). Signature domains (for example, Homeobox, Forkhead, T-box, HLH) were searched against the protein set using Pfam HMM models and hmmsearch of the hmmer3 package, with the database supplied ‘gathering’ threshold cutoffs75,76. Sequence hits were extracted and aligned with MAFFT (ref. 77) and a phylogeny reconstructed using RAxML with the LG model of protein evolution and gamma correction78.
Transcription factors were assigned via matches beneath the ‘gathering’ threshold to Pfam domains contained in the transcriptionfactor.org database79, with the addition of MH1, COE1_DBD, BTD, LAG1-DNAbind and HMG_Box Pfam models.
Ectopleura larynx proteins were predicted with Transdecoder, including Pfam hit retention, from Trinity assembled reads (v.2.4.0); SRA accessions SRR923510_1 and SRR923510_265,70.
Synteny analyses
Genes were ordered on their scaffolds (using the GFF files described in Supplementary Table 10) on the basis of the average of their start and end position. For each gene, the adjacent genes recorded, ignoring order and orientation but respecting boundaries between scaffolds (terminal genes had only one neighbour). Between-species comparisons were performed using the orthologous groups from OMA, to avoid ambiguity from one:many and many:many genes. When both members of an adjacent pair in one species were orthologous to the members of an adjacent pair in the other species, two genes were recorded as being involved in a CAPO (conserved adjacent pair of orthologues). A consecutive run of adjacent pairs (that is a conserved run of three genes) would thus be two pairs but count as three unique genes. Significance was assessed by performing the same analyses 100 times with a randomized Clytia gene order.
RNA-Seq analyses and stage-specific expression
RNA-Seq reads were aligned to the genome using STAR (v.2.5.3a) with default mapping parameters80. Counts of reads per gene were obtained using HTSeq-count81. Gene-level counts were further analysed using the DESeq2 R package82. An estimate of the mode of row geometric means (rather than the default median) was used to calculate size factors. PCA and heatmaps were generated using regularized logarithms of counts (DESeq2 ‘rlog’ with blind = F). Bootstrapped hierarchical clustering was performed with pvclust using the default parameters83. To identify genes whose expression is restricted to particular stages we used a two-step procedure. We first analysed absolute expression levels, using an approach outlined below and identified genes that were ‘on’ (as opposed to ‘off’) in a particular library. We then filtered this list to ensure that genes that were ‘on’ showed a statistically significant ‘up’ log-fold change of expression level, relative to their ‘off’ stages, using standard RNA-Seq approaches82. Planula stages were defined as any of early gastrula, 1-,2- or 3-day-old planula; polyp any of stolon, primary polyp, polyp head; medusa of any baby medusa, mature medusa or male medusa. The gonozooid library was ignored in this classification as inspection of its expressed genes (and PCA) indicates that it is, as expected, a composite of polyp and medusa stages. For the ‘on/off’ analysis, frequency plots of our log-transformed expression data revealed bimodal distribution patterns (see Fig. 3c and Supplementary 7). Following Hebenstreit and Teichmann40 we fitted the length normalized rlog-transformed gene expression data sets for each library, averaged over replicates, to a mixture of two Gaussian distributions using the mixmodel R package84. The total number of ‘on’ genes for a given library is estimated by multiplying the mixing proportion (lambda) of the ‘on’ (high expression) peak by the total number of genes fitted. Individual genes were defined as ‘on’ if they had a posterior probability >0.5 of coming from the more highly expressed distribution. The gene was then classified as ‘on’ in a stage (planula, polyp, medusa) if any of the component libraries of that stage (for example, EG, P1, P2 or P3 for planula) showed expression of that gene. Genes that were not exclusively ‘off’ or exclusively ‘on’ were then also filtered by a log-fold change analysis performed using all genes. Significant differences in gene expression were calculated via pairwise contrasts of all different ‘conditions’ (replicated libraries). To be considered ‘up’ in planula, polyp or medusa, a gene needed to be significantly up (lfc threshold = 0.0, alt hypothesis = ‘greater’) in at least one ‘condition’ of that stage relative to all ‘conditions’ of one or both of the other stages, requiring the DESeq2 adjusted P <0.001 across multiple pairwise comparisons. For example, if a gene was significantly more highly expressed in 1-day-old planula (P1) than all constituent medusa or polyp stages, it was considered ‘up’ in planula.
This combined approach addresses two issues. First, we avoid the choice of an arbitrary FPKM (fragments per kilobase per million mapped reads) type value as an indicator of expression. Our frequency-distribution based approach defines gene ‘on’ or ‘off’ states independently of the total numbers of distinct transcripts expressed in a given sample, unlike FPKM values which are a measure of concentration and so for similarly expressed genes will be relatively higher for ‘off’ genes in samples with low overall complexity. Second, log-fold change analyses in themselves are not reliable indicators of specificity in the sense that we are interested in, as they deal with relative expression levels: a gene could show a statistically significant difference and still be clearly expressed in both stages, if for example it has an expression level of 5 log units in stage a and 10 log units in stage b. Such differences are expected, owing to very different cellular composition between life-cycle stages. By combining these two approaches we identify genes with rigorous evidence for significant differential expression with a more easily interpretable biological meaning.
Gene Ontology term enrichment
Gene Ontology terms were assigned via sequence hits to the PANTHER database using the supplied ‘pantherScore2.0.pl’ program. Term enrichment was tested using the ‘Ontologizer’ software with a ‘Parent–Child–Union’ calculation (the default) and Bonferroni multiple testing correction85.
In situ hybridization
In situ hybridization probes were synthesized from multiple types of templates, either pGEM-T Easy plasmids (following one or two rounds of insert amplification), PCR products (reverse primer comprised a T7 promoter) or expressed sequence tag clones48; see Supplementary Table 11 for further details. In situ hybridization was performed, as previously described86, on 2-week-old female medusae. Images were taken on either Zeiss Axio Imager 2 or Olympus BX61 microscopes and processed with ImageJ 1.47v and Adobe Photoshop CS6.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequence data have been deposited at EBI under Bioproject accessions PRJEB28006 and PRJEB30490. Data downloads and a genome browser are available at http://marimba.obs-vlfr.fr/organism/Clytia/hemisphaerica (see Supplementary Section 2). There are no restrictions on data. A data archive for repeats, phylogeny and expression analysis is available at: https://doi.org/10.5281/zenodo.1470435.
References
Koizumi, O. et al. The nerve ring in cnidarians: its presence and structure in hydrozoan medusae. Zoology 118, 79–88 (2015).
Hyman, L. H. The Invertebrates: Protozoa Through Ctenophora, Vol 1 (McGraw-Hill, New York, 1940)..
Zapata, F. et al. Phylogenomic analyses support traditional relationships within cnidaria. PLoS ONE 10, e0139068 (2015).
Kayal, E. et al. Phylogenomics provides a robust topology of the major cnidarian lineages and insights on the origins of key organismal traits. BMC Evol. Biol. 18, 68 (2018).
Kraus, J. E. M., Fredman, D., Wang, W., Khalturin, K. & Technau, U. Adoption of conserved developmental genes in development and origin of the medusa body plan. EvoDevo 6, 23 (2015).
Sanders, S. M. & Cartwright, P. Interspecific differential expression analysis of RNA-Seq data yields insight into life cycle variation in hydractiniid hydrozoans. Genome Biol. Evol. 7, 2417–2431 (2015).
Sanders, S. M. & Cartwright, P. Patterns of Wnt signaling in the life cycle of Podocoryna carnea and its implications for medusae evolution in Hydrozoa (Cnidaria). Evol. Dev. 17, 325–336 (2015).
Leclère, L., Copley, R. R., Momose, T. & Houliston, E. Hydrozoan insights in animal development and evolution. Curr. Opin. Genet. Dev. 39, 157–167 (2016).
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007).
Shinzato, C. et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323 (2011).
Baumgarten, S. et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl Acad. Sci. USA 112, 11893–11898 (2015).
Chapman, J. A. et al. The dynamic genome of Hydra. Nature 464, 592–596 (2010).
Momose, T. et al. High doses of CRISPR/Cas9 ribonucleoprotein efficiently induce gene knockout with low mosaicism in the hydrozoan Clytia hemisphaerica through microhomology-mediated deletion. Sci. Rep. 8, 11734 (2018).
Houliston, E., Momose, T. & Manuel, M. Clytia hemisphaerica: a jellyfish cousin joins the laboratory. Trends Genet. 26, 159–167 (2010).
Quiroga Artigas, G. et al. A gonad-expressed opsin mediates light-induced spawning in the jellyfish Clytia. Elife 7, e29555 (2018).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 3, 543–548 (2017).
Derelle, R. et al. Convergent origins and rapid evolution of spliced leader trans-splicing in metazoa: insights from the ctenophora and hydrozoa. RNA 16, 696–707 (2010).
Kayal, E. et al. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol. Evol. 4, 1–12 (2012).
Nishihara, H., Plazzi, F., Passamonti, M. & Okada, N. MetaSINEs: broad distribution of a novel sine superfamily in animals. Genome Biol. Evol. 8, 528–539 (2016).
Pisani, D. et al. Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl Acad. Sci. USA 112, 15402–15407 (2015).
Whelan, N. V. et al. Ctenophore relationships and their placement as the sister group to all other animals. Nat. Ecol. Evol. 1, 1737–1746 (2017).
Feuda, R. et al. Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Curr. Biol. 27, 3864–3870.e4 (2017).
García-García, M. J. & Anderson, K. V. Essential role of glycosaminoglycans in Fgf signaling during mouse gastrulation. Cell 114, 727–737 (2003).
Fourrage, C., Swann, K., Gonzalez Garcia, J. R., Campbell, A. K. & Houliston, E. An endogenous green fluorescent protein–photoprotein pair in Clytia hemisphaerica eggs shows co-targeting to mitochondria and efficient bioluminescence energy transfer. Open Biol. 4, 130206 (2014).
Lowe, C. J. et al. Anteroposterior patterning in hemichordates and the origins of the chordate nervous system. Cell 113, 853–865 (2003).
Holland, L. Z. et al. Evolution of bilaterian central nervous systems: a single origin?. EvoDevo 4, 27 (2013).
Matus, D. Q. et al. Molecular evidence for deep evolutionary roots of bilaterality in animal development. Proc. Natl Acad. Sci. USA 103, 11195–11200 (2006).
Ryan, J. F. et al. Pre-bilaterian origins of the hox cluster and the hox code: evidence from the sea anemone, Nematostella vectensis. PLoS ONE 2, e153 (2007).
Rentzsch, F., Guder, C., Vocke, D., Hobmayer, B. & Holstein, T. W. An ancient chordin-like gene in organizer formation of Hydra. Proc. Natl Acad. Sci. USA 104, 3249–3254 (2007).
Genikhovich, G. & Technau, U. On the evolution of bilaterality. Development 144, 3392–3404 (2017).
Simakov, O. et al. Hemichordate genomes and deuterostome origins. Nature 527, 459–465 (2015).
Hartl, M. et al. Stem cell-specific activation of an ancestral myc protooncogene with conserved basic functions in the early metazoan Hydra. Proc. Natl Acad. Sci. USA 107, 4051–4056 (2010).
Irimia, M. et al. Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Res. 22, 2356–2367 (2012).
Chourrout, D. et al. Minimal protohox cluster inferred from bilaterian and cnidarian hox complements. Nature 442, 684–687 (2006).
Kamm, K., Schierwater, B., Jakob, W., Dellaporta, S. L. & Miller, D. J. Axial patterning and diversification in the cnidaria predate the Hox system. Curr. Biol. 16, 920–926 (2006).
Sullivan, J. C., Ryan, J. F., Mullikin, J. C. & Finnerty, J. R. Conserved and novel Wnt clusters in the basal eumetazoan Nematostella vectensis. Dev. Genes Evol. 217, 235–239 (2007).
Chiori, R. et al. Are Hox genes ancestrally involved in axial patterning? Evidence from the hydrozoan Clytia hemisphaerica (Cnidaria). PLoS ONE 4, e4231 (2009).
Shimeld, S. M., Degnan, B. & Luke, G. N. Evolutionary genomics of the Fox genes: origin of gene families and the ancestry of gene clusters. Genomics 95, 256–260 (2010).
Mazza, M. E., Pang, K., Reitzel, A. M., Martindale, M. Q. & Finnerty, J. R. A conserved cluster of three PRD-class homeobox genes (homeobrain, rx and orthopedia) in the Cnidaria and Protostomia. EvoDevo 1, 3 (2010).
Hebenstreit, D. et al. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 7, 497 (2011).
Mackie, G. O., Singla, C. L. & Stell, W. K. Distribution of nerve elements showing FMRFamide-like immunoreactivity in hydromedusae. Acta Zool. 66, 199–210 (1985).
Takeda, N. et al. Identification of jellyfish neuropeptides that act directly as oocyte maturation-inducing hormones. Development 145, dev156786 (2018).
Kerk, S. Y., Kratsios, P., Hart, M., Mourao, R. & Hobert, O. Diversification of C. elegans motor neuron identity via selective effector gene repression. Neuron 93, 80–98 (2017).
Jager, M., Quéinnec, E., Le Guyader, H. & Manuel, M. Multiple Sox genes are expressed in stem cells or in differentiating neuro-sensory cells in the hydrozoan Clytia hemisphaerica. EvoDevo 2, 12 (2011).
Rentzsch, F., Layden, M. & Manuel, M. The cellular and molecular basis of cnidarian neurogenesis. Wiley Interdiscip. Rev. Dev. Biol. 6, e257 (2017).
Sinigaglia, C., Busengdal, H., Leclère, L., Technau, U. & Rentzsch, F. The bilaterian head patterning gene six3/6 controls aboral domain development in a cnidarian. PLoS Biol. 11, e1001488 (2013).
Marlow, H. et al. Larval body patterning and apical organs are conserved in animal evolution. BMC Biol. 12, 7 (2014).
Chevalier, S., Martin, A., Leclère, L., Amiel, A. & Houliston, E. Polarised expression of FoxB and FoxQ2 genes during development of the hydrozoan Clytia hemisphaerica. Dev. Genes Evol. 216, 709–720 (2006).
Lapébie, P. et al. Differential responses to Wnt and PCP disruption predict expression and developmental function of conserved and novel genes in a cnidarian. PLoS Genet. 10, e1004590 (2014).
Bodo, F. & Bouillon, J. Étude histologique du développement embryonnaire de quelques hydroméduses de Roscoff: Phialidium hemisphaericum (L.), Obelia sp. Péron et Lesueur, Sarsia eximia (Allman), Podocoryne carnea (Sars), Gonionemus vertens Agassiz. Cah. Biol. Mar. 9, 69–104 (1968).
Leclère, L. & Rentzsch, F. RGM regulates BMP-mediated secondary axis formation in the sea anemone Nematostella vectensis. Cell Rep. 9, 1921–1930 (2014).
Martindale, M. Q., Pang, K. & Finnerty, J. R. Investigating the origins of triploblasty: ‘mesodermal’ gene expression in a diploblastic animal, the sea anemone Nematostella vectensis (phylum, Cnidaria; class, Anthozoa). Development 131, 2463–2474 (2004).
Steinmetz, P. R. H., Aman, A., Kraus, J. E. M. & Technau, U. Gut-like ectodermal tissue in a sea anemone challenges germ layer homology. Nat. Ecol. Evol. 1, 1535–1542 (2017).
Röttinger, E., Dahlin, P. & Martindale, M. Q. A framework for the establishment of a cnidarian gene regulatory network for ‘endomesoderm’ specification: the inputs of ß-catenin/TCF signaling. PLoS Genet. 8, e1003164 (2012).
Simion, P. et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 27, 958–967 (2017).
Saina, M., Genikhovich, G., Renfer, E. & Technau, U. BMPs and chordin regulate patterning of the directive axis in a sea anemone. Proc. Natl Acad. Sci. USA 106, 18592–18597 (2009).
Genikhovich, G. et al. Axis patterning by bmps: cnidarian network reveals evolutionary constraints. Cell Rep. 10, 1646–1654 (2015).
He, S. et al. An axial Hox code controls tissue segmentation and body patterning in Nematostella vectensis. Science 361, 1377–1380 (2018).
Manuel, M. Early evolution of symmetry and polarity in metazoan body plans. C. R. Biol. 332, 184–209 (2009).
Gold, D. A. et al. The genome of the jellyfish Aurelia and the evolution of animal complexity. Nat. Ecol. Evol. 3, 96–104 (2019).
Carré, D. & Carré, C. Origin of germ cells, sex determination, and sex inversion in medusae of the genus Clytia (Hydrozoa, leptomedusae): the influence of temperature. J. Exp. Zool. 287, 233–242 (2000).
Liu, Y., Schröder, J. & Schmidt, B. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 29, 308–315 (2013).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Huang, S., Kang, M. & Xu, A. HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579 (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Xue, W. et al. L_RNA_scaffolder: scaffolding genomes with transcripts. BMC Genomics 14, 604 (2013).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-Seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-Seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
TransDecoder (Github); http://transdecoder.github.io
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013-2015; http://www.repeatmasker.org
Train, C.-M., Glover, N. M., Gonnet, G. H., Altenhoff, A. M. & Dessimoz, C. Orthologous Matrix (OMA) algorithm 2.0: more robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference. Bioinformatics 33, i75–i82 (2017).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Wilson, D., Charoensawan, V., Kummerfeld, S. K. & Teichmann, S. A. DBD—taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 36, D88–D92 (2008).
Dobin, A. et al. STAR: ultrafast universal RNA-Seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15, 550 (2014).
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Benaglia, T., Chauveau, D., Hunter, D. & Young, D. mixtools: an R package for analyzing finite mixture models. J. Stat. Softw. 32, 1–29 (2009).
Bauer, S., Grossmann, S., Vingron, M. & Robinson, P. N. Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650–1651 (2008).
Sinigaglia, C., Thiel, D., Hejnol, A., Houliston, E. & Leclère, L. A safer, urea-based in situ hybridization method improves detection of gene expression in diverse animal species. Dev. Biol. 434, 15–23 (2018).
Kubota, S. The life-history of Clytia edwardsi (hydrozoa; campanulariidae) in Hokkaido, Japan. J. Fac. Sci. Hokkaido Univ. Ser. VI Zool. 21, 317–354 (1978).
Acknowledgements
We thank D. Carré, who first suggested that Clytia hemisphaerica would be a convenient cnidarian species for experimentation and isolated our founder adult medusae from the Villefranche plankton. We thank I. Mathieson (UPenn) and R. Mott (UCL) for statistical advice and S. Collet, L. Gissat and L. Gilletta for animal maintenance. Initial sequencing was funded directly by the Genoscope–CEA. Other funding was provided by the CORBEL European Research Infrastructure cluster project, grants from the Agence Nationale de la Recherche (nos. ANR-13-BSV2-0008 “OOCAMP” and ANR-13-PDOC-0016 “MEDUSEVO”), a Marie Curie training network (no. FP7-PEOPLE-2012-ITN 317172 “NEPTUNE”), a grant of the Austrian Science Fund (FWF; no. P27353) to U.T., EMBRC-France (no. ANR-10-INBS-0002), the André Picard Network, as well as core CNRS and Sorbonne University funding to the LBDV. Part of the imaging was performed at the Villefranche-sur-mer imaging platform (PIV).
Author information
Authors and Affiliations
Contributions
R.R.C., L.L. and E.H. conceived the study, contributed to the analyses and wrote the paper. R.R.C. assembled the genome and performed the initial RNA-Seq analysis. L.L., R.R.C. and C.S. prepared the figures. C.H. generated the Marimba database. All the authors contributed to data generation and/or analyses.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary Figures 1–8, Supplementary Tables 1–3 and 5–11.
Supplementary Table 4
Expression patterns and genome locations for all transcription factors and predicted genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leclère, L., Horin, C., Chevalier, S. et al. The genome of the jellyfish Clytia hemisphaerica and the evolution of the cnidarian life-cycle. Nat Ecol Evol 3, 801–810 (2019). https://doi.org/10.1038/s41559-019-0833-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-019-0833-2
This article is cited by
-
Genomic and single-cell analyses reveal genetic signatures of swimming pattern and diapause strategy in jellyfish
Nature Communications (2024)
-
Highly conserved and extremely evolvable: BMP signalling in secondary axis patterning of Cnidaria and Bilateria
Development Genes and Evolution (2024)
-
Nematostella vectensis exemplifies the exceptional expansion and diversity of opsins in the eyeless Hexacorallia
EvoDevo (2023)
-
Functional analysis in a model sea anemone reveals phylogenetic complexity and a role in cnidocyte discharge of DEG/ENaC ion channels
Communications Biology (2023)
-
The evolutionary history of Brachyury genes in Hydrozoa involves duplications, divergence, and neofunctionalization
Scientific Reports (2023)
