
Abstract
Giant tortoises are among the longest-lived vertebrate animals and, as such, provide an excellent model to study traits like longevity and age-related diseases. However, genomic and molecular evolutionary information on giant tortoises is scarce. Here, we describe a global analysis of the genomes of Lonesome George—the iconic last member of Chelonoidis abingdonii—and the Aldabra giant tortoise (Aldabrachelys gigantea). Comparison of these genomes with those of related species, using both unsupervised and supervised analyses, led us to detect lineage-specific variants affecting DNA repair genes, inflammatory mediators and genes related to cancer development. Our study also hints at specific evolutionary strategies linked to increased lifespan, and expands our understanding of the genomic determinants of ageing. These new genome sequences also provide important resources to help the efforts for restoration of giant tortoise populations.
Similar content being viewed by others


Main
Comparative genomic analyses leverage the mechanisms of natural selection to find genes and biochemical pathways related to complex traits and processes. Multiple works have used these techniques with the genomes of long-lived mammals to shed light on the signalling and metabolic networks that might play a role in regulating age-related conditions1,2. Similar studies on unrelated longevous organisms might unveil novel evolutionary strategies and genetic determinants of ageing in different environments. In this regard, giant tortoises constitute one of the few groups of vertebrates with an exceptional longevity: in excess of 100 years according to some estimates.
In this manuscript, we report the genomic sequencing and comparative genomic analysis of two long-lived giant tortoises: Lonesome George—the last representative of Chelonoidis abingdonii3, endemic to the island of Pinta (Galapagos Islands, Ecuador)—and an individual of Aldabrachelys gigantea, endemic to the Aldabra Atoll and the only extant species of giant tortoises in the Indian Ocean4 (Fig. 1a). Unsupervised and supervised comparative analyses of these genomic sequences add new genetic information on the evolution of turtles, and provide novel candidate genes that might underlie the extraordinary characteristics of giant tortoises, including their gigantism and longevity.

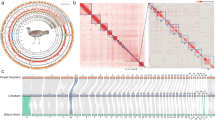
a, Satellite view of the Galapagos Islands (top; scale bar: 50 km) and Aldabra Atoll (bottom left; scale bar: 10 km), and pictures of C. abingdonii (middle) and A. gigantea (bottom right). Both pictures are from http://eol.jsc.nasa.gov. b, Demographic history of giant tortoises, inferred using a hidden Markov model approach as implemented in the PSMC model. The default mutation rate (μ) for humans of 2.5 × 10−8 and an average generation time (g) of 25 years were used in the calculations.
Results and discussion
The genome of Lonesome George was sequenced using a combination of Illumina and PacBio platforms (Supplementary Section 1.1). The assembled genome (CheloAbing 1.0) has a genomic size of 2.3 gigabases and contains 10,623 scaffolds with an N50 of 1.27 megabases (Supplementary Section 1.1 and Supplementary Tables 1–3). We also sequenced, with the Illumina platform, the closely related tortoise A. gigantea at an average read depth of 28×. These genomic sequences were aligned to CheloAbing 1.0.
TimeTree database estimations (http://www.timetree.org) indicate that Galapagos and Aldabra giant tortoises shared a last common ancestor about 40 million years ago, while both diverged from the human lineage more than 300 million years ago (Supplementary Section 1.4). A preliminary analysis of demographic history using the pairwise sequentially Markovian coalescent (PSMC)5 model showed that while the effective population size of C. abingdonii has been steadily declining for the past million years, with a slight uptick about 90,000 years ago, the population of Aldabra giant tortoises experienced substantial fluctuations over this period (Fig. 1b). Effective population size reconstructions for C. abingdonii lose statistical power at the million-year time frame, probably due to complete coalescence. In turn, this suggests that overall diversity in these giant tortoises must have been low throughout many generations. Together, these results prompt us to propose that the populations of these insular giant tortoises were vulnerable at the time of human discovery of the Galapagos Islands, probably elevating their extinction risk.
Using homology searches with known gene sets from humans and Pelodiscus sinensis (the Chinese soft-shell turtle), along with RNA sequencing (RNA-Seq) data from C. abingdonii blood and an A. gigantea granuloma, we automatically predicted a primary set of 27,208 genes from the genome assembly using the MAKER2 algorithm6. We then performed pairwise alignments between each of the primary predicted protein sequences and the UniProt databases for humans and P. sinensis, whose annotated sequences show relatively high quality when compared with data available for other turtles7. Using alignments spanning at least 80% of the longest protein and showing more than 60% identity, we constructed sets of protein families shared among these species. This preliminary analysis singled out several protein families that seem to have undergone moderate expansion in a common ancestor of C. abingdonii and A. gigantea. Almost all of these expansions were also confirmed in the genome of the related, long-lived tortoise Gopherus agassizii (Supplementary Section 1.2 and Supplementary Table 4). Most of these genes have been linked to exosome formation, suggesting that this process may have been important in tortoise evolution.
We also interrogated the predicted gene set for evidence of positive selection in giant tortoises. This analysis singled out 43 genes with evidence of giant-tortoise-specific positive selection (Supplementary Section 1.2, Supplementary Table 5 and Supplementary Fig. 1). This list includes genes with known roles in the dynamics of the tubulin cytoskeleton (TUBE1 and TUBG1) and intracellular vesicle trafficking (VPS35). Importantly, the analysis of genes showing evidence of positive selection also includes AHSG and FGF19, whose expression levels have been linked to successful ageing in humans8. The role of both factors in metabolism regulation9,10—another hallmark of ageing11,12—suggests that the specific changes observed in these proteins may have arisen to accommodate the challenges that longevity poses on this system. The list of genes with signatures of positive selection also features TDO2, whose inhibition has been proposed to protect against age-related diseases through regulation of tryptophan-mediated proteostasis13. In addition, we found evidence for positive selection affecting several genes involved in immune system modulation, such as MVK, IRAK1BP1 and IL1R2. Taken together, these results identify proteostasis, metabolism regulation and immune response as key processes during the evolution of giant tortoises via effects on longevity and resistance to infection.
Parallel to this automatic analysis, we used manually supervised annotation on more than 3,000 genes selected a priori for a series of hypothesis-driven studies on development, physiology, immunity, metabolism, stress response, cancer susceptibility and longevity (Supplementary Section 1.3 and Supplementary Fig. 2). We searched for truncating variants, variants affecting known motifs and variants whose human counterparts are related to known genetic diseases (Supplementary Section 1.3 and Supplementary Table 6). These variants were first confirmed with the RNA-Seq data. Then, more than 100 of the most interesting variants in terms of putative functional relevance were also validated by PCR amplification followed by Sanger sequencing. To this end, we used a panel of genomic DNA samples of 11 different species of giant tortoises endemic to different islands from the Galapagos Archipelago (Supplementary Section 1, Supplementary Table 7 and Supplementary Fig. 3).
The manually supervised annotation of development-related genes showed the complete conservation of the Hox gene set among giant tortoises, with the exception of HOXC3, which seems to have been lost in the radiation of Archelosauria14,15 (Supplementary Section 2, Supplementary Table 8 and Supplementary Fig. 4). BMP and GDF gene families were also found to be conserved, although the duplication event that gave rise to GDF1 and GDF3 in mammals did not occur in turtles, birds and crocodiles. In contrast, we found a duplication of the ParaHox gene CDX4 in giant tortoises, also present in other reptiles as well as avian reptiles (birds). This annotation also showed the duplication of WNT11 in turtles and chickens (but not in the lizard Anolis carolinensis), and the specific duplication of WNT4 in turtles. Given the roles of these duplicated genes and their conservation in most vertebrate species, they could prove to be useful candidates to study the morphological development of turtles, particularly in relation to shell formation. Of note, KDSR—one of the genes possibly under positive selection in giant tortoises—has been linked to hyperkeratinization disorders16. Also, in this regard, we annotated 30 β-keratins in C. abingdonii, 26 of which seem to be functional. These numbers are lower than those previously reported for β-keratins in other turtles17. Finally, we did not find in C. abingdonii or A. gigantea any functional orthologues of genes specifically involved in tooth development (such as ENAM, AMEL, AMBN, DSPP, KLK4 and MMP20). This finding confirms a pattern in the evolutionary molecular mechanisms for tooth loss, which seems to have been followed consistently and independently across vertebrates. Taken together, these results offer multiple candidates to study developmental traits in tortoises (Supplementary Section 2 and Supplementary Figs. 5–8).
In most species, the immune function is an evolutionary driver that is under strong selective pressure and has important implications in ageing and disease18. The specific components and functionality of immune system components in Reptilia, however, have not been extensively characterized beyond the major histocompatibility complex (MHC)19,20. Our detailed analysis of 891 genes involved in immune function consistently found duplications affecting immunity genes in giant tortoises compared with mammals (Supplementary Section 3, Supplementary Table 9 and Supplementary Figs. 9–13). We found a genomic expansion of PRF1 (encoding perforin) in giant tortoises and other turtles, compared with chickens (one copy), A. carolinensis (two copies) and most mammals (one copy). Both C. abingdonii and A. gigantea possess 12 copies of this gene (validated by Sanger sequencing), although three of them have been pseudogenized in C. abingdonii. In addition, we detected and validated, by Sanger sequencing, an expansion of the chymase locus, containing granzymes, in giant tortoises (Supplementary Section 3.1 and Supplementary Fig. 10). Both expansions are expected to affect cytotoxic T lymphocyte and natural killer functions, which play important roles in defence against both pathogens and cancer21,22. Other concurrent expansions involve APOBEC1, CAMP, CHIA and NLRP genes, which participate in viral, microbial, fungal and parasite defence, respectively. These results suggest that the innate immune system in turtles, and especially in giant tortoises, may play a more relevant role than in mammals, consistent with the less important role that adaptive immunity seems to play19. We found that class I and II MHC genes probably underwent a duplication event in a common ancestor between giant tortoises and painted turtles (Chrysemys picta bellii). We also annotated 40 class III MHC genes, thus confirming the conservation of this cluster in giant tortoises. The large number of MHC genes in giant tortoises is consistent with the suggestion that ancestors of archosaurs and chelonians did not possess a minimal essential MHC as found in the chicken genome20 (Supplementary Section 3.3, Supplementary Table 10 and Supplementary Figs. 14–16).
Giant tortoises are at the upper end of the size scale for extant Chelonii, and have often been used as an example of gigantism23. We analysed a series of genes involved in size regulation in vertebrates, most notably dogs (Supplementary Section 2, Supplementary Table 8 and Supplementary Fig. 6). Our results on genes related to growth hormone, the insulin-like growth factor (IGF) system and stanniocalcins suggest that these genes are well conserved; therefore, additional size determinants may exist in giant tortoises. As a complex phenotype, gigantism in tortoises is expected to be caused by interactions between different genetic and environmental factors. An interesting finding in this regard is the presence of several gene variants in tortoises (including G. agassizii) probably affecting the activities of glucose metabolism genes, such as MIF (p.N111C; expected to yield a locked trimer) and GSK3A (p.R272Q in the activation loop). Given the roles of these positions in the mammalian orthologues of these genes, tortoise-specific changes could point to differences in the regulation of glucose intake and tolerance (Supplementary Section 4, Supplementary Table 11, and Supplementary Figs. 17 and 18). We also found expansions and inactivations in other genes involved in energy metabolism. Thus, glyceraldehyde-3-phosphate dehydrogenase (GAPDH)—a glycolytic enzyme with a key role in energy production, as well as in DNA repair and apoptosis24—is expanded in giant tortoises. Conversely, the NLN gene encoding neurolysin is pseudogenized in tortoises. The loss of this gene in mice has been related to improved glucose uptake and insulin sensitivity25. Taken together, these results led us to hypothesize that genomic variants affecting glucose metabolism may have been a factor in the development of tortoises.
The analysis of genes related to the stress response has also highlighted several putative variants in giant tortoises affecting globins and DNA repair factors (Supplementary Section 5, Supplementary Tables 12 and 13, and Supplementary Figs. 19–22, 32 and 33). We found that, despite living terrestrially, giant tortoises conserve the hypoxia-related globin GbX26. Together with coelacanths, turtles, including giant tortoises, are the only organisms known to possess all eight different types of globins27. Consistent with this, we found in both giant tortoise genomes a variant in the transcription factor TP53 (p.S106E) that has been linked to hypoxia resistance in some mammals and fishes28. The presence of the same residue in Testudines strongly suggests a process of convergent evolution in the adaptation to hypoxia, probably driven by an ancestral aquatic environment, which left this footprint in the genomes of terrestrial giant tortoises.
An important trait of large, long-lived vertebrates is their need for tighter cancer protection mechanisms, as illustrated by Peto’s paradox29,30. In turn, this need for additional protection illustrates the deep relationship and interdependence between cancer and longevity (Fig. 2). Notably, tumours are believed to be very rare in turtles31. Therefore, we analysed more than 400 genes classified in a well-established census of cancer genes as oncogenes and tumour suppressors32. Although most presented a highly conserved amino acid sequence when compared with the sequences of other organisms, we uncovered alterations in several tumourigenesis-related genes (Fig. 2a, Supplementary Section 6, Supplementary Table 14 and Supplementary Figs. 23–29). First, we found that several putative tumour suppressors are expanded in turtles compared with other vertebrates, including duplications in SMAD4, NF2, PML, PTPN11 and P2RY8. In addition, the aforementioned expansion of PRF1, together with the tortoise-specific duplication of PRDM1, suggests that immunosurveillance may be enhanced in turtles. Likewise, we found giant-tortoise-specific duplications affecting two putative proto-oncogenes—MYCN and SET. Notably, the SET complex mediates oxidative stress responses induced by mitochondrial damage through the action of PRF1 and GZMA in cytotoxic T lymphocyte- and natural killer-mediated cytotoxicity33. Taken together, these results suggest that multiple gene copy-number alterations may have influenced the mechanisms of spontaneous tumour growth. Nevertheless, further studies are needed to evaluate the genomic determinants of putative giant-tortoise-specific cancer mechanisms.

a, Genes potentially implicated in C. abingdonii and A. gigantea longevity extension and cancer resistance, classified according to their putative role in the different hallmarks. Tables indicate copy-number variations and relevant variants of age-related genes and tumour suppressors found in C. abingdonii, A. gigantea and other species. Within these tables, numbers indicate gene copy numbers, and asterisks represent pseudogenization events. Dots in colours relating to each hallmark represent presence of the variant. b, Venn diagrams showing the relationships between cancer-, ageing- and immunity-related genes, as classified before annotation. Top, all of the genes related to each category that have been manually annotated, including the number of genes in each group. Bottom, those genes showing potentially interesting variations after annotation.
Finally, we selected, for manually supervised annotation, a set of 500 genes that may be involved in ageing modulation (Supplementary Section 7 and Supplementary Table 15). The extreme longevity of giant tortoises is expected to involve multiple genes affecting different hallmarks of ageing11. We found several alterations in the genomes of giant tortoises that may play a direct role in six of them, and impinge on other ageing hallmarks and processes, such as cancer progression34 (Fig. 2b). First, we identified changes in three candidate factors (NEIL1, RMI2 and XRCC6) related to the maintenance of genome integrity, a primary hallmark of ageing11 (Fig. 3a). Thus, we found and validated a duplication affecting NEIL1, a key protein involved in the base-excision repair process whose expression has been linked to extended lifespans in several species35. Likewise, RMI2 is duplicated in tortoises, suggesting an enhanced ability to resolve homologous recombination intermediates to limit DNA crossover formation in cells36. In a preliminary exploration of this hypothesis, we overexpressed NEIL1 and RMI2 in HEK-293T cells and exposed the infected cells to a sublethal dosage of H2O2 or ultraviolet light, monitoring DNA damage by western blot analysis at 24 and 48 h after treatment. As shown in Supplementary Figs. 22, 32 and 33, the expression of both genes results in reduced levels of phosphorylated histone H2AX and cleaved poly (ADP-ribose) polymerase (PARP), suggesting reduced levels of DNA damage37. In turn, this result is consistent with the hypothesis that NEIL1 and RMI2 levels may regulate the strength of DNA repair mechanisms. Also in relation to DNA repair mechanisms, we identified and validated a variant affecting XRCC6—encoding a helicase involved in non-homologous end joining of double-strand DNA breaks—which may affect a known sumoylation site (p.K556R). This lysine is conserved in diverse vertebrates but, notably, is changed in giant tortoises, and also in the naked mole rat (p.K556N), the longest-lived rodent, which suggests a putative process of convergent evolution (Fig. 3b). Since sumoylation is induced following DNA damage and plays a key role in DNA repair response and multiple regulatory processes38, this variant may reflect selective pressures acting on the regulation of the repair of double-strand DNA breaks in long-lived organisms (Supplementary Section 5.5).

a, Copy-number variations and putative function-altering point variants found in C. abingdonii, A. gigantea and closely related species. b, Alignments showing the variants highlighted in XRCC6 and DCLRE1B.
Regarding telomere attrition—another primary hallmark of ageing11—we uncovered in giant tortoises one variant in DCLRE1B (p.R498C) potentially affecting its binding interface with telomeric repeat binding factor 2 (TERF2) (Fig. 3b and Supplementary Section 7.2). This change, together with the aforementioned variants affecting DNA repair genes that may also impinge on telomere dynamics39,40,41, highlights the relevance of telomere maintenance as a regulatory mechanism of longevity in tortoises. Moreover, we found changes potentially affecting proteostasis (Fig. 2a). We independently found specific expansions of the elongation factor gene EEF1A1 in C. abingdonii, A. gigantea and G. agassizii, as described with the automatic annotation. Importantly, overexpression of EEF1A1 homologues in Drosophila melanogaster has been linked to an increased lifespan in this species42.
Over time, nutrient sensing deregulation—another hallmark of ageing—can result from alterations in metabolic control mechanisms and signalling pathways12. The aforementioned variant affecting the activation loop of GSK3A (Supplementary Section 4.1), which is present in C. abingdonii and all tested tortoises from the Galapagos Islands and Aldabra Atoll, as well as their continental outgroups, G. agassizii and C. picta bellii, may be involved in the maintenance of glucose homoeostasis. Interestingly, the inhibition of GSK3 can extend lifespan in D. melanogaster43. Likewise, the identified alterations in other giant tortoise genes implicated in glucose metabolism, such as the aforementioned inactivation of NLN, may provide interesting candidates to study nutrient sensing in these long-lived species (Supplementary Section 7.4).
Regarding the mitochondrial function, we found two variants (p.Q366M and p.M487T) potentially affecting the function of ALDH2, a mitochondrial aldehyde dehydrogenase involved in alcohol metabolism and lipid peroxidation, among other detoxification processes44. Notably, the p.Q366M variant, which may alter the NAD-binding site of ALDH2, is exclusively found in Galapagos giant tortoises, but not in their continental close relative Chelonoidis chilensis, nor in the more distantly related Aldabra or Agassiz’s tortoises. Thus, these changes could also alter the detoxification process and contribute to pro-longevity mechanisms. Together with the above described specific alterations in other genes of giant tortoises, such as NLN and GAPDH, which encode enzymes associated with mitochondrial functions45,46, these variants may also impinge on mitochondrial dysfunction, an antagonistic hallmark of ageing11 (Supplementary Section 7.5).
We have also found evidence in tortoises of some variants related to altered intercellular communication (Supplementary Section 7.6 and Supplementary Fig. 30), an integrative hallmark of ageing11. Thus, we have detected exclusively in C. abingdonii a premature stop codon affecting ITGA1 (p.R990*), an essential integrin involved in cell–matrix and cell–cell interactions. In addition, the aforementioned variant affecting MIF is also expected to cause the formation of inactivating interchain disulfide bonds, inhibiting intracellular signalling cascades47. Moreover, MIF deficiency reduces chronic inflammation in white adipose tissue and expands lifespan, especially in response to caloric restriction48,49. Finally, we have annotated a specific variant in IGF1R that is expected to affect the interaction between this receptor and the IGF1/2 growth factors50. Notably, a homology model of this region in IGF1R in C. abingdonii suggests that position 724 is located at the surface of the protein, and the presence of an aspartic acid residue changes the local electrostatic field (Fig. 4a). The extended lifespan in different species correlates with IGF signalling decrease51,52, which suggests that this unique change in IGF1R may provide an attractive target to study the cellular mechanisms underlying the exceptional lifespan of these animals. To explore the functional consequences of differential IGF1 signalling caused by the p.N724D variant found in the IGF1 receptor (IGF1R), we infected HEK-293T cells with pCDH, pCDH-IGF1RWT and pCDH-IGF1RN724D plasmids. Cells expressing the mutant receptor showed an attenuation of IGF1 signalling, compared with those expressing the wild-type protein, measured as a significant reduction in the phosphorylation levels of IGF1R at 5 min (95% confidence interval of difference: 0.1119–1.5330, t = 2.454, P = 0.026) and 10 min (95% confidence interval of difference: 0.1991–1.6200, t = 2.714, P = 0.0153) after IGF1 treatment (Fig. 4b, Supplementary Section 7.6.2 and Supplementary Fig. 31). According to a two-way analysis of variance, the exogenous IGF1R form accounted for 16.07% of total variation (F1,4 = 20.91, P = 0.0102), while time accounted for 44.23% of total variation (F3,12 = 6.57, P = 0.0071). Interestingly, we also found in tortoises a short deletion in the coding region of IGF2R that results in the loss of two amino acids. The fact that IGF2R variants have been associated with human longevity53 opens the possibility that the variant found in tortoises could also contribute to increasing the lifespan of these long-lived animals.

a, Alignment of IGF1R around residue p.N724 in C. abingdonii, A. gigantea and other representative species. The predicted electrostatic surfaces of human (top right) and modelled C. abingdonii (bottom right) IGF1R around the same residue are shown for comparison. Negatively charged areas are depicted in red, while positively charged areas are depicted in blue. b, Western blot analysis and densitometry quantification of the phospho-IGF1R (pIGF1R)/total IGF1R ratio at 5, 10 and 20 min intervals after IGF1 addition in HEK-293T cells infected with pCDH, pCDH-IGF1RWT and pCDH-IGF1RN724D plasmids. Bars indicate means ± s.e.m. *P < 0.05, Fisher’s least significant difference test (n = 3 independent experiments).
In summary, in this work, we report the preliminary characterization of giant tortoise genomes. We complemented the automatic annotation of genomes from two giant tortoise species with a hypothesis-driven strategy using manually supervised annotation of a large set of genes. The analysis of the resulting sequences offers candidate genes and pathways that may underlie the extraordinary characteristics of these iconic species, including their development, gigantism and longevity. A better understanding of the processes that we have studied may help to further elucidate the biology of these species and therefore aid the ongoing efforts to conserve these dwindling lineages. Lonesome George—the last representative of C. abingdonii, and a renowned emblem of the plight of endangered species—left a legacy including a story written in his genome whose unveiling has just started.
Methods
Genome sequencing and assembly
We obtained DNA from a blood sample from Lonesome George—the last member of C. abingdonii. This DNA was sequenced, using the Illumina HiSeq 2000 platform, from a 180-base pair-insert paired-end library, a 5-kilobase (kb)-insert mate-pair library and a 20-kb-insert mate-pair library. These libraries were assembled with the AllPaths algorithm54 for a draft genome containing 64,657 contigs with an N50 of 74 kb. Then, we scaffolded the contigs with SSPACE version 3.0 (ref. 55) using the long-insert mate-pair libraries. Finally, we filled the gaps with PBJelly version 15.8.24 (ref. 56) using the reads obtained from 18 BioPac cells. This step yielded 10,623 scaffolds with an N50 of 1.27 megabases, for a final assembly 2.3 gigabases long. Then, we soft-masked repeated regions using RepeatMasker (http://www.repeatmasker.org) with a database containing chordate repeated elements (included in the software) as a reference. Additionally, we assessed the completeness of assembly by their estimated gene content, using Benchmarking Universal Single-Copy Orthologs (BUSCO version 3.0.0)57, which tested the status of a set of 2,586 vertebrata genes from the comprehensive catalogue of orthologues58. We also performed RNA-Seq from C. abingdonii blood and A. gigantea granuloma, and aligned the resulting reads to the assembled genome using TopHat59 (version 2.0.14). Finally, we obtained whole-genome data from A. gigantea with one Illumina lane of a 180-base pair paired-end library. The resulting reads were aligned to the C. abingdonii genome with BWA60 (version 0.7.5a). Raw reads from C. abingdonii were also aligned to the genome for manual curation of the results. All work on field samples was conducted at Yale University under Institutional Animal Care and Use Committee permit number 2016-10825, Galapagos Park Permit PC-75-16 and Convention on International Trade in Endangered Species number 15US209142/9.
Genome annotation
Using the genome assembly of C. abingdonii and the RNA-Seq reads from C. abingdonii and A. gigantea, we performed de novo annotation with MAKER2. The algorithm was also fed both human and P. sinensis reference sequences, and performed two runs in a Microsoft Azure virtual machine (Supplementary Table 16). In parallel, we used selected genes from the human protein database in Ensembl as a reference to manually predict the corresponding homologues in the genome of C. abingdonii using the BATI algorithm (Blast, Annotate, Tune, Iterate)61. Briefly, this algorithm allows a user to annotate the position and intron/exon boundaries of genes in novel genomes from tblastn results. In addition, tblastn results are integrated to search for novel homologues in the explored genome. Sequencing data have been deposited at the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra), with comments showing which regions were filled with the BioPac reads and therefore may contain frequent errors.
Effective population size changes and diversity
We reconstructed changes in the effective population over time using the PSMC model5 in the following way: the reads of both individuals were aligned to the reference assembly using bwa mem (version 0.7.15-r1140). We then constructed pseudodiploid sequences using variant calls generated with SAMtools and BCFtools62, requiring minimal base and mapping qualities of 30. We additionally masked out any region with coverage below 36 or above 216 for the C. abingdonii sample, and below 8 or above 52 for the A. gigantea sample, as a function of their respective genome-wide average coverage. The resulting sequences were used to run 100 PSMC bootstrap replicates per individual, using the following parameters: -N25 -t15 -r5 -p ‘4 + 25*2 + 4 + 6’. The result was averaged and scaled to real time assuming a mutation rate (μ) of 2.5 × 10−8 and a generation time (g) of 25 years.
Expansion of gene families
To detect expansion of gene families, we aligned pairwise all the predicted proteins from the automatic annotation to the UniProt63 database of human proteins and the UniProt database of P. sinensis proteins using BLAST64 (version 2.6.011). Then, we used in-house Perl scripts to group these proteins in one-to-one, one-to-many and many-to-many orthologous relationships. Only alignments spanning at least 80% of the longer protein, and with more than 60% identities, were considered. Finally, we interrogated the resulting database to find families with C. abingdonii-specific expansions and curated the results manually. This way, we constructed extended orthology sets that may contain more than one sequence per species. These sets recapitulate most of the known families, although some of these families appear split according to sequence similarity.
Phylogenetic, evolutionary and structural analyses
Next, we assessed evidence for signatures of positive selection affecting the predicted set of genes. For this purpose, we used databases from the human (Homo sapiens), mouse (Mus musculus), dog (Canis lupus familiaris), gecko (Gekko japonicus), green anole lizard (A. carolinensis), python snake (Python bivittatus), common garter snake (Thamnophis sirtalis), Habu viper (Trimeresurus mucrosquamatus), budgerigar (Melopsittacus undulatus), zebra finch (Taeniopygia guttata), flycatcher (Ficedula albicollis), duck (Anas platyrhynchos), turkey (Meleagris gallopavo), chicken (Gallus gallus), Chinese soft-shell turtle (P. sinensis), green sea turtle (Chelonia mydas) and painted turtle (C. picta bellii) to generate pairwise alignments of all available genes one by one. To this end, we used BLAST and simple in-house Perl scripts (https://github.com/vqf/LG), which allowed us to group the genes by identity (focusing only on those presenting one-to-one orthology). We then discarded those groups in which there were more than three species missing (always excluding those in which C. abingdonii was missing). This way, we obtained 1,592 groups of sequences (similar to other studies). We then aligned them with PRANK version 150803 using the codon model and analysed the alignments with codeml from the PAML package65. To search for genes with signatures of positive selection affecting genes specific to C. abingdonii, we executed two different branch models—M0, with a single ω0 value (where ω represents the ratio of non-synonymous to synonymous substitutions) for all the branches (nested), and M2a, with a foreground ω2 value exclusive for C. abingdonii and a background ω1 value for all the other branches. As a control, the second model was repeated using P. sinensis as the foreground branch. Genes with a high ω2 value (>1) and a low ω1 value (ω1 < 0.2 and ω1 ~ ω0) in C. abingdonii, but not in P. sinensis (Supplementary Section 1.2 and Supplementary Tables 5 and 17), were then considered to be under positive selection. After this, we used the M8 model to assess the individual importance of every site in these positively selected genes, obtaining a list of sites of special interest in this evolutionary effect. These results were compared with those of the Aldabra tortoise through alignments, to evaluate which of these important residues were altered (Supplementary Table 18). Homology models were performed with SWISS-MODEL66 from the closest template available. The results were inspected and rendered with DeepView version 4.0.1. Electric potentials were calculated with DeepView using the Poisson–Boltzmann computation method. Figures were generated with PovRay (http://povray.org).
Functional analyses
HEK-293T cells were infected with pCDH, pCDH-NEIL1, pCDH-RMI2 or pCDH-NEIL1 + pCDH-RMI2 in the case of repair studies, and pCDH, pCDH-IGF1RWT or pCDH-IGF1RN724D in the case of IGF1R analyses. For the repair studies, we isolated clones of infected HEK-293T cells with proper expression levels of NEIL1 and RMI2. Cells were exposed to ultraviolet light (20 J m−2) or H2O2 (500 μM) 24 and 48 h before being lysed in NP-40 lysis buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM EDTA pH 8 and 1% NP-40, and supplemented with protease inhibitor cocktail (cOmplete, EDTA-free; Roche), as well as phosphatase inhibitors (PhosSTOP; Roche/NaF; Merck). For the IGF1R variant analyses, cells were serum starved for 14 h, then treated with 100 nM IGF1 for 5, 10 and 20 min before lysis in the same buffer. Equal amounts of protein were resolved by 8 to 13% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to PVDF membranes (GE Healthcare Life Sciences). Membranes were blocked for 1 h at room temperature with TBS-T (0.1% Tween 20) containing 5% bovine serum albumin. Immunoblotting was performed with primary antibodies diluted 1:500 to 1:1000 in TBS-T and 1% bovine serum albumin and incubated overnight at 4 °C. The primary antibodies used were: anti-phospho-Histone H2AX (Ser139) (EMD Millipore; 05-636, clone JBW301, lot 2854120), anti-PARP (Cell Signaling Technology; 9542S, rabbit polyclonal, lot 15), anti-FLAG (Cell Signaling Technology; 2368S, rabbit polyclonal, lot 12), anti-IGF1R (Abcam; ab182408, clone EPR19322, lot GR312678-8), anti-IGF1R (p Tyr1161) (Novus Biologicals; NB100-92555, rabbit polyclonal, lot CJ36131), anti-β-actin (Sigma–Aldrich, A5441, clone AC-15, lot 014M4759) and anti-α-tubulin (Sigma–Aldrich, T6074, clone B-5-1-2, lot 075M4823V). After washing with TBS-T, membranes were incubated with secondary antibodies conjugated with IRDye 680RD (LI-COR Biosciences; 926-68071, polyclonal goat-anti-rabbit, lot C41217-03; and 926-32220, polyclonal goat-anti-mouse, lot C00727-03) or IRDye 800CW (LI-COR Biosciences; 926-32211, polyclonal goat-anti-rabbit, lot C60113-05; and 926-32210, polyclonal goat-anti-mouse, lot C50316-03) for 1 h at room temperature. Protein bands were scanned on an Odyssey infrared scanner (LI-COR Biosciences). Band intensities were quantified by ImageJ and used to calculate the phospho-IGF1R/IGF1R ratio in the case of the IGF1R assay. In each replicate, cells were infected independently. For the samples from ultraviolet treatment, Flag (RMI2) was detected on the same samples used for the remaining western blots shown in this panel, run in parallel on an identical blot. Similarly, for the samples from H2O2 treatment, the western blots shown were carried out with the same samples run in parallel in three identical blots (one for PARP and actin, a second for Flag (NEIL1 and RMI2) and a third for pH2AX). Each sample contained one replicate. Statistical comparisons consisted of two-way analysis of variance performed using GraphPad Prism 7.0 software. Differences were considered statistically significant when P < 0.05. Effect sizes are expressed as group sum-of-squares divided by the total sum-of-squares (R2). At each time point, both groups were also compared with Fisher’s least significant difference test (uncorrected; α = 0.05).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The scripts for manual annotation (BATI) can be accessed at http://degradome.uniovi.es/downloads.html. Custom scripts used to produce multiple alignments for positive selection and copy-number studies are freely available at https://github.com/vqf/LG.
Data availability
Data supporting the findings of this study are available within the paper and its Supplementary Information. Sequencing data have been deposited at the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with BioProject accession number PRJNA416050. The accession number of the assembled genomic sequence is PKMU00000000. MAKER2-predicted protein sequences can be downloaded from https://github.com/vqf/LG.
References
Kim, E. B. et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011).
Keane, M. et al. Insights into the evolution of longevity from the bowhead whale genome. Cell Rep. 10, 112–122 (2015).
Nicholls, H. The legacy of Lonesome George. Nature 487, 279–280 (2012).
Kehlmaier, C. et al. Tropical ancient DNA reveals relationships of the extinct Bahamian giant tortoise Chelonoidis alburyorum. Proc. R. Soc. B 284, 20162235 (2017).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome annotation and curation using MAKER and MAKER‐P. Curr. Protoc. Bioinformatics 48, 1–39 (2014).
Wang, Z. et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat. Genet. 45, 701–706 (2013).
Sanchis-Gomar, F. et al. A preliminary candidate approach identifies the combination of chemerin, fetuin-A, and fibroblast growth factors 19 and 21 as a potential biomarker panel of successful aging. Age 37, 9776 (2015).
Pal, D. et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 18, 1279–1285 (2012).
Kir, S. et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 331, 1621–1624 (2011).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
López-Otín, C., Galluzzi, L., Freije, J. M., Madeo, F. & Kroemer, G. Metabolic control of longevity. Cell 166, 802–821 (2016).
Van der Goot, A. T. et al. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl Acad. Sci. USA 109, 14912–14917 (2012).
Crawford, N. G. et al. A phylogenomic analysis of turtles. Mol. Phylogenet. Evol. 83, 250–257 (2015).
Chiari, Y., Cahais, V., Galtier, N. & Delsuc, F. Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria). BMC Biol. 10, 65 (2012).
Boyden, L. M. et al. Mutations in KDSR cause recessive progressive symmetric erythrokeratoderma. Am. J. Hum. Genet. 100, 978–984 (2017).
Li, Y. I., Kong, L., Ponting, C. P. & Haerty, W. Rapid evolution of beta-keratin genes contribute to phenotypic differences that distinguish turtles and birds from other reptiles. Genome Biol. Evol. 5, 923–933 (2013).
Barreiro, L. B. & Quintana-Murci, L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet. 11, 17–30 (2010).
Zimmerman, L. M., Vogel, L. A. & Bowden, R. M. Understanding the vertebrate immune system: insights from the reptilian perspective. J. Exp. Biol. 213, 661–671 (2010).
Balakrishnan, C. N. et al. Gene duplication and fragmentation in the zebra finch major histocompatibility complex. BMC Biol. 8, 29 (2010).
Dotiwala, F. et al. Killer lymphocytes use granulysin, perforin and granzymes to kill intracellular parasites. Nat. Med. 22, 210–216 (2016).
Voskoboinik, I., Whisstock, J. C. & Trapani, J. A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 (2015).
Jaffe, A. L., Slater, G. J. & Alfaro, M. E. The evolution of island gigantism and body size variation in tortoises and turtles. Biol. Lett. 7, 558–561 (2011).
Chuang, D. M., Hough, C. & Senatorov, V. V. Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 45, 269–290 (2005).
Cavalcanti, D. M. et al. Neurolysin knockout mice generation and initial phenotype characterization. J. Biol. Chem. 289, 15426–15440 (2014).
Corti, P. et al. Globin X is a six-coordinate globin that reduces nitrite to nitric oxide in fish red blood cells. Proc. Natl Acad. Sci. USA 113, 8538–8543 (2016).
Schwarze, K., Singh, A. & Burmester, T. The full globin repertoire of turtles provides insights into vertebrate globin evolution and functions. Genome Biol. Evol. 7, 1896–1913 (2015).
Zhao, Y. et al. Codon 104 variation of p53 gene provides adaptive apoptotic responses to extreme environments in mammals of the Tibet plateau. Proc. Natl Acad. Sci. USA 110, 20639–20644 (2013).
Caulin, A. F. & Maley, C. C. Peto’s paradox: evolution’s prescription for cancer prevention. Trends Ecol. Evol. 26, 175–182 (2011).
Chiari, Y., Glaberman, S. & Lynch, V. J. Insights on cancer resistance in vertebrates: reptiles as a parallel system to mammals. Nat. Rev. Cancer 18, 525 (2018).
Garner, M. M., Hernandez-Divers, S. M. & Raymond, J. T. Reptile neoplasia: a retrospective study of case submissions to a specialty diagnostic service. Vet. Clin. North Am. Exot. Anim. Pract. 7, 653–671 (2004).
Futreal, P. A. et al. A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 (2004).
Martinvalet, D., Zhu, P. & Lieberman, J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22, 355–370 (2005).
Gorbunova, V., Seluanov, A., Zhang, Z., Gladyshev, V. N. & Vijg, J. Comparative genetics of longevity and cancer: insights from long-lived rodents. Nat. Rev. Genet. 15, 531–540 (2014).
MacRae, S. L. et al. DNA repair in species with extreme lifespan differences. Aging 7, 1171–1184 (2015).
Daley, J. M., Chiba, T., Xue, X., Niu, H. & Sung, P. Multifaceted role of the Topo III⍺–RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 42, 11083–11091 (2014).
Ivashkevich, A., Redon, C. E., Nakamura, A. J., Martin, R. F. & Martin, O. A. Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 327, 123–133 (2012).
Cremona, C. A. et al. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol. Cell 45, 422–432 (2012).
Wang, Y., Ghosh, G. & Hendrickson, E. A. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl Acad. Sci. USA 106, 12430–12435 (2009).
Tong, A. S. et al. ATM and ATR signaling regulate the recruitment of human telomerase to telomeres. Cell Rep. 13, 1633–1646 (2015).
Ribes-Zamora, A., Indiviglio, S. M., Mihalek, I., Williams, C. L. & Bertuch, A. A. TRF2 interaction with Ku heterotetramerization interface gives insight into c-NHEJ prevention at human telomeres. Cell Rep. 5, 194–206 (2013).
Shikama, N., Ackermann, R. & Brack, C. Protein synthesis elongation factor EF-1 alpha expression and longevity in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 91, 4199–4203 (1994).
Castillo-Quan, J. I. et al. Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep. 15, 638–650 (2016).
Ohta, S., Ohsawa, I., Kamino, K., Ando, F. & Shimokata, H. Mitochondrial ALDH2 deficiency as an oxidative stress. Ann. NY Acad. Sci. 1011, 36–44 (2004).
Serizawa, A., Dando, P. M. & Barrett, A. J. Characterization of a mitochondrial metallopeptidase reveals neurolysin as a homologue of thimet oligopeptidase. J. Biol. Chem. 270, 2092–2098 (1995).
Tristan, C., Shahani, N., Sedlak, T. W. & Sawa, A. The diverse functions of GAPDH: views from different subcellular compartments. Cell. Signal. 23, 317–323 (2011).
Fan, C. et al. MIF intersubunit disulfide mutant antagonist supports activation of CD74 by endogenous MIF trimer at physiologic concentrations. Proc. Natl Acad. Sci. USA 110, 10994–10999 (2013).
Verschuren, L. et al. MIF deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ. Res. 105, 99–107 (2009).
Harper, J. M., Wilkinson, J. E. & Miller, R. A. Macrophage migration inhibitory factor-knockout mice are long lived and respond to caloric restriction. FASEB J. 24, 2436–2442 (2010).
Whittaker, J. et al. Alanine scanning mutagenesis of a type 1 insulin-like growth factor receptor ligand binding site. J. Biol. Chem. 276, 43980–43986 (2001).
Kenyon, C. J. The genetics of ageing. Nature 464, 504–512 (2010).
Brohus, M., Gorbunova, V., Faulkes, C. G., Overgaard, M. T. & Conover, C. A. The insulin-like growth factor system in the long-lived naked mole-rat. PLoS ONE 10, e0145587 (2015).
Soerensen, M. et al. Human longevity and variation in GH/IGF-1/insulin signaling, DNA damage signaling and repair and pro/antioxidant pathway genes: cross sectional and longitudinal studies. Exp. Gerontol. 47, 379–387 (2012).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Zdobnov, E. M. et al. OrthoDBv9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 45, D744–D749 (2017).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Quesada, V., Velasco, G., Puente, X. S., Warren, W. C. & López-Otín, C. Comparative genomic analysis of the zebra finch degradome provides new insights into evolution of proteases in birds and mammals. BMC Genomics 11, 220 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Biasini, M. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014).
Acknowledgements
We thank J. R. Obeso for support, J. M. Freije, X. S. Puente, R. Valdés-Mas, F. G. Osorio, D. López-Velasco, A. Corrales, P. Salinas, D. Rodríguez, A. López-Soto, A. R. Folgueras and M. Mittelbrunn for helpful comments and advice, M. Garaña, O. Sanz, J. Isla and A. Marcos (Microsoft) for computing facilities, and F. Rodríguez, D. A. Puente and S. A. Miranda for excellent technical assistance. We also acknowledge generous support from J. I. Cabrera. We thank Banco Santander for funding a short stay of S.F.-R. and D.C.-I. at Yale University. V.Q. is supported by grants from the Principado de Asturias and Ministerio de Economía y Competitividad, including FEDER funding. L.F.K.K. is supported by an FPI fellowship associated with BFU2014-55090-P (FEDER). T.M.-B. is supported by MINECO BFU2017-86471-P (MINECO/FEDER, UE), an NIH U01 MH106874 grant, the Howard Hughes International Early Career programme, Obra Social ‘La Caixa’ and Secretaria d’Universitats i Recerca, and CERCA Programme del Departament d’Economia i Coneixement de la Generalitat de Catalunya. C.L.-O. is supported by grants from the European Research Council (DeAge; ERC Advanced Grant), Ministerio de Economía y Competitividad, Instituto de Salud Carlos III (RTICC) and Progeria Research Foundation. The Instituto Universitario de Oncología is supported by Fundación Bancaria Caja de Ahorros de Asturias. We also thank staff at the Galapagos National Park and Galapagos Conservancy for logistic and financial support.
Author information
Authors and Affiliations
Contributions
V.Q. and J.G.P.-S. performed the automatic analysis of genomes. S.F.-R. coordinated the manual genomic annotation, which was performed by J.G.P.-S., O.S.-F., D.C.-I., M.G.A., M.A.-V., D.C., P.M., J.R.A., I.T.-G., D.R.-V. and M.P.-T. S.F.-R. and D.C.-I. performed the validation of the identified genomic variants. G.B. coordinated the functional analyses of the identified genomic variants, which were carried out by O.S.-F., D.C.-I., M.G.A., M.A.-V., D.C., P.M., J.R.A. and I.T.-G. J.M. helped to screen the wild samples for SNP validation, and contributed to results interpretation. M.Q., L.B.B., J.P.G., Y.C., S.G., C.C., B.R.E., S.J.G., D.L.E., R.C.G., M.A.R. and N.P. contributed to early data collection and analyses. W.T., D.O.R. and J.P.G. helped to obtain material-securing permits and biological samples. K.P.W. partly supported data collection and supervised the initial analysis. Z.-F.J. prepared DNA and RNA samples for genomic analyses and conducted raw data quality checks. L.F.K.K. and T.M.-B. performed population history and diversity studies. V.Q., A.C. and C.L.-O. directed the research, analysed the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Figures and Tables
Supplementary Data
Table S16. Summary of average characteristics of the automatic annotation set of C. abingdonii compared to sets of other species. Table S17. Genes with signatures of positive selection in C. abingdonii. Table S18. Positions of interest within the genes with signatures of positive selection. Table S19. microRNAs identified in C. abingdonii's genome. Table S20. Primers used for validation of gene alterations identified in C. abingdonii's genome. Table S21. Expansion events with coverage and identity for nucleotide and amino acid sequences compared to one of the paralogues
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quesada, V., Freitas-Rodríguez, S., Miller, J. et al. Giant tortoise genomes provide insights into longevity and age-related disease. Nat Ecol Evol 3, 87–95 (2019). https://doi.org/10.1038/s41559-018-0733-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-018-0733-x
This article is cited by
-
Exploring Aeromonas dhakensis in Aldabra giant tortoises: a debut report and genetic characterization
BMC Microbiology (2024)
-
Insights into aging mechanisms from comparative genomics in orange and silver roughies
Scientific Reports (2024)
-
Linking telomere dynamics to evolution, life history and environmental change: perspectives, predictions and problems
Biogerontology (2024)
-
Genetics of human telomere biology disorders
Nature Reviews Genetics (2023)
-
The molecular evolution of genes previously associated with large sizes reveals possible pathways to cetacean gigantism
Scientific Reports (2023)
