
Abstract
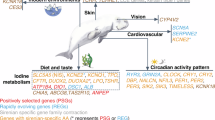
Icefishes (suborder Notothenioidei; family Channichthyidae) are the only vertebrates that lack functional haemoglobin genes and red blood cells. Here, we report a high-quality genome assembly and linkage map for the Antarctic blackfin icefish Chaenocephalus aceratus, highlighting evolved genomic features for its unique physiology. Phylogenomic analysis revealed that Antarctic fish of the teleost suborder Notothenioidei, including icefishes, diverged from the stickleback lineage about 77 million years ago and subsequently evolved cold-adapted phenotypes as the Southern Ocean cooled to sub-zero temperatures. Our results show that genes involved in protection from ice damage, including genes encoding antifreeze glycoprotein and zona pellucida proteins, are highly expanded in the icefish genome. Furthermore, genes that encode enzymes that help to control cellular redox state, including members of the sod3 and nqo1 gene families, are expanded, probably as evolutionary adaptations to the relatively high concentration of oxygen dissolved in cold Antarctic waters. In contrast, some crucial regulators of circadian homeostasis (cry and per genes) are absent from the icefish genome, suggesting compromised control of biological rhythms in the polar light environment. The availability of the icefish genome sequence will accelerate our understanding of adaptation to extreme Antarctic environments.
Similar content being viewed by others


Main
Antarctic icefishes inhabit the Earth’s coldest marine environment. These remarkable animals are the only vertebrates that lack functional red blood cells and functional haemoglobin genes; they are ‘white blooded’1,2. Icefish blood carries oxygen solely in physical solution, resulting in an oxygen-carrying capacity per unit of blood volume of less than 10% of that in closely related red-blooded Antarctic notothenioid fishes1,3. Blackfin icefish (Chaenocephalus aceratus), along with 5 other species among the 16 recognized species of icefishes (Channichthyidae) within the notothenioid teleosts, also lack cardiac myoglobin4,5,6. Icefishes evolved mechanisms that appear to compensate for loss of these oxygen-binding proteins, including enormous hearts with increased stroke volume relative to body size7, enhanced vascular systems4, and changes in mitochondrial density and morphology4.
Ancestral notothenioids were red-blooded but had no myoglobin in their skeletal muscle; they lived on the ocean floor and lacked a buoyancy-generating swim bladder. As Antarctica cooled, finally reaching −1.9 °C in the high Antarctic about 10–14 million years ago (Ma)8, ecological niches opened into which notothenioids radiated due to adaptive changes for cold tolerance9, including antifreeze glycoproteins (AFGPs) in larvae and adults10,11, and ice-resistant egg chorion proteins surrounding embryos12. Notothenioids, living in constant cold, evolved a substantially non-conventional heat-shock response13,14. From these benthic ancestors, eight notothenioid taxa, including the icefishes, evolved to exploit the food-rich water column through increased buoyancy, which was achieved by reducing densely mineralized elements such as bones and scales15,16,17 and increasing deposits of lipids18. Some icefishes became ambush feeders9,19 concomitant with craniofacial adaptations, and possibly a decrease in metabolic oxygen demand7,17,20. To help investigate the genomic basis for these extreme evolutionary adaptations, we sequenced the genome of the blackfin icefish.
Results
Genome assembly and annotation
The genome of a female Antarctic blackfin icefish from the Antarctic Peninsula (Supplementary Figs. 1 and 2) was sequenced by single-molecule real-time technology with a PacBio Sequel instrument, yielding ~90× genome coverage and a 13-kilobase average read length (Supplementary Table 1). The genome size was estimated, by k-mer analysis using Jellyfish software, to be 1.1 gigabase pairs (Supplementary Fig. 3). The FALCON-Unzip assembled genome contained 3,852 contigs totalling 1.06 gigabase pairs with a contig N50 size of 1.5 megabase pairs (Mb) (Table 1). Evaluation of the genome for completeness based on BUSCO21 identified 89.9% complete and 3.6% fragmented genes from the 4,584-gene Actinopterygii dataset (Supplementary Table 2). The icefish genome contains 30,773 inferred protein-coding genes based on combined ab initio gene prediction, homology searching and transcript mapping (Table 1 and Supplementary Tables 3 and 4). Small-RNA transcriptomics from 5 tissues facilitated annotation of microRNAs (miRNAs), identifying 290 miRNA genes that produced 334 unique mature miRNAs (Table 1 and Supplementary Tables 5 and 6). The icefish genome contains 50.4% repetitive sequences, most of which (47.4% of the total genome) are transposable elements (Supplementary Table 7 and Supplementary Note 1). Inferring the history of repeat elements by calculating the relative age of transposable element copies through Kimura distance analyses and comparisons with other teleosts (Supplementary Note 1) revealed a recent burst of DNA transposons and long and short interspersed elements. This result is consistent with the hypothesis22 that exposure to strong environmental changes, such as cooling to sub-zero temperatures and a series of glaciation and deglaciation cycles, led to massive mobilization of transposable elements.
Genetic linkage map and genome assembly integration
To make a chromonome (a chromosome length genome assembly)23, we constructed a genetic map for blackfin icefish. RAD-tag sequencing24 produced 20 million reads each for male and female parents, and an average of 2.4 million reads for each of 83 individual progeny. Stacks software25 identified 60,038 RAD-tags, of which 56,256 (93.7%) were present in at least 10 progeny. Of 7,215 polymorphic RAD-tags, 4,952 (55.8%) were present in at least 60 of 83 progeny and 4,023 localized to the male map, the female map or both at a minimum logarithm of the odds (LOD) of 12. JoinMap 4.1 assigned markers to 24 linkage groups (Supplementary Fig. 4, accession SRP118539)—one for each cytogenetic chromosome26. Because the map showed that each icefish chromosome was an orthologue of each medaka chromosome, we numbered icefish linkage groups to match their medaka counterparts. C. aceratus linkage group 6 (Cac6) had the most markers (202) and Cac2 had the fewest (131). Cac21 was the longest (65.8 cM) and Cac12 was the shortest (46.7 cM). Chromonomer software (http://catchenlab.life.illinois.edu/chromonomer) aligned contigs to the genetic map. Of 3,852 contigs in the assembly, 1,063 (27%) aligned on the genetic map by at least one marker for a total length of 820 Mb of the 1,065 Mb (77%) assembly. Only one contig was chimeric (Ice_000013): one end mapped to Cac7 and the other to Cac14 (Supplementary Fig. 4).
Synolog27 displayed conserved syntenies, revealing that each icefish chromosome is orthologous to a single chromosome in both medaka (Oryzias latipes, Ola; Fig. 1a) and European sea bass (Dicentrarchus labrax, Dla; Supplementary Fig. 5a). We detected a single small internal translocation (Supplementary Fig. 5d,e), but no reciprocal chromosomal translocations were found in the lineages of icefishes, sea bass and medaka since their lineages diverged ~113 Ma28. Chromosome stability in teleost fish is remarkable compared with mammals, where, for example, different deer species have between 3 and 40 haploid chromosomes and different rodents have between 5 and 51 (ref. 29). Although the blackfin icefish retains the ancestral chromosome number, many Antarctic notothenioids do not; for example, different species in the genus Notothenia have 13, 12 or 11 chromosomes rather than the ancestral 24 due to centromeric fusions of entire ancestral chromosomes30.

a, Gene content in icefish chromosomes supports a one-to-one correspondence between icefish and medaka chromosomes. Each line represents orthologous genes in icefish and medaka, colour-coded by icefish chromosome. The few lines that cross linkage groups (LGs) probably represent paralogues. b, A comparison of orthologous gene orders in icefish LG12 (Cac12) and medaka LG12 (Ola12) illustrates icefish-specific chromosome inversions and transpositions (see text). Each line represents orthologous genes in the icefish and medaka chromosome, colour-coded by icefish genomic scaffold. Conserved syntenic blocks are labelled 1–8. c, Comparison of orthologous gene order in Cac12 and European sea bass LG19 (Dla19). Conserved syntenic blocks are labelled 1–8. d, Comparison of orthologous gene order between sea bass Dla19 and medaka Ola12 reveals that most chromosome rearrangements occurred after the divergence of the icefish lineage from the sea bass lineage. M, megabase position along the chromosome.
Although orthologous chromosomes in icefish, sea bass and medaka chromosomes share gene content, gene order was often not well conserved. For example, Cac12 and Ola12 contain multiple conserved syntenic blocks (Fig. 1b, blocks 1–8) rearranged by inversions and transpositions. Most of those blocks have the same order in Ola12 and Dla12, showing that rearrangements occurred in the icefish lineage after it separated from the sea bass lineage. Conserved blocks ‘4’ and ‘5’ appear in the opposite order in sea bass and stickleback chromosomes (Fig. 1c and Supplementary Fig. 5b), and comparisons between sea bass and medaka (Fig. 1d) or stickleback (Supplementary Fig. 5c) suggest an inversion in the medaka lineage. Other lineage-specific rearrangements in Cac12 are evident, and analysis of other chromosomes confirms that, despite the paucity of translocations over more than 100 Myr (Fig. 2a), multiple rearrangements within chromosomes occurred in the icefish lineage after it separated from the sea bass lineage (Supplementary Fig. 6).

a, Phylogenetic tree and gene family gain-and-loss analysis, including the number of gained gene families (+) and lost gene families (−). Blue numbers specify divergence times between lineages. The red dotted line indicates the appearance of Antarctic ice sheets (35 Ma), which allowed the circum-Antarctic current to form after the opening of the Drake Passage. Subsequent cooling of the Southern Ocean drove local extinction of most fish taxa and adaptive radiation of the Antarctic notothenioid suborder. E, Eocene; M, Miocene; O, Oligocene; P, Palaeocene. b, Inferring icefish population history by PSMC analysis. The left y axis represents the demographic history of C. aceratus (red line). During the Plio-Pleistocene (3–0.9 Ma), which is shaded blue, Antarctic sea-surface temperatures dropped by around 2.5 °C, judged by a proxy for marine palaeo-temperature changes based on oxygen isotope ratios91,92 (right y axis). Concomitant decreases in marine temperatures (black line) probably allowed the cold-adapted C. aceratus populations to increase in size. The green shading represents the mid-Pleistocene transition, during which temperature fluctuations were large. g, generation time; μ, mutation rate.
Phylogenomics and genome expansion
A comparison of genome sequences by OrthoMCL showed that the blackfin icefish has 18,636 of 24,159 orthologous gene clusters identified in 13 teleosts. A genome-wide set of 3,718 one-to-one orthologues provided a phylogenetic tree of 13 teleosts using maximum likelihood (Supplementary Tables 8 and 9). According to the time-calibrated phylogeny, the common ancestor of the three Antarctic fishes with genome sequences (C. aceratus, Parachaenichthys charcoti (Charcot’s dragonfish) and Notothenia coriiceps (bullhead notothen)) diverged from the stickleback lineage ~77 Ma, and icefishes diverged from the dragonfish lineage ~7 Ma (Fig. 2a and Supplementary Fig. 7). Gene family analysis identified a core set of 9,647 gene families that were shared among 6 represented fishes (three Antarctic species, stickleback, medaka and zebrafish) and 445 blackfin icefish-specific gene families (Supplementary Fig. 8).
The icefish has 373 significantly expanded and 346 significantly contracted gene families based on the z score of gene count differences among 13 teleosts (Supplementary Tables 10 and 11). The blackfin icefish lineage experienced the largest gene family turnover among the 13 species after it diverged from the dragonfish (significant gains: 280 genes; significant losses: 6 genes) (Fig. 2a and Supplementary Table 12). Gene families with a significant number of genes gained were enriched for sensory perception (Supplementary Note 2), oxidoreductase activity and ion binding (Supplementary Table 13). Forty genes appeared to be positively selected specifically in the icefish lineage after divergence from the dragonfish lineage (Supplementary Table 14). Positively selected genes were enriched in two functional categories: oxidoreductase activity (presumably related to life without haemoglobin) and lipid binding (presumably related to buoyancy increase connected to changes from a strictly benthic lifestyle) (Supplementary Table 15).
Genomic variation and population history
We identified 9,365,677 heterozygous single nucleotide polymorphisms in the genome of the sequenced icefish female, resulting in a frequency of heterozygous sites in the sequenced fish of 8.79 × 10−3, which is greater than other individual genomes of marine fish such as the Atlantic cod (2.09 × 10−3)31 and ocean sunfish (0.78 × 10−3)32. Analysis using the pairwise sequentially Markovian coalescent (PSMC) model33 suggested two epochs that shaped icefish demographic history. First, icefish populations appeared to reach maximum size ~1 Ma at the end of the Plio-Pleistocene cooling event (3.0–0.9 Ma), after Antarctic ocean surface temperatures had dropped by 2.5 °C34. Adaptations made during the slow cooling of the Plio-Pleistocene may have allowed the icefish lineage time to achieve its maximum effective population size. Second, icefish populations appeared to decline during temperature fluctuations in the mid-Pleistocene transition (~1.2–0.55 Ma)35 (Fig. 2b), which probably presented a physiological burden for the thermally sensitive icefish35.
Expansion of AFGP and zona pellucida gene families
AFGP genes, which evolved from trypsinogen genes36, were tandemly duplicated in icefish as they are in Antarctic toothfish (Dissostichus mawsoni)37 (Fig. 3a). Our results show that the Antarctic fish AFGP–trypsinogen locus is situated between mitochondrial ribosomal protein L (mrpl) and E3 ubiquitin-protein ligase CBL (cbl), consistent with the location of the trypsinogen gene in several percomorph teleosts (Fig. 3a). A low-coverage Illumina-based draft assembly of the C. aceratus genome annotated 4 copies of AFGP genes38, whereas our results revealed 11 copies of AFGP genes adjacent to 10 tandem copies of trypsinogen genes and 2 copies of trypsinogen-like protease genes.

a, AFGP and trypsinogen gene loci. The pink-shaded area indicates the trypsinogen gene locus. kbp, kilobase pair. b, Zona pellucida c5 (zpc5) locus. c, sod3 gene cluster. Genomic neighbourhoods are shown within representative sequenced teleost genomes. Each arrow indicates a complete gene orientated in the (5′ → 3′) direction. d. Phylogenetic analysis of vertebrate sod3 genes. Divergence times were calculated by applying the mutation rate formula μ = D/2t = 3.28 × 10−9. Ca, C. aceratus (icefish); Dr, D. rerio (zebrafish); Ga, G. aculeatus (stickleback); Nc, N. coriiceps (bullhead notothen); Ol, O. latipes (medaka); Pf, P. Formosa (Amazon molly); Tr, T. rubripes (fugu); Xm, X. maculatus (platyfish).
Antarctic fish embryos do not appear to express AFGP genes12,39, which raises the question of how these embryos resist freezing12. Zona pellucida egg-coat proteins play roles in fertilization and preventing polyspermy in mammals40, and provide thickness and hardness to fish eggshells41. Zona pellucida proteins from Antarctic toothfish depress the melting point of ice12. The zona pellucida protein family expanded extensively in the C. aceratus genome: 131 zona pellucida genes, including 109 tandemly duplicated genes on 20 contigs (Supplementary Table 16), fell into 11 subfamilies based on sequence similarities (Supplementary Table 17). In contrast with our icefish sequence, 2 other Antarctic species—the bullhead notothen N. coriiceps42 and dragonfish P. charcoti43—had only 18 or 30 zona pellucida genes, respectively, and only 16–35 zona pellucida genes appeared in 7 non-Antarctic teleosts based on Ensembl queries. Furthermore, transcripts of only 18 zona pellucida genes were found in the Antarctic toothfish12. Blackfin icefish had many more copies of the zpax1, zpc1, zpc2 and zpc5 genes than other fish (Supplementary Table 16). For instance, 18 zpc5 genes were tandemly duplicated in a single contig (Ice_000281) that also contained two zpc3 paralogues (Fig. 3b). Another contig (Ice_000114) had five zpc3 and three zpc5 genes, which suggests conserved synteny of these paralogues, in agreement with their location in the genomes of stickleback and medaka (Supplementary Fig. 9). The ovaries and liver express zona pellucida genes in vertebrates44, and most C. aceratus zona pellucida genes were strongly expressed in the ovaries, similar to three other Antarctic fish12 (Supplementary Fig. 10), although transcription of several zona pellucida genes was detected in other C. aceratus organs. It is possible that the extra-ovarian expression observed for some zona pellucida paralogues represents adaptive, C. aceratus-specific neofunctionalization45 of this expanded gene family.
Genes for oxygen-binding proteins
Most teleost genomes have two globin gene clusters that arose during the teleost genome duplication: the LA cluster (with lcmt1 and aqp8 on one side and rhbdf1b on the other) and the MN cluster (flanked by mpg and nprl3 on one side and kank2 on the other)46. Within each teleost hb cluster, α- and β-chain genes generally alternate, in contrast with mammals, in which α- and β-gene clusters reside on different chromosomes46. The loss of hbb genes and pseudogenation of hba genes is an icefish synapomorphy; 15 of 16 icefish species retained only a 3′ fragment of an α-globin gene2. The sixteenth species retained an intact but unexpressed hba gene fused to two β-pseudogenes, which was probably inherited from ancestors due to incomplete lineage sorting2. The results show that the residual α-globin fragment in blackfin icefish mapped to the LA cluster, and the blackfin icefish genome possesses no trace of the MN cluster, although surrounding genes were preserved intact (Supplementary Fig. 11). In contrast, intact genes for myoglobin47, cytoglobin48 and neuroglobin49 appear in the C. aceratus genome in the context of conserved synteny among teleosts (Supplementary Fig. 12). Our sequence provides the substrate to learn the molecular genetic mechanisms that inhibit myoglobin expression, which remain to be elucidated.
Oxidative stress
Some icefishes, including C. aceratus, are more sensitive to oxidative stress than red-blooded notothenioids are50,51,52. The volume of polyunsaturated-fatty-acid-rich mitochondria per volume of skeletal or cardiac muscle cell in icefishes is approximately twice as large as in red-blooded Antarctic fishes, which may make icefishes more susceptible to reactive oxygen species (ROS) formation and lipid peroxidation at current environmental temperatures52. Furthermore, the lower thermal tolerance of icefishes relative to red-blooded notothenioids may be due to increased protein and lipid damage in icefish cardiac muscle50. If disrupted, the respiratory chain in icefish mitochondria generates more ROS than red-blooded Antarctic fish51. Finally, levels of antioxidants in icefishes are low relative to red-blooded notothenioids52. These data suggest that some icefishes are probably under selective pressure to enhance their antioxidant defence systems53.
Gene families associated with ROS homeostasis (Supplementary Table 18), including those encoding superoxide dismutase (SOD) and NAD(P)H:quinone acceptor oxidoreductase (NQO), were expanded in the C. aceratus genome. We found that blackfin icefish has five sod genes (sod1, sod2 and three tandemly repeated copies of sod3) compared with just three sod genes typical for other percomorph teleosts (Fig. 3c and Supplementary Table 19). Mutation rate analysis suggested that the sod3 gene duplicates arose as recently as ~2.3 Ma (Fig. 3d). Because the multiple sod3 genes appear to encode extracellular SOD3 enzymes (each protein possesses an apparent secretory signal peptide), an understanding of their roles in extracellular versus intracellular ROS homeostasis will require further study.
The expansion of nqo1 genes in the icefish genome was striking: we found a total of 33 genes, in contrast with the 2–10 nqo1 genes annotated in most fish genomes (Supplementary Fig. 13). Teleosts generally have two loci containing nqo1 genes in the genomic contexts vang-nqo1s-ackr3 and il17-nqo1-gabarapl; both regions appear to have been ancestrally linked, as they are today in pufferfish (Takifugu rubripes) and medaka, and the icefish nqo1 genes conformed to this pattern. However, C. aceratus possessed 26 additional nqo genes on 3 contigs that did not appear to be part of the 2 conserved nqo1 loci (Supplementary Fig. 13). In addition, the icefish is the only sequenced teleost to have two tandem copies of 8-oxoguanine DNA glycosylase (ogg1), which encodes a protein that excises from DNA a modified base that arises from reactive oxygen damage, whereas other sequenced teleost genomes have just one ogg1 copy (Supplementary Fig. 14).
Circadian adaptation to extremely fluctuating photoperiods
Polar species inhabit an environment with extreme annual fluctuations of day length, raising questions regarding the role of circadian rhythm genes in these organisms. The cry and per genes regulate a phylogenetically conserved circadian feedback loop by reciprocal transcriptional controls54. Teleost genomes have various numbers of cry genes (for example, seven in zebrafish and five in stickleback)55. Although icefish maintained the genomic structure around cry and per genes that is strongly conserved in teleost genomes, cry1, cry2, per2a and per3 sequences appeared to be specifically deleted in icefish evolution (Fig. 4a and Supplementary Table 20). The icefish genome possesses only three cry genes—the smallest number identified in any teleost. Although the bullhead notothen and dragonfish genome assemblies are incomplete, they also possess and lack the same circadian rhythm genes as blackfin icefish; thus, available evidence promotes the hypothesis that the extremes of winter darkness and summer light may have reduced the utility of, and hence decreased the pressure to retain, some circadian rhythm regulators in Antarctic fish. Behavioural studies on Antarctic icefishes and other notothenioid species will be necessary to validate this hypothesis.

a,b, Genomic structures and syntenic comparisons of the period genes per2a (a) and per3 (b). c,d, Cryptochrome gene clusters for the cry1ab (c) and cry2 (d) are shown within representative sequenced teleost genomes.
Concluding remarks
Here, we report a high-quality genome assembly and linkage map, which together provide a chromonome for the Antarctic blackfin icefish. This assembly reveals the remarkable stability of teleost chromosome contents across at least 110 Myr, with all 24 chromosomes mutually orthologous in medaka, European sea bass and blackfin icefish. Comparative analysis of the icefish chromonome revealed more inversions and intrachromosomal transpositions when comparing icefish and European sea bass than between the more anciently diverged European sea bass and medaka. Whether this rate change accompanied the chilling of Antarctica or is restricted to the icefish radiation requires comparisons with equivalently high-quality genome sequences for other notothenioid fish. The icefish genome sequence places into a genomic context the expansion of genes in the AFGP/trypsinogen/trypsinogen-like protease locus, the expansion of presumably ice-protective zona pellucida-encoding genes, and the loss of active haemoglobin genes. Also, we provide evidence for the expansion of gene families involved in the cellular redox state, including sod3 and nqo1, which might represent evolutionary adaptations to ROS production associated with mitochondrial expansion in this white-blooded clade. Uniquely, we find that some genetic regulators of circadian rhythms (cry and per paralogues) that are present in all or most other teleosts are missing from the icefish genome, fitting a hypothesis that extremes in polar day lengths altered circadian regulation in Antarctic fish. Finally, the blackfin icefish genome provides an elegant natural model to facilitate exploration of genomic contributions to a wide range of evolutionary, ecological, metabolic, developmental and biochemical features of Antarctic fish as they adapted to the extreme low temperatures, high oxygen levels and greatly fluctuating day lengths of Antarctica.
Methods
Sample collection for genome sequencing
Antarctic blackfin icefish C. aceratus (length: ~30 cm) (Supplementary Fig. 1) were collected from depths of 20–30 m in Marian Cove, near King Sejong Station, on the northern Antarctic Peninsula (62° 14′ S, 58° 47′ W) (Supplementary Fig. 2) in January 2017 using a baited hook and line. Local water temperatures averaged 1.6 ± 0.8 °C. Genomic DNA was isolated from a single female specimen. Additional C. aceratus were collected for construction of a genetic linkage map, and for miRNA sequencing in May of 2012–2015 by bottom trawling from the Antarctic Research and Supply Vessel Laurence M. Gould at depths of 160–200 m southwest of Low Island (Antarctic Specially Protected Area 152, Western Bransfield Strait; latitudes 63° 15′ S–63° 30′ S; longitudes 62° 00′ W–62° 45′ W, bounded on the northeast by Low Island)). These fish were transported alive to Palmer Station, Antarctica, where they were maintained in seawater aquaria at −1–0 °C following best practices for maintaining icefishes in captivity56.
Genome sequencing and de novo assembly
Genomic DNA of a single female icefish (Supplementary Fig. 1) was extracted from muscle tissue. Genomic DNA libraries were prepared according to the manufacturer’s instructions, and the libraries were sequenced using a PacBio Sequel System using P6-C4 sequencing chemistry (DNA Link). The average coverage of single-molecule real-time sequences was ~90-fold and the average subread length was 10.6 kilobases from genomic DNA libraries. The FALCON-Unzip assembler provided a de novo assembly57. We used BUSCO to evaluate genome completion using the 4,584-Actinopterygii gene dataset21. For genome size estimation from DNA libraries, k-mer analysis was performed using Jellyfish58 (Supplementary Table 3).
Organ-specific RNA sequencing
The RNeasy Mini Kit (Qiagen) was used, according to the manufacturer’s instructions, to extract total RNA from 12 tissues (the brain, eye, gill, heart, intestine, kidney, liver, muscle, ovary, skin, spleen and stomach) from the individual used for genome sequencing (Supplementary Table 3). The quality of the total RNA was evaluated using an Agilent Bioanalyzer (integrity values ≥ 8). Transcriptome library construction and sequencing were performed using an Illumina HiSeq 2500 system. Illumina paired-end reads (2 × 100) from each organ were mapped to icefish genomic contigs using TopHat59, and the transcriptome of each tissue was assembled using Cufflinks (http://cole-trapnell-lab.github.io/cufflinks).
Gene annotation
We used MAKER to perform genome annotation60. We constructed a de novo repeat library using RepeatModeler (version 1.0.3)61, including the RECON and RepeatScout62 software with default parameters. Tandem Repeats Finder63 was used to predict consensus sequences, classifying information for each repeat and tandem repeat, including simple repeats, satellites and low-complexity repeats. Repetitive elements were identified using RepeatMasker64. Subsequently, the repeat-masked genomes with BLASTn and protein information from tBLASTx were used for ab initio gene prediction with SNAP software65. Reference proteins from teleosts with sequenced genomes (Danio rerio, Gasterosteus aculeatus, Gadus morhua, Tetraodon nigroviridis, T. rubripes, Astyanax mexicanus, O. latipes, Poecilia formosa, Oreochromis niloticus and Xiphophorus maculatus) and two sequenced Antarctic fishes (P. charcoti and N. coriiceps) were included in the comparative analyses with transcripts of C. aceratus (Supplementary Table 3). Exonerate software, which provides integrated information for SNAP annotation, was applied to polish MAKER alignments. Next, MAKER selected and revised final gene models considering all information. MAKER predicted a total of 30,773 icefish genes. To identify non-coding RNAs in icefish contigs, we used the Infernal software package (version 1.1)66 and covariance models from the Rfam database67. Putative transfer RNA genes were identified using tRNAscan-SE68, which uses a covariance model that scores candidates based on their sequence and predicted secondary structures.
miRNA sequencing and annotation
Organs for miRNA annotation originated from the same male specimen of C. aceratus used in the study of erythropoietic miRNAs in Antarctic icefishes69. Samples of pronephric (head) kidney, pectoral girdle bone, heart ventricle, pectoral adductor muscle and skeletal muscle were dissected and stored in RNAlater at −80 °C until further use at the University of Oregon. Procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Oregon (10–26) and the IACUC of Northeastern University (12–0306R).
Total RNAs were extracted using the Zymo Research Direct-zol RNA MiniPrep kit according to the manufacturer’s instructions. Five tissue-specific small RNA libraries were then prepared and barcoded using the Bioo Scientific NEXTflex Small RNA sequencing kit with 15 PCR cycles, and sequenced by Illumina HiSeq 2500 at the University of Oregon Genomics and Cell Characterization Core Facility. Raw single-end 50-nucleotide-long reads were deposited in the National Center for Biotechnology Information Short Read Archive under accession number SRP069031. Reads from the five libraries were processed together using the bioinformatic tool Prost! (https://github.com/uoregon-postlethwait/prost)70. Briefly, raw reads were trimmed from adaptor sequences, filtered for quality using the FASTX-Toolkit, bioinformatically filtered for length between 17 and 25 nucleotides, filtered for a minimum of 5 identical reads, and grouped by genomic location. Groups of sequences were then annotated against mature and hairpin sequences present in miRBase release 21 (ref. 71), the extended zebrafish miRNA annotation72, the spotted gar annotation73 and the three-spined stickleback annotation. Gene nomenclature follows recent conventions74, including those for zebrafish75.
Construction of the C. aceratus genetic linkage map
Animals and in vitro fertilization
The C. aceratus linkage map was produced from Low Island specimens obtained in 2012. Eggs from a single female and sperm from a single male were stripped, gametes were mixed, and progeny from this single in vitro fertilization event were maintained at about −1 °C until embryos were 2 months old, when they were euthanized and stored in 100% EtOH at −20 °C. We euthanized male and female parents by MS-222 overdose, then collected samples of muscle, liver, fin and spleen, which we stored in 100% EtOH at −20 °C. From each individual embryo and each individual parent, we isolated genomic DNA using the DNeasy Blood & Tissue Kit (Qiagen). The University of Oregon IACUC approved all protocols (13–27RR).
Creation of RAD-tag libraries
Genomic DNA was purified from the male and female parents and 91 of their progeny. RAD-tag libraries were created as described24,30. DNA from each embryo was digested with high-fidelity SbfI (New England Biolabs) restriction enzyme overnight and each sample was separately barcoded with P1 and P2 adaptor ligations overnight, using 91 different 5-nucleotide barcodes. Pooled, size-selected DNA (50 ng) was amplified by PCR for 12 cycles, and the PCR product was gel purified by excising a 200–500 base pair (bp) fraction. Libraries were sequenced on the University of Oregon’s Illumina HiSeq 2000 to obtain 100-nucleotide single-end reads.
Marker genotyping
Sequenced reads from the Illumina runs were sorted by barcode, allowing up to one mismatched nucleotide in the barcode sequence. Reads containing uncalled bases and those that had an average Phred quality score that fell below 10 over 15% of the read length were discarded. Retained reads were genotyped using Stacks software version 1.30 (ref. 25). Progeny with fewer than 1,000,000 reads were excluded from Stacks analysis. The parameters for Stacks provided to denovo_map.pl were: a minimum of 20 reads for a stack (-m 20), up to 4 differences when merging stacks into loci (-M 4) and up to 2 fixed differences when merging loci from the parents into the catalogue (-n 2). Stacks exported data into JoinMap 4.1 (ref. 76) for linkage analysis.
Map construction
Of the 91 F1 individual progeny sequenced, 8 fish had too little coverage or too many missing genotypes (>20%) to use for mapping. Linkage analysis was performed with JoinMap 4.1 (ref. 76), with markers present in at least 60 of the 83 remaining individuals. Markers were initially grouped in JoinMap 4.1 using the ‘independence LOD’ parameter under ‘population grouping’ with a minimum LOD value of 12.0. Markers that remained unlinked at LOD < 12 were excluded. Marker ordering was performed using the maximum-likelihood algorithm in JoinMap 4.1 with default parameters. Putative double recombinants were identified using the ‘genotype probabilities’ feature in JoinMap 4.1 and by visual inspection of the colourized graphical genotypes. After visual inspection of the individual sequences in Stacks, markers were corrected as needed. For example, if a double recombinant was a homozygote with a small number of reads, the genotype was eliminated because it might represent a heterozygote that by chance lacked sufficient depth to provide a sequence for the second allele. Likewise, if a double recombinant was a heterozygote with only one sequence for the second allele, the genotype was eliminated because the second sequence might represent a sequencing error. The new dataset with corrected genotypes was loaded again into JoinMap 4.1, and linkage analysis was repeated until JoinMap identified no suspicious genotypes. The ‘expected recombination count’ feature in JoinMap 4.1 was used to identify individuals with more recombination events than expected. Visual inspection of the marker order was performed, and when necessary, the marker order was manually optimized by moving a marker or group of markers to a new position that reduced the total number of recombination events.
Integration of the genome assembly and linkage map
The nucleotide sequence of the 4,023 polymorphic markers localized on the genetic linkage map was used as a search query against the sequence of assembled icefish contigs using GSNAP77, requiring unique alignments, and allowing up to 10% mismatches (-m 0.1). Markers from the genetic linkage map were integrated with the contigs from the icefish genome assembly to create a chromosome level assembly (or chromonome) using the software Chromonomer (http://catchenlab.life.illinois.edu/chromonomer/) as described27.
Analysis of conserved syntenies
We used J. Catchen’s software Synolog27 to visualize the location of orthologous and paralogous genes among the genomes of icefish, medaka, European sea bass and stickleback, and to identify regions of conserved synteny between the different species.
Comparative genomics analyses
We identified orthologous gene clusters using the OrthoMCL78 pipeline, which applies the Markov clustering algorithm. Default options were used in all steps for genome sequences from 13 species (Supplementary Table 8). Analysis utilized available well-annotated and well-assembled genomes of species selected to represent clades critical for the analyses. For icefish, we used coding sequences based on the MAKER annotation pipeline. The orthologous gene clusters were categorized from one-to-one to many-to-many. Single-copy genes and species-specific genes were considered as singletons. Phylogenetic tree construction was performed based on one-to-one, single-copy orthologous genes. The sequences of protein-coding genes were aligned using the Probabilistic Alignment Kit (PRANK)79 with the codon alignment option. Poorly aligned regions with gaps were removed using Gblocks80 with a codon model for subsequent procedures. The maximum-likelihood method was applied to construct a phylogenetic tree using RAxML with 1,000 bootstraps, and divergence times were calibrated with TimeTree (median estimates of the pairwise divergence time for D. rerio and G. morhua: 230.4 Ma)81 using MCMCtree implemented in PAML packages82. The likelihood analysis for gene gain and gene loss was identified using CAFÉ 4.0 (ref. 83) with P < 0.05. The separate birth (λ) and death (μ) rates were estimated using the lambdamu command implemented in CAFÉ 4.0, with –s and –t options.
Positively selected genes
We identified 6,715 orthologous groups shared by 13 teleosts. A total of 3,718 single-copy gene families were used to construct a phylogenetic tree and to estimate times since lineage divergence using the methods described above. Using PRANK, orthologous gene sequences were aligned, and poorly aligned sequences with gaps were removed using Gblocks80. Alignments showing less than 40% identity and genes shorter than 150 bp were eliminated in subsequent procedures. Using the Codeml programme, the values of dN (ratio of nonsynonymous), dS (ratio of synonymous) and ω (average dN/dS ratio) were estimated for each gene implemented in the PAML package with the free-ratio model82 under F3X4 codon frequencies. The orthologues with ω ≤ 5 and dS ≤ 3 were retained84. We applied basic and branch-site models to define positively selected genes, and likelihood ratio tests were used to remove genes under relaxation of selective pressure. To identify functional categories and pathways that were enriched in the positively selected genes, we performed the Blast2GO enrichment test85 with the Fisher’s exact test (cutoff: P ≤ 0.05).
Inference of demographic history
The PSMC analysis33, based on a hidden Markov model, has been used to estimate the history of effective population sizes based on genome-wide heterozygous sequence data32,86. To obtain consensus diploid sequences, error-corrected reads from the female icefish used for genome sequencing were aligned back to the reference genome using BWA-MEM (BWA version 0.7.15)87 with default settings. Heterozygous variants were called using SAMtools mpileup (version 1.2) with bcftools call (version 1.2) and vcfutils.pl vcf2fq pipeline (https://github.com/lh3/psmc) with the following settings: the excessive mismatch containing reads filtering (-C) was set to 50, the minimum read depth (-d) was set to 8 (which is one-third of the average genome coverage of 24) and the maximum read depth (-D) was set to 49 (which is twice the average genome coverage). Genome-wide consensus fastq files were converted to PSMC modelling input files (psmcfa) using the fq2psmcfa tool provided in the PSMC package. Inference of population history was carried out using the psmc tool with the options ‘-N25 -t15 -r5 -p “4 + 25*2 + 4 + 6’ and plotted setting the generation time to 7 (because C. aceratus first spawns eggs at an age of 6–8 years) and a mutation rate of 3.28 × 10−9 (which is the rate obtained in this study). The mutation rate was estimated based on the Jukes–Cantor distance (D) between C. aceratus and P. charcoti among 3,718 one-to-one orthologous genes using the mutation rate formula μ = D/2t = 3.28 × 10−9 (D = 0.011 and t = 6.7 Myr were obtained in this study).
Gene family analyses
Genes belonging to families of interest were curated using manual gene search methods. Target gene sequences from the genomes of several teleost fishes were used to directly search the icefish genome database. The protein sequences of these manually curated genes were aligned using Clustal Omega88 and MUSCLE89. Phylogenetic trees were constructed using FastTree90 or RAxML with 1,000 bootstraps. The timing of gene duplication events was estimated using the mutation rate formula μ = D/2t = 3.28 × 10−9.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The Antarctic blackfin icefish C. aceratus genome and transcriptome data have been deposited in the NCBI database as BioProject PRJNA420419. RAD-tag sequences of all mapped individuals are available online at the NCBI Sequence Read Archive (accession number SRP118539).
References
Ruud, J. T. Vertebrates without erythrocytes and blood pigment. Nature 173, 848–850 (1954).
Near, T. J., Parker, S. K. & Detrich, H. W.III A genomic fossil reveals key steps in hemoglobin loss by the Antarctic icefishes. Mol. Biol. Evol. 23, 2008–2016 (2006).
Holeton, G. F. Oxygen uptake and circulation by a hemoglobinless Antarctic fish (Chaenocephalus aceratus Lonnberg) compared with three red-blooded Antartic fish. Comp. Biochem. Physiol. 34, 457–471 (1970).
Sidell, B. D. & O’Brien, K. M. When bad things happen to good fish: the loss of hemoglobin and myoglobin expression in Antarctic icefishes. J. Exp. Biol. 209, 1791–1802 (2006).
Moylan, T. J. & Sidell, B. D. Concentrations of myoglobin and myoglobin mRNA in heart ventricles from Antarctic fishes. J. Exp. Biol. 203, 1277–1286 (2000).
Grove, T. J., Hendrickson, J. W. & Sidell, B. D. Two species of Antarctic icefishes (genus Champsocephalus) share a common genetic lesion leading to the loss of myoglobin expression. Polar Biol. 27, 579–585 (2004).
Hemmingsen, E. A. Biology of Antarctic Fish 191–203 (Springer, Berlin & Heidelberg, 1991).
Kennett, J. P. Cenozoic evolution of Antarctic glaciation, the circum-Antarctic Ocean, and their impact on global paleoceanography. J. Geophys. Res. 82, 3843–3860 (1977).
Eastman, J. T. Antarctic Fish Biology: Evolution in a Unique Environment (Academic Press, New York, 1993).
DeVries, A. The role of antifreeze glycopeptides and peptides in the freezing avoidance of Antarctic fishes. Comp. Biochem. Physiol. 90B, 611–621 (1988).
Cheng, C. H. & Detrich, H. W. 3rd Molecular ecophysiology of Antarctic notothenioid fishes. Phil. Trans. R. Soc. Lond. B 362, 2215–2232 (2007).
Cao, L. et al. Neofunctionalization of zona pellucida proteins enhances freeze-prevention in the eggs of Antarctic notothenioids. Nat. Commun. 7, 12987 (2016).
Hofmann, G. E., Buckley, B. A., Airaksinen, S., Keen, J. E. & Somero, G. N. Heat-shock protein expression is absent in the Antarctic fish Trematomus bernacchii (family Nototheniidae). J. Exp. Biol. 203, 2331–2339 (2000).
Place, S. P. & Hofmann, G. Constitutive expression of a stress-inducible heat shock protein gene, hsp70, in phylogenetically distant Antarctic fish. Polar Biol. 28, 261–267 (2005).
Near, T. J., Jones, C. D. & Eastman, J. T. Geographic intraspecific variation in buoyancy within Antarctic notothenioid fishes. Antarct. Sci. 21, 123–129 (2009).
Eastman, J. T., Witmer, L. M., Ridgely, R. C. & Kuhn, K. L. Divergence in skeletal mass and bone morphology in Antarctic notothenioid fishes. J. Morphol. 275, 841–861 (2014).
Albertson, R. C. et al. Molecular pedomorphism underlies craniofacial skeletal evolution in Antarctic notothenioid fishes. BMC Evol. Biol. 10, 4 (2010).
Hagen, W., Kattner, G. & Friedrich, C. The lipid compositions of high-Antarctic notothenioid fish species with different life strategies. Polar Biol. 23, 785–791 (2000).
Kock, K.-H. Antarctic icefishes (Channichthyidae): a unique family of fishes. A review, part I. Polar Biol. 28, 862–895 (2005).
Hu, Y. et al. Evolution in an extreme environment: developmental biases and phenotypic integration in the adaptive radiation of Antarctic notothenioids. BMC Evol. Biol. 16, 142 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Auvinet, J. et al. Mobilization of retrotransposons as a cause of chromosomal diversification and rapid speciation: the case for the Antarctic teleost genus Trematomus. BMC Genomics 19, 339 (2018).
Braasch, I. et al. A new model army: emerging fish models to study the genomics of vertebrate evo‐devo. J. Exp. Zool. B 324, 316–341 (2015).
Amores, A., Catchen, J., Ferrara, A., Fontenot, Q. & Postlethwait, J. H. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics 188, 799–808 (2011).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3 1, 171–182 (2011).
Morescalchi, A. et al. A multiple sex-chromosome system in Antarctic ice-fishes. Polar Biol. 11, 655–661 (1992).
Small, C. M. et al. The genome of the Gulf pipefish enables understanding of evolutionary innovations. Genome Biol. 17, 258 (2016).
Steinke, D., Salzburger, W. & Meyer, A. Novel relationships among ten fish model species revealed based on a phylogenomic analysis using ESTs. J. Mol. Evol. 62, 772–784 (2006).
Scherthan, H. Chromosome Numbers in Mammals. In eLS (John Wiley & Sons, Chichester, 2012); https://doi.org/10.1002/9780470015902.a0005799.pub3
Amores, A., Wilson, C. A., Allard, C. A. H., Detrich, H. W. & Postlethwait, J. H. Cold fusion: massive karyotype evolution in the Antarctic bullhead notothen Notothenia coriiceps. G3 7, 2195–2207 (2017).
Star, B. et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210 (2011).
Pan, H. et al. The genome of the largest bony fish, ocean sunfish (Mola mola), provides insights into its fast growth rate. Gigascience 5, 36 (2016).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
McKay, R. et al. Antarctic and Southern Ocean influences on Late Pliocene global cooling. Proc. Natl Acad. Sci. USA 109, 6423–6428 (2012).
Hayward, B. W., Kawagata, S., Grenfell, H. R., Sabaa, A. T. & O’Neill, T. Last global extinction in the deep sea during the mid‐Pleistocene climate transition. Paleoceanography 22, PA3103 (2007).
Chen, L., DeVries, A. L. & Cheng, C.-H. C. Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc. Natl Acad. Sci. USA 94, 3811–3816 (1997).
Nicodemus-Johnson, J., Silic, S., Ghigliotti, L., Pisano, E. & Cheng, C. H. C. Assembly of the antifreeze glycoprotein/trypsinogen-like protease genomic locus in the Antarctic toothfish Dissostichus mawsoni (Norman). Genomics 98, 194–201 (2011).
Baalsrud, H. T. et al. De novo gene evolution of antifreeze glycoproteins in codfishes revealed by whole genome sequence data. Mol. Biol. Evol. 35, 593–606 (2018).
Cziko, P. A., Evans, C. W., Cheng, C.-H. C. & DeVries, A. L. Freezing resistance of antifreeze-deficient larval Antarctic fish. J. Exp. Biol. 209, 407–420 (2006).
Wassarman, P. M. Zona pellucida glycoproteins. J. Biol. Chem. 283, 24285–24289 (2008).
Sano, K. et al. Comparison of egg envelope thickness in teleosts and its relationship to the sites of ZP protein synthesis. J. Exp. Zool. B 328, 240–258 (2017).
Shin, S. C. et al. The genome sequence of the Antarctic bullhead notothen reveals evolutionary adaptations to a cold environment. Genome Biol. 15, 468 (2014).
Ahn, D.-H. et al. Draft genome of the Antarctic dragonfish, Parachaenichthys charcoti. GigaScience 6, 1–6 (2017).
Wu, T. et al. Bioinformatic analyses of zona pellucida genes in vertebrates and their expression in Nile tilapia. Fish Physiol. Biochem. 44, 435–449 (2018).
Force, A. et al. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545 (1999).
Opazo, J. C., Butts, G. T., Nery, M. F., Storz, J. F. & Hoffmann, F. G. Whole-genome duplication and the functional diversification of teleost fish hemoglobins. Mol. Biol. Evol. 30, 140–153 (2013).
Small, D. J., Moylan, T., Vayda, M. E. & Sidell, B. D. The myoglobin gene of the Antarctic icefish, Chaenocephalus aceratus, contains a duplicated TATAAAA sequence that interferes with transcription. J. Exp. Biol. 206, 131–139 (2003).
Cuypers, B. et al. Antarctic fish versus human cytoglobins—the same but yet so different. J. Inorg. Biochem. 173, 66–78 (2017).
Cheng, C. H. C., di Prisco, G. & Verde, C. Cold-adapted Antarctic fish: the discovery of neuroglobin in the dominant suborder Notothenioidei. Gene 433, 100–101 (2009).
Mueller, I. A. et al. Exposure to critical thermal maxima increases oxidative stress in hearts of white- but not red-blooded Antarctic notothenioid fishes. J. Exp. Biol. 215, 3655–3664 (2012).
O’Brien, K. M. & Mueller, I. A. The unique mitochondrial form and function of Antarctic channichthyid icefishes. Integr. Comp. Biol. 50, 993–1008 (2010).
Mueller, I. A., Grim, J. M., Beers, J. M., Crockett, E. L. & O’Brien, K. M. Inter-relationship between mitochondrial function and susceptibility to oxidative stress in red- and white-blooded Antarctic notothenioid fishes. J. Exp. Biol. 214, 3732–3741 (2011).
Klein, R. D. et al. Antioxidant defense system and oxidative status in Antarctic fishes: the sluggish rockcod Notothenia coriiceps versus the active marbled notothen Notothenia rossii. J. Therm. Biol. 68, 119–127 (2017).
Shearman, L. P. et al. Interacting molecular loops in the mammalian circadian clock. Science 288, 1013–1019 (2000).
Liu, C. et al. Molecular evolution and functional divergence of zebrafish (Danio rerio) cryptochrome genes. Sci. Rep. 5, 8113 (2015).
Le François, N. R. et al. Characterization and husbandry of wild broodstock of the blackfin icefish Chaenocephalus aceratus (Lönnberg 1906) from the Palmer Archipelago (Southern Ocean) for breeding purposes. Polar Biol. 40, 2499–2516 (2017).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-3.0 (Institute for Systems Biology, 2017); http://www.RepeatMasker.org
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Gardner, P. P. et al. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39, D141–D145 (2010).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Desvignes, T., Detrich, H. W. & Postlethwait, J. H. Genomic conservation of erythropoietic microRNAs (erythromiRs) in white-blooded Antarctic icefish. Mar. Genom. 30, 27–34 (2016).
Batzel, P., Desvignes, T., Sydes, J., Eames, B. F. & Postlethwait, J. H. Prost!, a tool for miRNA annotation and next generation smallRNA sequencing experiment analysis. Zenodo (2018). https://doi.org/10.5281/zenodo.1937101
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73 (2014).
Desvignes, T., Beam, M. J., Batzel, P., Sydes, J. & Postlethwait, J. H. Expanding the annotation of zebrafish microRNAs based on small RNA sequencing. Gene 546, 386–389 (2014).
Braasch, I. et al. The spotted gar genome illuminates vertebrate evolution and facilitates human–teleost comparisons. Nat. Genet. 48, 427–437 (2016).
Desvignes, T. et al. miRNA nomenclature: a view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet. 31, 613–626 (2015).
Bradford, Y. et al. ZFIN: enhancements and updates to the zebrafish model organism database. Nucleic Acids Res. 39, D822–D829 (2011).
Van Oojien, J. W. et al. JoinMap 4: software for the calculation of genetic linkage maps in experimental populations (Kyazma B.V., 2006). https://www.kyazma.nl/index.php/JoinMap/
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Li, L., Stoeckert, C. J. Jr & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Löytynoja, A. & Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl Acad. Sci. USA 102, 10557–10562 (2005).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320 (2014).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Li, C. et al. Two Antarctic penguin genomes reveal insights into their evolutionary history and molecular changes related to the Antarctic environment. Gigascience 3, 27 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Sievers, F. et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Lisiecki, L. E. & Raymo, M. E. A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 20, PA1003 (2005).
Jouzel, J. et al. Orbital and millennial Antarctic climate variability over the past 800,000 years. Science 317, 793–796 (2007).
Acknowledgements
We acknowledge the support provided by the 30th Korea Antarctic overwintering members, and we extend special thanks to D.-W. Han for icefish sampling. We acknowledge logistical support provided by staff at the Division of Polar Programs of the National Science Foundation, personnel of the Antarctic Support Contract group, and captains and crews of the Antarctic Research and Supply Vessel Laurence M. Gould. This is contribution #386 from the Marine Science Center at Northeastern University. This work was supported by Korea Polar Research Institute Polar Genome 101 project grant PE18080 (to H.P.), the Deutsche Forschungsgemeinschaft and Hagler Institute of Advanced Study at Texas A&M University (to M.S.), National Institutes of Health grant R01AG031922 from the National Institute on Aging (to J.H.P. and H.W.D.), grants 5R01OD011116 and R24RR032670 (to J.H.P.) from the National Institutes of Health Office of the Director, and National Science Foundation grants ANT-0944517, PLR-1247510 and PLR-1444167 from the Division of Polar Programs (to H.W.D.) and PLR-1543383 (to J.H.P. and H.W.D.).
Author information
Authors and Affiliations
Contributions
H.W.D., J.H.P. and H.P. conceived the study. B.-M.K., S.K., D.-H.A., J.-H.K., I.-C.K., A.A., T.D., P.B., J.S., T.T., C.W., J.H.P., H.W.D., W.C.W. and J.M.C. conducted the experimental work and sequenced the genomes and transcriptomes. B.-M.K., S.K., J.H.L., S.G.L., H.L., J.L. and H.-W.K. analysed the genome data. H.W.D., A.A., T.D. and J.H.P. performed the C. aceratus cross and produced the genetic map. B.-M.K., S.K., W.C.W., M.S., H.W.D., J.H.P. and H.P. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figures 1–3 and 5–17 and Supplementary Tables 1–21
Supplementary Figure 4
Genetic linkage map for the blackfin icefish
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, BM., Amores, A., Kang, S. et al. Antarctic blackfin icefish genome reveals adaptations to extreme environments. Nat Ecol Evol 3, 469–478 (2019). https://doi.org/10.1038/s41559-019-0812-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-019-0812-7
This article is cited by
-
De novo assembly and annotation of the Patagonian toothfish (Dissostichus eleginoides) genome
BMC Genomics (2024)
-
An Antarctic lichen isolate (Cladonia borealis) genome reveals potential adaptation to extreme environments
Scientific Reports (2024)
-
Novel mitochondrial genome rearrangements including duplications and extensive heteroplasmy could underlie temperature adaptations in Antarctic notothenioid fishes
Scientific Reports (2023)
-
De novo transcriptome assembly of an Antarctic nematode for the study of thermal adaptation in marine parasites
Scientific Data (2023)
-
Genomic adaptation of giant viruses in polar oceans
Nature Communications (2023)

{kind=link}