
Abstract
Intraspecific diversity promotes evolutionary change, and when partitioned among geographic regions or habitats can form the basis for speciation. Marine species live in an environment that can provide as much scope for diversification in the vertical as in the horizontal dimension. Understanding the relevant mechanisms will contribute significantly to our understanding of eco-evolutionary processes and effective biodiversity conservation. Here, we provide an annotated genome assembly for the deep-sea fish Coryphaenoides rupestris and re-sequencing data to show that differentiation at non-synonymous sites in functional loci distinguishes individuals living at different depths, independent of horizontal spatial distance. Our data indicate disruptive selection at these loci; however, we find no clear evidence for differentiation at neutral loci that may indicate assortative mating. We propose that individuals with distinct genotypes at relevant loci segregate by depth as they mature (supported by survey data), which may be associated with ecotype differentiation linked to distinct phenotypic requirements at different depths.
Similar content being viewed by others

Main
While longitudinal and latitudinal habitat transitions have been proposed to define marine communities and promote intraspecific differentiation1,2,3, little is known about the importance of transitions along ocean depth gradients4,5, although substantial changes in species assemblages with depth have been recorded (for example, ref. 6), and relatively narrow depth ranges may distinguish closely related species (for example, refs 7,8). Understanding the relevant mechanisms will contribute significantly to our understanding of eco-evolutionary processes and the origin of marine biodiversity. We chose the roundnose grenadier (Coryphaenoides rupestris) as a model system because it is a widespread species that can inhabit a comparatively broad range of depths9 from ~180 m to 2,600 m. It is a batch spawner, producing up to 69,000 pelagic eggs per female10. It has a spawning season peaking in autumn11 (recent observations were in September at 1,500 m; ref. 12), preys on fish, cephalopods and invertebrates in both benthic and pelagic habitats13, and shows minor genetic differentiation across its geographic range14,15,16.
Adaptation to habitat can occur among populations within a species, and if disruptive selection is associated with assortative mating has the potential to promote incipient speciation through ecological processes in sympatry17. When environmental change exposes new habitats and niche potential, adaptive radiations may rapidly generate a new lineage of species18,19. To the extent that differential selection can retain polymorphisms within or among populations, this may facilitate the process of adaptive radiation. Here, we focus on one of the key habitat transitions in the oceans—between the photic mesopelagic region and the aphotic regions below (together with the more contiguous changes associated with increasing depth). There is the potential for species (such as the roundnose grenadier), whose habitat range extends across this boundary or along the depth gradient, to experience differential selective pressures. We tested hypotheses about adaptation to these deep-sea habitats using genome sequence data together with data on the ecology and life history of the subject species. We found that juvenile fish of this species are found primarily in relatively shallow depths (near the transition between the mesopelagic and bathypelagic zones) and then migrate as they mature to different depths, and this is strongly associated with their genotype at a set of functional loci. In particular, all adults below ~1,800 m share the same homozygous genotype at each locus. There is evidence for strong selection maintaining this difference, but no clear evidence for differentiation driven by assortative mating.
Results and discussion
We produced an annotated reference genome for C. rupestris with a total length of 0.829 gigabase pairs, a mean depth of 104× and an N50 of 159,738 (see Supplementary Methods for details). We used this draft genome to map 60 additional genomes sequenced to a mean depth of ~6×, representing a transect from 750 m to 1,800 m over a horizontal sampling range of 25 km (Supplementary Fig. 1), collected on the same day. Manhattan plots using generalized linear model (GLM) and FST metrics (Fig. 1a and Supplementary Fig. 2) consistently showed the same pattern of outliers for comparisons between 1,800 m and shallower sites along the transect, although they were the most pronounced for the comparison between 1,000 m and 1,800 m. Among 25 genomic regions showing clusters of outlier single nucleotide polymorphisms (SNPs) exceeding the Manhattan plot threshold correction for multiple tests (a total of 346 outliers; Fig. 1a), 9 SNPs coded for non-synonymous changes within 6 genes, and genotypes were strongly correlated with habitat depth (Fig. 2). These 9 non-synonymous sites were on 5 contigs and surrounded by multiple significant outliers on the same contig (for example, the non-synonymous sites on contig 1041 have 32 significant outliers nearby on the same contig). Principal component analysis (PCA) for all 346 outliers also showed a strong correlation with depth, while the remaining 5.9 million non-outlier SNPs did not (Fig. 3a), and pairwise FST values for a subset of 44,650 neutral loci were not significantly different from zero (see Methods and Supplementary Table 1). Simulations showed this number of loci to be sufficient to detect an FST of 0.0007 with a power of 0.86 (see Methods). An independent assessment of genomic regions associated with habitat depth (cacti plots; Fig. 1b and Supplementary Table 2) reinforced the evidence for selection. Although some catch data have previously been interpreted to suggest diurnal and seasonal vertical migrations13, these strong genomic associations instead suggest considerable adult fidelity to specific habitat depths. Temporary migrations during spawning to an intermediate depth (~1,500 m) are possible based on recent data12.

a, Negative log10 of P values based on the GLM for individuals at 750 m versus 1,500 m, and 1,000 m versus 1,800 m. The horizontal lines mark Bonferroni (red) and FDR (blue) corrections for multiple comparisons, with 346 outliers in the 1,000 m versus 1,800 m comparison above the red line. Alternating blue and red dots show the transition from one contig to another. b, Localized phylogenetic patterns within the genome (‘cacti’). The majority of the genome exhibits neutral genetic divergence (as exemplified by cactus 4), while one cactus (cactus 5), which accounts for less than 1% of the genome, displays all but one 1,800 m sample clustering in the same lineage (see Supplementary Table 2).

The six coding regions are indicated, and contig references are given. Each column represents an SNP, colour coded for homozygote (blue or yellow) and heterozygote (green). Each row represents an individual, arranged by habitat depth. N denotes an unresolved genotype at that position for that individual.

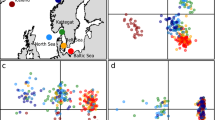
a, PCA for 60 genomes along transect 1 for non-outlier (above) and outlier (below) SNPs. b, PCA plots for neutral (above) and outlier (below) SNPs from RAD-seq data for the Hebrides region. Support for sequential principal components is shown in the bottom left of each plot.
Evidence for linkage disequilibrium due to population mixture and a two-locus Wahlund effect20 further reinforces the pattern of differentiation at outlier loci for comparisons between 1,800 m and shallower depths (Supplementary Fig. 3). To determine whether the observed linkage disequilibrium was driven by concerted evolution or mixture linkage disequilibrium, we therefore needed to restrict our analyses to just one depth (which greatly attenuates the effects from mixture linkage disequilibrium; Supplementary Fig. 3). Comparing the decay profiles for linkage disequilibrium associated with physical linkage for the samples from 1,000 m, we find that outliers associated with habitat depth show elevated linkage disequilibrium compared with non-outliers, and that the non-synonymous SNPs show the strongest effect (Fig. 4), suggesting coordinated inheritance among loci.

Linkage disequilibrium, assessed as R2, is plotted against map distance along contigs for outlier loci associated with depth (from GLM analysis, above the Bonferonni threshold). Non-synonymous changes within coding genes (type 1), other outliers associated with depth (type 2) and a sample of the remaining SNPs not associated with depth (neutral) are shown. Comparisons among contigs are given for outliers showing the R2 distribution only (50% of distribution within the boxes).
To test the relationship between genotypes and habitat depth in other geographic locations, we used the polymerase chain reaction to screen four of the nine outlier loci coding for non-synonymous changes (representing different contigs and the strongest outlier SNPs; Supplementary Fig. 2) in nine widely distributed populations (see Methods). We found a consistent association between genotype and depth for all locations (Fig. 5). We also generated SNP loci from restriction-site-associated DNA sequencing analyses (RAD-seq; see Methods) and distinguished putative neutral loci from outlier loci using the FDist method. Focusing on four sample sites near the Hebrides (see Supplementary Fig. 1), we found ordination clustering by depth for outlier loci (121 SNPs), but no clear pattern for neutral loci (11,992 SNPs; Fig. 3b). This set of outlier loci, which was chosen based on a signal for positive selection in general rather than a specific association with depth, shared just one SNP with the 346 outliers associated with depth identified from the re-sequencing data. However, 42 of the 121 RAD-seq outlier SNPs were on 16 contigs where re-sequencing outlier SNPs were also found. Individuals sampled from 1,800 m clustered together on the PCA plots for outlier loci (Fig. 3), while individuals from shallower water were less consistent, with samples falling into multiple clusters (including the 1,800 m cluster). This may be expected from the analysis illustrated in Fig. 2, where some individuals at shallower depths are homozygous for the ‘deep-water’ alleles, and may reflect temporary movement associated with seasonal spawning concentrations at ~1,500 m (ref. 12).

The proportion of individuals at each depth with the ‘deep-adapted’ allele is shown (pooling heterozygotes and homozygotes).
For the 60 re-sequenced genomes along the depth transect (Supplementary Fig. 1), there was a highly significant pattern of homozygote excess at the nine outlier loci with non-synonymous changes (Supplementary Table 3). Homozygote excess was not more generally observed, as most of the 5.9 million non-outlier loci were consistent with Hardy–Weinberg equilibrium (HWE) (3.7% divergent after correction for false discovery), and non-outlier loci out of HWE were more likely to show heterozygous excess (while the 346 outliers mostly showed homozygote excess; Supplementary Table 3). In the contig showing the most non-synonymous changes, with two outlier loci associated with three non-synonymous changes (contig 1041), we detected five RAD-seq outlier SNPs (out of 4,989 SNPs derived from 207 samples across 9 populations) and assessed their genotypes across the full geographic range. For samples taken at ≥1,800 m, one allele dominated at each of these loci, while the same allele was less frequent among samples from <1,800 m (Supplementary Table 4). In each case there was significant homozygote excess, consistent with under-dominance and disruptive selection (P < 0.00001; Supplementary Table 4).
We considered juvenile fish separately because trawl survey data demonstrate that juveniles of this species are predominantly found in waters at 1,200 m or less (Supplementary Fig. 4). We collected 96 juveniles from 1,000 m near the 60-genome transect location (transect 2; Supplementary Fig. 1) and genotyped these individuals at the four non-synonymous outlier SNPs we genotyped in the adults. The allele frequencies of juveniles differed significantly from the adults captured at the same depth (Fig. 5, Supplementary Table 4 and Supplementary Fig. 5), showing roughly the same proportion of genotypes associated with adults from 1,800 m as with adults from 1,000 m (Fig. 5). Genotype frequencies in juveniles also showed significant deviation from HWE at all four loci in a pattern consistent with disruptive selection (Supplementary Table 4). Disruptive selection has been proposed as an important driver of ecological speciation when coupled with assortative mating21,22,23. However, as illustrated in Fig. 3, for non-outlier SNPs there was no clear evidence for differentiation among samples taken at different depths from the same geographic region, and differentiation would be expected if there was sustained assortative mating. This assumes that over time (sufficient time for concerted evolution in this case; see Fig. 4), assortative mating would result in differentiation at neutral loci through genetic drift. There is also no evidence of distinct breeding aggregations separated by depth12,19.
Cohorts tracked from samples collected by trawl (see Methods) in eight different years indicate an ontogenetic depth migration (Supplementary Fig. 4). A latent-class model (two-component mixture model; see Methods) applied to a subset of high-density age classes (>299 individuals per km2) revealed two ontogenetic trajectories with a proportion of fish remaining at ~1,000 m, while the rest migrated to depths of ≥1,500 m before settling (Fig. 6), although a proportion of the fish at 1,500 m may represent a temporary spawning aggregation12. Together, the data indicate deep and shallower water specialists with distinct genotypes (especially at 1,800 m or deeper; see Fig. 2) that sort by habitat as they mature. Note that while the latent-class modelling suggests at least two ontogenetic strategies associated with depth, sampling effects and the movement of individuals during spawning make the characterization of these strategies imprecise, especially with respect to settlement depth (see Methods). Disruptive selection could be maintaining genotypes associated with adult habitats with different phenotypic requirements24. This may be associated with the partitioning of available habitat (frequency dependence) or the evolution of distinct strategies with similar fitness levels25. There may be assortative mating, but not to a level leading to differentiation that is detectable by our high-resolution analyses, or it may have begun very recently. Alternatively, the trajectory may not be towards speciation, but instead in support of maintaining both phenotypes in the population.

Two strategies identified by the latent-class model. Median trajectories of the ontogenetic depth migration strategies identified by the latent-class model are indicated by bold lines (blue and red) with 95% confidence intervals (shaded areas). The point size and colour reflects the density of individuals (high, red and large; low, small and yellow). This model output was based on samples where density exceeded 299 individuals per km2. CPUE, catch per unit effort.
We considered the putative function of loci associated with habitat depth. For the Rho-associated protein kinase 1 (ROCK1) locus, there were three non-synonymous changes resulting in two proximate amino acid substitutions (threonine, aspartic acid to valine, glycine in the ‘Smc’ region). Various studies have described functions associated with energy metabolism at this locus26,27. However, a gene ontology analysis for all 69 genes associated with the 346 depth-correlated outlier SNPs detected from the Manhattan plot analyses found 29 gene ontology terms significantly over-represented in the outlier gene list (Supplementary Table 5), and these showed little association with metabolic processes. A given locus had up to 14 SNPs associated with it (one within 30 Kb, 15 within 10 Kb and the rest having SNPs within introns or exons). Of the 29 terms, 16 were associated with development or morphogenesis (Supplementary Table 5). This may be consistent with a more general trend for deep-demersal species to show distinct phenotypes compared with related species at shallower depths10, although intraspecific morphological variation with depth has not yet been reported for this genus. There is also no well-studied, closely related species to serve as a reference for gene ontology terms, so proposed functions should be interpreted with caution.
A key difference between habitats at 1,000 m and below 1,800 m is access to the deep-scattering layer within the mesopelagic zone. The deep-scattering layer is a mid-water (200–1,000 m) mass of small fishes, cephalopods, crustaceans and zooplankton that provides a rich source and variety of prey items28,29,30. It should be possible for C. rupestris to feed there at a relatively high trophic level (and some data are consistent with this31; Supplementary Fig. 6), although the predation risk may also be relatively high. In deeper water, the benthic and pelagic systems become increasingly decoupled, resulting in low particulate organic carbon influx and lower food availability, so feeding may be more closely associated with the benthos and from a less diverse prey resource. We compared stable isotope data for carbon and nitrogen. Fish from ≥1,800 m were 13C enriched and 15N depleted compared with samples from 1,000 or 1,050 m (multivariate analysis of variance, F = 6.43, P = 0.003; Supplementary Fig. 6). These data support our genetic and life-history data, indicating two strategies: one in shallower water with access to the relatively abundant resources of the deep-scattering layer, but potentially greater predation risk, and another in relatively deep water feeding on distinct prey (suggested by carbon isotope data).
Our data reveal genomic differences associated with ecological specializations that are probably related to the anatomical and physiological requirements of the different environments and behavioural strategies, and indicate that fish adopt a habitat suited to their genotype as they mature. This type of vertical population structuring has important implications for fisheries management in general, and is not currently recognized for stock definition. For C. rupestris in particular, the Northeast Atlantic stock is currently assessed as single unit with a total allowable catch of around 2,000 tonnes. From 2017, the European Union has prohibited bottom trawling at depths greater than 800 m (http://data.consilium.europa.eu/doc/document/ST-11142-2016-INIT/en/pdf). While this management strategy protects the deep population of C. rupestris, it means that the entire total allowable catch must now be drawn solely from the shallow population, possibly leading to over-exploitation.
The evolutionary implications are that both temporal (developmental) and spatial factors (associated with habitat characteristics) affect the spatial distribution of diversity established by natural selection, and that this process is not necessarily associated with incipient speciation promoted by assortative mating. Species maintaining intraspecific diversity by strong selection (perhaps for partitioning resources with different phenotypic requirements) could be predisposed to rapid speciation in a conducive environment. Across the broader phylogeny for this genus, a number of species other than C. rupestris inhabit a similarly wide depth range, especially those species also found in the abyss, while other species show narrower, non-overlapping habitat depth ranges6. The primary phylogenetic division in this genus is between abyssal and non-abyssal species, with species within those lineages radiating at similar times6. This suggests that in some cases habitat depth has led to divisions in alpha taxonomy within this genus; however, our data for C. rupestris suggest instead a system whereby conspecific ecotypes associated with habitat depth are being maintained by disruptive selection at a set of loci in linkage disequilibrium.
Methods
Data reporting
Sample sizes for the detection of outliers were determined based on published simulation analyses that consider power in the context of the depth of sequence and the number of samples (for example, ref. 32). For the analysis of alternative ontogenetic strategies, to achieve model convergence, only data for which the estimated density of fish was greater than 299 individuals per km2 were kept, corresponding to 81,549 out of 176,733 fish. There were no randomized experiments, and investigators were not blinded to allocation for outcome assessment.
Sampling
Samples were obtained by trawl fishing for this and earlier projects33,34. Muscle tissue or fin clips were collected as soon after landing as possible and stored in 20% dimethyl sulfoxide saturated with salt or 95% ethanol, and long-term at −20 °C. Details of specific sample sets are provided in the Supplementary Methods.
Genome sequencing
Illumina libraries were constructed for short-read, mate-pair and genome sampling (RAD-seq)35 using genomic DNA extracted using a phenol chloroform protocol36, and sequenced on Illumina HiSeq 2000 and 2500 machines. Additional sequencing was undertaken on a PacBio sequencer using single molecule real-time technology. Genome assembly and annotation, together with the genome re-sequencing protocols, strategy for RAD-seq sequencing and data analyses were performed as in refs 37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55 and are described in detail in the Supplementary Methods.
SNP calling and outlier detection
We aligned 60 re-sequenced genomes from transect 2 (Supplementary Fig. 1) to our newly assembled and annotated genome for C. rupestris, as described in the Supplementary Methods. SNPs were called using SAMtools version 1.3 (mpileup command with -q 20 -Q 10 –ugf flags) and BCFtools (call command with –bvcg flags)56. We subsequently removed indels using VCFtools (-gzvcf)57 and filtered low-quality SNPs using the filter command (-e ‘QUAL < 30’ -s ‘LOW_QUAL’). This pipeline resulted in 23,883,346 biallelic SNPs.
Outlier detection
We tested for outliers using the GLM implemented in the software TASSEL version 5.0 (ref. 58). First, we ordered the SNPs in the vcf file, from the SAMtools pipeline above, using the SortGenotypeFilePlugin command. We then converted the file to a Hapmap format to speed up the downstream pipeline. Using the -filterAlign plugin, the Hapmap file was filtered for the minimum number of individuals scored per site (MinCount was set to 48; 80% of the individuals in the dataset) and a minor allele frequency of 0.05. After filtering, the total number of SNPs retained from the original 23,883,346 SNPs was 5,929,065. Next, this file was merged with the trait file, in which each sample is assigned to sampling depth, using the -intersect command. To identify outlier loci, we ran the GLM analysis using 1,000 permutations using the FixedEffectLMPlugin command. This function performs association analysis using a least-squares fixed-effects linear model and uses F tests to test for the association between the segregating site (SNP genotype) and trait (depth distribution). In addition, we calculated FST for each SNP across each pairwise depth comparison (for example, 1,000 m versus 1,500 m) using VCFtools.
The R package qqman59 was used to visualize the Manhattan plots (Fig. 1 and Supplementary Fig. 2). We corrected for multiple comparisons on the GLM plots using the Bonferroni type I error correction60 and considered only those SNPs with alpha values below the threshold 8.43 × 10−9 to be outliers. Furthermore, we calculated the false discovery rate (FDR)61 using the programme largeQvalue62. This programme implements the algorithms of refs 63,64,65, and takes a set of P values and maps these onto q values. We chose a FDR of 0.01, resulting in a threshold of 2.8 × 10−5. We used the default values of lambda λ (0, 0.9 and 0,05), 100 bootstraps and the ‘--robust flag’ command. Using the more conservative Bonferroni approach, we detected 346 outliers from the comparison between 1,000 m and 1,800 m. All other loci were considered to be neutral in the context of selection associated with depth (5,928,719). We used the software PLINK version 1.07 (ref. 66) to calculate observed and expected heterozygosity for both neutral and outlier loci, identifying loci out of HWE using an exact test67. HWE based on a deficit or excess of heterozygotes was calculated using the formula FIS = 1 – (Hobs/Hexp), where FIS is Wright’s inbreeding coefficient, Hobs is the observed heterozygosity and Hexp is the expected heterozygosity at HWE, and the inbreeding index values for each SNP were assessed. The outlier SNPs detected by GLM were mapped against the annotated C. rupestris draft genome using JBrowse version 1.12.3 (ref. 68).
Linkage disequilibrium calculation
We measured linkage disequilibrium by calculating the squared correlation coefficient between unphased genotypes, using PLINK version 1.90b3.45 (ref. 69). We started with three datasets: ‘neutral’ loci (a subsample of SNPs that showed no significant correlation with depth); ‘type 1’ outliers (the 9 SNPs that are differentiated between 1,000 m and 1,800 m, and which code for non-synonymous changes within coding genes); and ‘type 2’ outliers (the remaining 337 SNPs that are differentiated between 1,000 m and 1,800 m). To limit operation size for the neutral dataset, we randomly selected a subset of 4,000 SNPs from the vcf file using in-house scripts. For each outlier dataset, we ran two calculations: one for all pairwise comparisons between loci occurring on the same contig and one for all pairwise comparisons for loci occurring on different contigs. Single population datasets were selected using VCFtools. We tested for a two-locus Wahlund effect (where a linear relationship indicates an effect20) using R by comparing the R2 (the square of the correlation coefficient between the two indicator variables) linkage disequilibrium estimate with the product of FST for locus 1 and FST for locus 2. All plots were constructed using R. This test was undertaken for the comparison between 1,800 m and shallower sample sites combined, as this was expected to show the strongest effect (Supplementary Fig. 3; Pearson’s product moment correlation = 0.56, 95% confidence = 0.54–0.58). We then ran the same test for just a single depth (1,000 m), but basing the FST product calculation on the full dataset as before20 (Pearson’s product moment correlation = 0.07, 95% confidence = 0.05–0.11). We chose 1,000 m because at that depth there were 16 fish sampled (rather than 14 at each of 750 m and 1,500 m), and at 1,800 m many relevant loci (including all detected non-synonymous loci) are fixed, prohibiting the calculation of linkage disequilibrium.
Screening for non-synonymous changes
To investigate the correlation of genotype with depth over a broader geographic range, we identified four outlier loci coding for non-synonymous changes associated with depth that represented different contigs and strong correlations, and designed a polymerase chain reaction strategy to genotype individuals at each non-synonymous SNP (see Supplementary Methods for details). We screened 224 adult fish (including the 60 re-sequenced genomes) from 9 geographic populations ranging in habitat depth from 500 m to 2,230 m from locations in the eastern and central North Atlantic and North Sea (see Supplementary Fig. 1 and Fig. 5). Adults from transect 1, juveniles from transect 2, and adults from 1,000 m and 1,800 m from transect 2 (Supplementary Fig. 1) were screened for the presence of the ‘deep-water’ adapted alleles at the same 4 outlier loci screened elsewhere. The proportions are compared in Supplementary Fig. 5 and confirm that the same adult fish correlations between genotype and depth are seen at both transect locations.
Genotypes were compared against HWE expectations using a 3 × 2 chi-square contingency test with one degree of freedom. We estimate allele frequencies p and q from the population, and then determine the three genotype frequencies (all loci were biallelic) from p and q. Since q = 1 – p and genotype frequencies are determined as p2, 2pq and q2, only p is able to vary and so there is only one degree of freedom. Further research on the extent to which differential gene expression or plasticity may be associated with living in different habitat depths would be useful but logistically challenging for species that cannot be easily maintained in experimental conditions.
Cactus-based analyses (SAGUARO)
We ran the software SAGUARO70, which combines a hidden Markov model with a self-organizing map, to detect boundaries between genomic regions that, based on programme-generated matrices of pairwise genomic distances, are characterized by different phylogenetic histories70. Each region or ‘cactus’ in the genome was computed from the data without any a priori input hypothesis. We ran SAGUARO on all 5,929,065 SNPs derived from the 60 re-sequenced genomes (after TASSEL filtering). VCF files were converted into binary ‘feature’ files and SAGUARO was run for ten iterations in which cacti were added, and two cycles per iteration, in which cacti were optimized and assigned to sites. SAGUARO identified 5 cacti that accounted for nearly 75% of the genome (Supplementary Table 1). Four of these five cacti did not result in population-specific clusters and resembled the PCA of the neutral datasets (Fig. 1). Only one—cactus 5—could distinguish the population at 1,800 m covering 0.155% of the genome (Fig. 1 and Supplementary Table 1).
Comparison of putative populations at outlier and non-outlier loci
PCAs were conducted on both the 5,928,719 non-outlier SNPs from the 60 re-sequenced genomes and the 346 outlier SNPs using the Principal Components Plug-in for TASSEL. The genotypes were converted to numeric scores using the Numeric Genotype function and the missing data imputed to the mean score for each site. PCAs were then performed using eigenvalue decomposition of the covariate matrix, and eigenvectors were calculated from a singular value decomposition of the data. The number of principal components was set by default to five. The full set of SNPs from the re-sequencing analysis was randomly subsampled for 50,000 SNPs using the shuf command in Linux and 44,650 neutral loci identified in Lositan71 (50,000 simulations, forced mean FST, confidence interval = 0.95). The four depths were compared for FST at these neutral loci using Arlequin version 3.5 (ref. 72) (results shown in Supplementary Table 1). We tested the power of this FST analysis by running simulations in R (1,000 replicates). Briefly, we calculated Weir and Cockerham’s multi-locus FST for 44,650 loci and 15 individuals per population to generate two distributions—one for a specific FST level and the other for panmixia—and calculated the power from the overlap between these distributions.
Outlier detection in Hebrides RAD-seq data
We used two methods to identify outlier loci among the four populations in the Hebrides region. First, we ran the FDist outlier approach as implemented in Lositan71, which incorporates heterozygosity and simulates a distribution for neutrally distributed markers. This method has been shown to have lower rates of type 1 and 2 errors compared with the FST outlier method implemented in Arlequin version 3.5 (ref. 72). We ran 50,000 simulations under the infinite alleles model with the option of neutral mean FST and forced mean FST. We employed a 95% confidence interval and a false discovery rate of 0.05. With this method, we detected 121 outlier loci. Next, we used the more conservative outlier loci detection methods of BayeScan version 2.1 (ref. 73) to detect loci under divergent selection. We ran the programme with the default sample size of 5,000 and a thinning interval of 10. We ran 20 pilot runs each of 5,000 iterations and an additional burn-in of 50,000. We conducted runs with prior odds for the neutral model set at 100 and a FDR of 0.05, which resulted in 122 outliers. There was a >90% overlap (110 loci) between the Lositan and BayeScan outliers.
PCA of Hebrides RAD-seq data
Based on the results of Lositan for the four Hebrides populations, we divided the SNPs into outlier and non-outlier datasets. We evaluated population clustering among the four populations using the PCA method implemented in the R package Adegenet version 2.0 (ref. 74). Adegenet converts the diploid allele information from the Stacks output for each individual into a data frame and uses those data to perform the PCA.
Screening of nine populations for outlier SNPs on contig 1041
We conducted an outlier analysis using Lositan on the ‘nine populations’ file of 4,989 SNPs. We ran 50,000 simulations under the infinite alleles model with the option of neutral mean FST and forced mean FST. We employed a 95% confidence interval and a false discovery rate of 0.05. With this method, we detected 341 outliers. Of these, five mapped to contig 1041 (which had the largest number of strong outliers from the comparison among 60 re-sequenced genomes). We extracted the genotypes for these five outlier loci and determined the allele frequencies across the dataset.
Gene ontology analysis
We searched for gene ontology terms using the FatiGO tool on the Babelomics 5 platform (http://babelomics.bioinfo.cipf.es/). FatiGO75 is an enrichment test whereby two lists of genes are compared to detect significant over-representation of functional annotations in the subject compared with the reference list. In this case, we compared the set of the 69 genes (Supplementary Table 4) associated with the 346 depth-correlated outlier SNPs detected from the Manhattan plot analyses and found within 30 kilobases of a coding gene, against the full list of 15,114 genes identified from our C. rupestris genome annotation that were associated with gene name, gene description and gene ontology terms using InterProScan. Gene ontology term functions were identified with reference to the human database (chosen due to the relatively complete level of annotation and functional analysis and the lack of a suitable reference from a closely related species). A Fisher’s exact test for 2 × 2 contingency tables (testing for over-representation in list 1) was used to check for significance. Significance was corrected for multiple testing using Benjamini and Hochberg’s FDR-controlling procedure60. Gene ontology biological processes, molecular function, cellular component, GOSlim gene ontology annotation, InterPro and the Genome-Scale Metabolic Network database were searched, filtering terms by 5–500 annotated identifications in each database.
Stable isotope analysis
For analysis of carbon and nitrogen stable isotopic ratios (δ13C and δ15N denotes isotopic ratios of 13/12 C and 14/15 N relative to known standards), white muscle tissue that had been stored in 95% ethanol from 25 C. rupestris samples from shallower than 1,100 m (including 16 from 1,000 m sequenced for genomes and 9 from a nearby site) and another 25 from 1,800 m or deeper (including 16 from 1,800 m sequenced for genomes and 9 from a nearby site) were subjected to lipid extraction preparation. Briefly, tissue pieces of approximately 0.25 cm3 were (1) finely diced and sonicated in 1 ml of 3:1 dichloromethane:methanol solution for 15 min, then (2) centrifuged at 3,000 rpm for 10 min before excess solution was removed. Steps 1 and 2 were repeated three times. The remaining solid sample was then sonicated in 1 ml deionized water for 15 min before being centrifuged at 3,000 rpm for 10 min. Excess water was removed and samples were air-dried at 50°C for 48 h. Dried samples were then mechanically powdered and 0.4 mg of each sample was sealed into tin capsules for analysis. Carbon and nitrogen isotope analyses of the samples was performed at the Stable Isotope Biogeochemistry Laboratory, Durham University, using a ECS 4010 Nitrogen / Protein Analyzer (Costech Analytical) connected to a Delta V Advantage Isotope Ratio Mass Spectrometer (Thermo Fisher Scientific). Carbon isotope ratios are corrected for 17O contribution and reported in standard δ notation in per mil (‰) relative to Vienna Pee Dee Belemnite. Isotopic accuracy was monitored through routine analyses of in-house standards, which were stringently calibrated against international standards (for example, United States Geological Survey (USGS) 40, USGS 24, International Atomic Energy Agency (IAEA) 600, IAEA CH3, IAEA CH7, IAEA N1 and IAEA N2). This provided a total linear range in δ13C between –46 ‰ and +3 ‰, and between –4.5 ‰ and +20.4 ‰ for δ15N. Analytical uncertainty in δ13C and δ15N was typically ±0.1 ‰ or better for replicate analyses of the international standards and <0.2 ‰ for replicate sample analysis. Total organic carbon was obtained as part of the isotopic analysis using an internal standard (glutamic acid, 40.82% C, 9.52% N). We tested for differences in δ13C and δ15N using the Wilks’ lambda multivariate analysis of variance using the programme Minitab version 14.0. Multivariate analysis of variance is an extension of the analysis of variance and tests for the difference in two or more vectors of means.
Modelling ontogenetic depth migration strategies
Size distributions were transformed into age distributions using the length at age table provided in ref. 76 and a multinomial logistic model as described by ref. 77.
Ontogenetic depth migration
To test whether C. rupestris shows an ontogenetic depth migration78,79 from shallow as juveniles to deeper as adults, we assigned individuals to age cohorts and modelled depth change over years. Cohorts of fish spawned between 1996 and 2003 were used to assess depth change over the first 12 years of life. Depth at capture was described using an asymptotic regression model with age as the independent variable (Bayesian JAGS model80) and executed in R 3.2.3 using the R package rjags (https://cran.r-project.org/web/packages/rjags/index.html). Based on model comparisons of deviance criteria, the final model was formalized as:
\({\rm{Depth}}_{i,C}\sim {\rm{dnorm}}({\rm{mu}}1_{i,C},{\rm{tau}}1_C)\)
The depth observed for individual ‘i’ from cohort C is drawn from a normal distribution with parameters mu1i,C and tau1C (1/tau1C is the cohort-specific deviance), where:
Asym is the asymptotic depth, R0 is the depth at age 0 and lrcC is a cohort-specific parameter corresponding to the rate of depth change with age. A hierarchical structure was adopted for the cohort-specific parameters lrcC and tau1C, with priors assigned to a normal and a gamma distribution, respectively, and non-informative uniform hyperparameters. Non-informative uniform priors were used for all the other parameters.
Alternative ontogenetic strategies
To assess the possibility of different ontogenetic depth migration strategies (and eventual adult settlement depth), depth at age data were modelled using a latent-class model (a two-component mixture model) where each fish belongs to one of two unobserved (latent) classes or strategies; that is, one shallower and one deeper (Fig. 6). To achieve model convergence, it was necessary to reduce the asymptotic function to two parameters and use a subset of the data for which the estimated density of fish was greater than 299 individuals per km2 (this corresponded to 74 hauls out of 120, and 81,549 fish). As before, the Bayesian model was executed in R 3.2.3 using the R package rjags and formalized as:
The depth observed for individual ‘i’ belonging to strategy S is drawn from a normal distribution with parameters mui,S and tauS (1/tauS is the strategy-specific deviance), where:
aS and bS are strategy-dependent parameters and, to give an asymptotic shape to the process, f(Agei) is defined as:
The proportions of individuals belonging to the two strategies were drawn from a Dirichlet distribution. A non-informative Dirichlet prior was used with the two parameters set to 1 and uniform priors were used for aS, bS and tauS. Note that, while we can infer that two strategies are present in different proportions, due to unequal survey coverage at all depths, we cannot estimate with certainty the proportion of each strategy. Furthermore, the assumption of only two strategies and the lower absolute densities observed at the greatest depths (1,800–2,000 m) might reduce the model’s capacity to identify deeper strategies and/or underestimate the depth at which individuals settle. It is also possible that the greater representation at 1,500 m may be a consequence of both populations sharing a spawning site at 1,500 m (ref. 12). Samples were collected during the spawning period and so the distribution could reflect this and result in an underestimation of the depth at settlement, particularly for the deep strategy.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
Sequence data have been deposited at GenBank under accession codes PRJNA417902 for the reference genome, SRP129631 for the 60 re-sequenced individuals and PRJNA430030 for the RAD-seq data. Figures 1–4 have associated source data, as do Supplementary Figs. 2–6. There are no restrictions on data availability.
References
Bowen, B. W. et al. Comparative phylogeography of the ocean planet. Proc. Natl Acad. Sci. USA 113, 7962–7969 (2016).
Piacenza, S. E. et al. Patterns and variation in benthic biodiversity in a large marine ecosystem. PLoS ONE 10, e0135135 (2015).
Briggs, J. C. & Bowen, B. W. A realignment of marine biogeographic provinces with particular reference to fish distributions. J. Biogeogr. 39, 12–30 (2012).
Rex, M. A. & Etter, R. J. Deep-Sea Biodiversity: Pattern and Scale. (Harvard Univ. Press: Cambridge, 2010.
Stuart, C. T. et al. CeDAMar global database of abyssal biological sampling. Aquat. Biol. 4, 143–145 (2008).
Gaither, M. R. et al. Depth as a driver of evolution in the deep sea: insights from grenadiers (Gadiformes: Macrouridae) of the genus Coryphaenoides. Mol. Phylogenet. Evol. 104, 73–82 (2016).
Mindel, B. L., Neat, F. C., Trueman, C. N., Webb, T. J. & Blanchard, J. L. Functional, size and taxonomic diversity of fish along a depth gradient in the deep sea. PeerJ 4, e2387 (2016).
Jennings, R. M., Etter, R. J. & Ficarra, L. Population differentiation and species formation in the deep sea: the potential role of environmental gradients and depth. PLoS ONE 8, e77594 (2013).
Coad, B. W. & Reist, J. D. Annotated List of the Arctic Marine Fishes of Canada (Fisheries and Oceans Canada, Winnipeg, 2004).
Allain, A. Reproductive strategies of three deep-water benthopelagic fishes from the northeast Atlantic Ocean. Fish. Res. 51, 165–176 (2001).
Bergstad, O. A. Distribution, population structure, growth and reproduction of the roundnose grenadier Coryphaenoides rupestris (Pisces: Macrouridae) in the deep waters of the Skagerrak. Mar. Biol. 107, 25–37 (1990).
Neat, F. C. Aggregating behaviour, social interactions and possible spawning in the deep-water fish Coryphaenoides rupestris. J. Fish Biol. 91, 975–980 (2017).
Haedrich, R. L. Pelagic capture of the epibenthic rattail Coryphaenoides rupestris. Deep-Sea Res 21, 977–979 (1974).
White, T. A., Stamford, J. & Hoelzel, A. R. Local selection and population structure in a deep-sea fish, the roundnose grenadier (Coryphaenoides rupestris). Mol. Ecol. 19, 216–226 (2010).
Knutsen, H., Jorde, P. E., Bergstad, O. A. & Skogen, M. Population genetic structure in a deepwater fish Coryphaenoides rupestris: patterns and processes. Mar. Ecol. Prog. Ser. 460, 233–246 (2012).
Longmore, C. et al. Otolith geochemistry indicates life-long spatial population structuring in a deep-sea fish, Coryphaenoides rupestris. Mar. Ecol. Prog. Ser. 435, 209–224 (2011).
Nosil, P. Ecological Speciation (Oxford Univ. Press, Oxford, 2012).
Seehausen, O. African cichlid fish: a model system in adaptive radiation research. Proc. R. Soc. B 273, 1987–1998 (2006).
Nevado, B., Atchison, G. W., Hughes, C. E. & Filatov, D. A. Widespread adaptive evolution during repeated evolutionary radiations in New World lupins. Nat. Comm. 7, 12384 (2016).
Waples, R. S. Testing for Hardy–Weinberg proportions: have we lost the plot? Heredity 106, 1–19 (2015).
Dieckmann, U. & Doebeli, M. On the origin of species by sympatric speciation. Nature 400, 354–357 (1999).
Bolnick, D. I. & Fitzpatrick, B. M. Sympatric speciation: models and empirical evidence. Annu. Rev. Ecol. Evol. Syst. 38, 459–487 (2007).
Getz, W. M., Salter, R., Seidel, D. P. & Hooft, P. Sympatric speciation in structureless environments. BMC Evol. Biol. 16, 50 (2016).
Rice, W. R. Disruptive selection on habitat preference and the evolution of reproductive isolation: a simulation study. Evolution 38, 1251–1260 (1984).
Østman, B., Lin, R. & Adami, C. Trade-offs drive resource specialization and the gradual establishment of ecotypes. BMC Evol. Biol. 14, 113 (2014).
Huang, H. et al. ROCK1 in AgRP neurons regulates energy expenditure and locomotor activity in male mice. Endocrinology 154, 3660–3670 (2013).
Zhou, X. et al. ROCK1 reduces mitochondrial content and irisin production in muscle suppressing adipocyte browning and impairing insulin sensitivity. Sci. Rep. 6, 29669 (2016).
Porteiro, F. M. & Sutton, T. Midwater fish assemblages and seamounts. In: Seamounts: Ecology, Fisheries, and Conservation Series 12 (eds Pitcher, T. J. et al.) 101–116 (Blackwell Publishing, Oxford, 2007).
Robinson, C. Mesopelagic zone ecology and biogeochemistry—a synthesis. Deep-Sea Res. II 57, 1504–1518 (2010).
Trueman, C., Johnston, G., O’Hea, B. & MacKenzie, K. Trophic interactions of fish communities at midwater depths enhance long-term carbon storage and benthic production on continental slopes. Proc. R. Soc. B 281, 20140669 (2014).
Mauchline, J. & Gordon, J. Diets and bathymetric distributions of the macrourid fish of the Rockall Trough, northeastern Atlantic Ocean. Mar. Biol. 81, 107–121 (1984).
Fumagalli, M. Assessing the effects of sequencing depth and sample size in population genetic inferences. PLoS ONE 8, e379667 (2013).
Gordon, J. D. M. & Bergstad, O. A. Species composition of demersal fish in the Rockall Trough, north-eastern Atlantic, as determined by different trawls. J. Mar. Biol. Assoc. UK 72, 213–230 (1992).
Priede, I. G. et al. The ecosystem of the Mid-Atlantic Ridge at the sub-polar front and Charlie–Gibbs Fracture Zone; ECO-MAR project strategy and description of the sampling programme 2007–2010. Deep-Sea Res. II 98, 220–230 (2013).
Peterson, P. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7, e37135 (2012).
Hoelzel, A. R. Molecular Genetic Analysis of Populations: A Practical Approach (Oxford Univ. Press, Oxford, 1998).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Joshi, N. A. & Fass, J. N. sickle: A Windowed Adaptive Trimming Tool for FASTQ Files Using Quality Version 1.33 (2011); https://github.com/najoshi/sickle
Birol, I. et al. De novo transcriptome assembly with ABySS. Bioinformatics 25, 2872–2877 (2009).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652 (2011).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0. (2010); http://www.repeatmasker.org/RepeatModeler/
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3 1, 171–182 (2011).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Rousset, F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106 (2008).
Lischer, H. E. L. & Excoffier, L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012).
Beaumont, M. A., . & Nichols, R. A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. B 263, 1619–1626 (1996).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Turner, S. D. qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. Preprint at https://www.biorxiv.org/content/early/2014/05/14/005165 (2014).
Simes, R. J. An improved Bonferroni procedure for multiple tests of significance. Biometrika 73, 751–754 (1986).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Brown, A. A. largeQvalue: a program for calculating FDR estimates with large datasets. Preprint at https://www.biorxiv.org/content/early/2015/03/18/010074 (2015).
Storey, J. D. A direct approach to false discovery rates. J. R. Stat. Soc. B 64, 479–498 (2002).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445 (2003).
Storey, J. D., Taylor, J. E. & Siegmund, D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J. R. Stat. Soc. B 66, 187–205 (2004).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Wigginton, J. E., Cutler, D. J. & Abecasis, G. R. A note on exact tests of Hardy–Weinberg equilibrium. Am. J. Hum. Genet. 76, 887–893 (2005).
Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J. & Holmes, I. H. JBrowse: a next-generation genome browser. Genome Res. 19, 1630–1638 (2009).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Zamani, N. et al. Unsupervised genome-wide recognition of local relationship patterns. BMC Genomics 14, 347 (2013).
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A. & Luikart, G. LOSITAN: a workbench to detect molecular adaptation based on a F ST-outlier method. BMC Bioinformatics 9, 323 (2008).
Narum, S. R. & Hess, J. E. Comparison of F ST outlier tests for SNP loci under selection. Mol. Ecol. Resour. 11, 184–194 (2011).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Al-Shahrour, F., Díaz-Uriarte, R. & Dopazo, J. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics 20, 578–580 (2004).
Lorance, P., Dupouy, H. & Allain, V. Assessment of the roundnose grenadier (Coryphaenoides rupestris) stock in the Rockall Trough and neighbouring areas (ICES sub-areas V–VII). Fish. Res. 51, 151–163 (2001).
Gerritsen, H. D., McGrath, D. & Lordan, C. A simple method for comparing age–length keys reveals significant regional differences within a single stock of haddock (Melanogrammus aeglefinus). ICES J. Mar. Sci. 63, 1096–1100 (2006).
Lin, H.-Y., Shiao, J.-C., Chen, Y.-G. & Iizuka, Y. Ontogenetic vertical migration of grenadiers revealed by otolith microstructures and stable isotopic composition. Deep-Sea Res. I 61, 123–130 (2012).
Trueman, C.N., Chung, M. T. & Shores, D. Ecogeochemistry potential in deep time biodiversity illustrated using a modern deep-water case study. Philos. Trans. R. Soc. B 371, 20150223 (2016).
Plummer, M. JAGS: a program for analysis of Bayesian graphical models using Gibbs sampling. In Proc. 3rd Int. Workshop on Distributed Statistical Computing, March 20–22, Vienna, Austria (2003). https://www.r-project.org/conferences/DSC-2003/
Acknowledgements
Funding was provided by the Natural Environment Research Council (UK) under grant number NE/K005359/1. The authors acknowledge the Mid-Atlantic Ridge Ecosystem project (led by M. Priede) from which many of the North Atlantic samples were acquired, as well as the crew of the research vessel Scotia and especially F. Burns, who facilitated collection of the transect samples in 2015. We thank O. A. Bergstad and S. Guldborg at the Institute of Marine Research in Arendal, and M. Girard, P. Lorance and E. Jones for the provision of samples. We thank W. Bleby for assistance in the laboratory, and R. Waples, O. Gaggiotti, M. Priede and E. Notarianni for helpful comments on manuscript drafts. The data reported in this paper are tabulated in the Supplementary Information and archived in the GenBank database. Genome sequencing and bioinformatic analyses were facilitated by DBS Genomics and the Hamilton HPC facility in Durham, and the Centre for Genome Research in Liverpool.
Author information
Authors and Affiliations
Contributions
A.R.H. conceived the study and contributed to the analyses. A.R.H. and M.R.G. wrote the paper with input from all authors. M.R.G., G.A.G., M.d.J., F.S., F.N., T.R., D.M., D.R.G., N.H., X.L., J.K., A.L., M.H. and S.H. contributed to data generation and analyses.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary References, Supplementary Tables 1–10, Supplementary Figures 1–7.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gaither, M.R., Gkafas, G.A., de Jong, M. et al. Genomics of habitat choice and adaptive evolution in a deep-sea fish. Nat Ecol Evol 2, 680–687 (2018). https://doi.org/10.1038/s41559-018-0482-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-018-0482-x
This article is cited by
-
Gene-associated markers as a genomic and transcriptomic resource for a highly migratory and apex predator shark (Isurus oxyrinchus)
Marine Biology (2022)
-
Facilitating population genomics of non-model organisms through optimized experimental design for reduced representation sequencing
BMC Genomics (2021)
-
Applying genomic data to seagrass conservation
Biodiversity and Conservation (2021)
-
Linking genomics and fish conservation decision making: a review
Reviews in Fish Biology and Fisheries (2020)
-
Conservation of adaptive potential and functional diversity
Conservation Genetics (2019)
