
Abstract
Adaptation to specialized diets often requires modifications at both genomic and microbiome levels. We applied a hologenomic approach to the common vampire bat (Desmodus rotundus), one of the only three obligate blood-feeding (sanguivorous) mammals, to study the evolution of its complex dietary adaptation. Specifically, we assembled its high-quality reference genome (scaffold N50 = 26.9 Mb, contig N50 = 36.6 kb) and gut metagenome, and compared them against those of insectivorous, frugivorous and carnivorous bats. Our analyses showed a particular common vampire bat genomic landscape regarding integrated viral elements, a dietary and phylogenetic influence on gut microbiome taxonomic and functional profiles, and that both genetic elements harbour key traits related to the nutritional (for example, vitamin and lipid shortage) and non-nutritional (for example, nitrogen waste and osmotic homeostasis) challenges of sanguivory. These findings highlight the value of a holistic study of both the host and its microbiota when attempting to decipher adaptations underlying radical dietary lifestyles.
Similar content being viewed by others


Main
The order Chiroptera (bats) exhibits a wide variety of dietary specializations, and includes the only three obligate blood-feeding mammalian species, the vampire bats (family Phyllostomidae, subfamily Desmodontinae). Blood is a challenging dietary source because it consists of an ~78% liquid phase and a dry-matter phase consisting of ~93% proteins and only ~1% carbohydrates1, providing very low levels of vitamins2, and potentially containing blood-borne pathogens. Vampire bats have evolved numerous key physiological adaptations to this lifestyle, for which the associated genomic changes have not yet been fully characterized due to the lack of an available reference genome. These adaptations include morphological specializations (such as claw-thumbed wings and craniofacial changes including sharp incisors and canines), infrared sensing capacity3 for the identification of easily accessible blood vessels in prey4, and renal adaptations to the high protein content in its diet5 (such as a high glomerular filtration rate and effective urea excretion). Furthermore, given the high risk of exposure to blood-borne pathogens, another important trait in the vampire bat is its immune system6.
Besides genomic adaptations, host-associated microbiota may play an additional, possibly equally important, role in the evolution of vertebrate dietary specialization7. Although the functional role of the vampire bat gut microbiome has not been studied, analyses of obligate invertebrate sanguivorous organisms8 have shown that the gut microbiota contributes to blood meal digestion9, provision of nutrients absent from blood10 and to immunological protection11. Studies on mammals have shown that the gut microbiome is a key aspect of an organism’s digestive capacities (energy harvest, nutrient acquisition and intestinal homeostasis)12, and that it also affects phenotypes related to the immune and neuroendocrine systems13,14. Furthermore, changes in the gut microbiome are associated with diseases such as diabetes, obesity, irritable bowel syndrome and inflammatory bowel disease15,16,17. In response to the growing awareness of the key roles that host–microbiome relationships can play across the spectra of life, various studies have advocated for the ‘hologenome’ concept18,19,20. In brief, this argues that natural selection acts on both the host and its microbiome (together forming the holobiont); thus, evolutionary studies should incorporate both. The extreme dietary adaptation of vampire bats provides a suitable model to investigate the effect of selection across the genome and microbiome, and thus allows exploration of the role of host-associated microbiome in the evolution of specialized diets.
Results and discussion
Here, we explore the contributions of both the common vampire bat’s nuclear genome and gut microbiome to its adaptation to obligate sanguivory. To this end, we generated both the common vampire bat genome and fecal metagenomic data sets as a proxy to study its gut microbiome, as well as faecal metagenomic data sets from other non-sanguivorous bat species. We used these data sets for comparative genomic and metagenomic analyses. Specifically, we analysed the common vampire bat genomic landscape, the ratio of substitution rates at non-synonymous and synonymous sites (dN/dS), putative gene loss and gene family expansion/contraction, and computational predictions of the functional impact of amino-acid substitutions. We also performed microbial taxonomic and functional profiling, identified the microbial taxonomic and functional core of the common vampire bat, and identified enriched microbial taxa and functions. Following a hologenomic approach, we identified elements in both the host genome and microbiome that could have played relevant roles in adaptation to sanguivory.
Genomic landscape
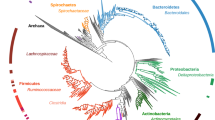
We sequenced and de novo assembled the ~2 gigabase (Gb) common vampire bat genome using Illumina sequencing technology (Supplementary Information 1). The genome is smaller than that of other mammals, but similar to previously reported bat genomes21. The initial assembly (~100 × mean coverage, scaffold N50 = 5.5 Mb and N90 = 933 kb; Supplementary Figs. 1 and 2) was subsequently improved using the in vitro proximity ligation-based technology for assembly contiguity refinement developed by Dovetail Genomics22. We obtained a final high-quality assembly with scaffold N50 = 26.9 Mb and N90 = 9.46 Mb, contig N50 = 36.6 kb and N90 = 8.8 kb (Supplementary Table 1, Supplementary Fig. 3 and Supplementary Information 1). We used our annotated common vampire bat genome (see Methods—Protein-coding gene and functional annotation) for comparative genomic analyses with publicly available bat genomes and other mammalian genomes (Supplementary Table 2 and Fig. 1).

Species with a light blue circle were used for comparison of transposable elements. In golden colour are the silhouettes of the species used for the identification of putative lost genes and their corresponding number of putatively lost genes compared to the human genome. The number of gene families under expansion (green) and contraction (red) in Chiroptera using E. europaeus as an outgroup are indicated. Estimated dates of nodes (in million years) are indicated in black. The gut microbiomes of bats with different diets derive from R. aegyptiacus (blue, frugivorous), M. gigas (green, carnivorous), R. ferrumequinum (grey, insectivorous) and the common vampire bat (red, sanguivorous). H. sapiens, Homo sapiens; E. helvum, Eidolon helvum.
Repetitive elements can significantly contribute to genome evolution. Thus, for a genomic landscape characterization, we first compared transposable elements in the common vampire bat genome to those within the genomes of non-phyllostomid bats with other diets: the carnivorous Megaderma lyra (greater false vampire bat, Megadermatidae), the insectivorous Pteronotus parnellii (Parnell’s moustached bat, Mormoopidae) and the frugivorous Pteropus vampyrus (large flying fox, Pteropodidae) (Fig. 1, Supplementary Table 3 and Supplementary Note 1). We identified a 1.6- to 2.26-fold higher copy number of the MULE-MuDR transposon in the common vampire bat genome relative to the other bat genomes. The high mutagenic capacity of MULE-MuDR has been demonstrated to have played a critical role in the evolution of some plants23. Furthermore, transposable elements in general may cause structural or functional changes within the genome and alter epigenetic regulation of the genes into which they are inserted24. Therefore, we explored whether these elements might have also played a role in the evolution of sanguivory by analysing their genomic location in the nuclear genome of the common vampire bat. We found that the identified common vampire bat transposable elements, MULE-MuDR elements in particular, were located within genomic regions enriched for gene ontology (GO) functions related to the challenges of sanguivory, such as antigen processing and presentation, defence response to viruses, lipid metabolism, and vitamin metabolism (Supplementary Information 2).
A sanguivorous diet facilitates exposure to blood-borne viruses that could lead to an increase in genomic invasion by retroviral and non-retroviral endogenous viral elements (EVEs). Thus, we next characterized their presence in the common vampire bat genome. Compared to previously published EVE studies on non-Chiropteran mammals, the common vampire bat exhibits a greater diversity of non-retroviral EVEs in terms of the number of integrations, including endogenized viral genes from avian Bornaviridae and Parvoviridae/Dependovirus. However, these findings are not restricted to vampire bats and are similar to those in other bat species25 (Supplementary Note 2, Supplementary Fig. 4 and Supplementary Information 3). Surprisingly, and in contrast to the prior expectations given its sanguivorous diet, the diversity of endogenous retroviral elements (ERVs) in the common vampire bat is very low compared to other bat species26. The only proviral elements detected were DrERV27 and DrgERV, both present in low copy numbers (Supplementary Note 3, Supplementary Fig. 5 and Supplementary Information 4). We hypothesize that genome colonization by ERVs could have been restricted by the genomic adaptations in the common vampire bat genome against ERV insertion and proliferation. In support of our hypothesis, we identified expansion of the anti-retroviral gene TRIM5 family (Viterbi P = 0.00088, Supplementary Information 5 and Supplementary Note 4).
Genomic adaptations to sanguivory
Feeding specialization often requires morphological and physiological adaptations in traits such as the sensory apparatus (for example, infrared sensing), locomotion, digestion, kidney function and immunity (Fig. 2a and Supplementary Information 6). For example, it has been shown that vampire bats have a loss of sweet taste genes and reduction of bitter taste genes28. In agreement, such genes were also identified in our putative gene loss analysis (Supplementary Information 7 and Supplementary Note 5). It is likely that the function of those genes is related to sanguivory, because sweet and bitter taste receptor genes influence glucose homeostasis in humans29. Interestingly, we found that the common vampire bat bitter taste receptor gene TAS2R3 has experienced episodic positive selection and shows two species-specific positively selected sites (PSSs) on topological domains, one of them having a potential impact on protein function (PROVEAN score = −4.4) (Supplementary Table 4 and Supplementary Fig. 6). Among the enriched GOs of the differentially evolving genes, we identified functions related to the regulation of RNA splicing, which could be relevant to sanguivory given that D. rotundus produces submandibular tissue-specific splicing isoforms to counteract the prey’s response to injury30 (Supplementary Information 8 and Supplementary Note 6). Regarding the recruitment of alternatively spliced forms, the ganglion-specific splicing of TRPV1 has been found to underlie the vampire bat’s infrared sensing ability31. Interestingly, we found that PRKD1, which directly modulates the product of TRPV132, is positively selected and exhibits species-specific PSSs (branch-site test P = 3.39 × 10−10, branch test P = 1.39 × 10−6, Supplementary Information 8). These examples suggest an important role of alternative splicing as a form of regulatory evolution fundamental to sanguivory (Supplementary Note 7). However, it is clear that despite the number of detected genomic features related to sanguivory adaptation, they alone cannot address all of the challenges posed by this diet (Fig. 2b).

a, Adaptational contributions to sanguivory accountable to genomic changes alone (blue labels). b, Adaptational contributions to sanguivory within a hologenomic context (blue labels for host genes; red labels for gut microbial traits).
Gut microbiome diet and phylogenetic influence
We generated Illumina shotgun metagenome data in order to compare the gut microbiomes of 13 faecal samples from common vampire bats with those of non-sanguivorous non-phyllostomid bats; specifically, eight frugivorous Rousettus aegyptiacus (Egyptian fruit bat, Pteropodidae), five insectivorous Rhinolophus ferrumequinum (greater horseshoe bat, Rhinolophidae) and five carnivorous Macroderma gigas (ghost bat, Megadermatidae) bats (Supplementary Information 9). We obtained a median of 15.8 Gb of sequencing data (~37.6 million 100-bp paired-end reads) for each dietary category. After filtering low-quality bases, adaptor sequences and bat-genome-derived reads, we obtained a median of 2.77 Gb of high-quality data for each species, totalling 86.73 Gb of data. We identified taxa and functions present only in the common vampire bat microbiome (gut microbiome core), as well as taxa and functions that exhibit statistically significant differences in abundance or contribution to variation between the different microbiomes (Supplementary Information 6 and 8, Supplementary Tables 5 and 6 and Supplementary Note 8).
It has been observed previously that similarity in the taxonomic composition of vertebrate gut microbiomes (including bats) can be influenced by the diet and the phylogenetic relationships of the respective host species33. Overall, the common vampire bat microbiome taxonomic composition is more similar to that of the insectivorous and carnivorous bats than to that of the frugivorous bat. This may reflect a phylogenetic influence on the microbiome taxonomic profile (Fig. 3a, Supplementary Fig. 7 and Supplementary Notes 9 and 10). In contrast, the vampire bat microbiome is strikingly different to that of the compared bats at the functional level, which was characterized by the KEGG annotations of the microbial non-redundant gene catalogues assembled from the metagenomic data sets. While there is little differentiation between the functional gut microbiomes of carnivorous, insectivores and frugivorous bats, the common vampire bat functional gut microbiome is almost completely distinct, and exhibits the least intra-species variation between the samples (Fig. 3b, Supplementary Fig. 8 and Supplementary Table 7). This suggests that the functional profile is less influenced by phylogeny than the taxonomic profile, and that the common vampire bat gut microbiome harbours a set of functions specialized to its extreme diet (Supplementary Note 11). Subsequently, we analysed the comparative genomic and metagenomic results in a hologenomic framework to demonstrate how both components contribute to adaptation to sanguivory (Fig. 4, Supplementary Figs. 9–12, Supplementary Tables 7–9 and Supplementary Information 8 and 10).

D. rotundus (sanguivorous, red), R. ferrumequinum (insectivorous, black), M. gigas (carnivorous, green) and R. aegyptiacus (frugivorous, blue). a, Euclidean distance dendrogram of the microbial presence/absence identifications at the species taxonomical level. b, Euclidean distance dendrogram from the UniProt-identified abundance functions from the normalized samples.

a, GOs of the single-copy orthologous genes with a dN/dS ratio that is statistically higher or lower in the common vampire bat in comparison to the other bats. b, KOs from the common vampire bat gut microbiome functional core. c, Genes with a dN/dS ratio that is statistically higher or lower in the common vampire bat in comparison to the other bats and that are directly associated with sanguivory challenges. d, Taxa and genes from the common vampire bat gut microbiome core directly associated with the adaptation to sanguivory.
The hologenomic framework of sanguivory
Viscosity and subsequent coagulation represent a challenge for ingestion and digestion of blood. Besides developing potent anticoagulants in its saliva34, the common vampire bat hologenome addresses this challenge in various ways. For instance, REG4, involved in metaplastic responses of the gastrointestinal epithelium, was found to be under ongoing positive selection (M8a/M8 test P = 0.047) with possible functional impact on its carbohydrate-binding capacity, including binding of the anticoagulant heparin (Supplementary Table 4 and Supplementary Information 8). Furthermore, we identified genes in the common vampire bat microbial functional core from pathways for degradation of heparan sulfate and dermatan sulfate, both being polysaccharides involved in blood coagulation (Supplementary Information 10). We also identified an enrichment in the common vampire bat microbial gene l-asparaginase (Fisher’s P = 0.00027), which decreases protein synthesis of coagulation factors35 (Supplementary Information 11).
Besides specialized digestion, sanguivory poses other challenges related to the poor nutritional value of blood itself, as well as to the side effects that may arise due to the blood components being the sole dietary source (Fig. 2b). We identified elements in both the genome and gut microbiome that might be involved in solving each of these challenges discussed next.
Hologenomic solutions to nutritional challenges
Low nutrient availability
Obligate sanguivory requires adaptation to very low levels of some nutrients, such as essential amino acids and the vitamin B complex36,37, and very high levels of others, such as salt38. Our data clearly demonstrate how both the host and its associated gut microbiome have dealt with these challenges. We found the gene LAMTOR5 to be positively selected in the common vampire bat genome (false discovery rate (FDR)= 2.02 × 10−7, Supplementary Information 8). This gene is involved in the response to nutrient starvation39, suggesting that the common vampire bat metabolism has adapted to the low nutrient content available in blood. Similarly, we identified KOs in the common vampire bat microbial core related to energy and carbohydrate metabolism (Fig. 4b and Supplementary Information 11). For example, when compared to the other bats, we identified an enrichment in the common vampire bat microbial genes involved in response to low nutrient availability (RelA/SpoT family protein, Fisher’s P = 0.0064, and guanosine pentaphosphate, Fisher’s P = 0.0018), and enzymes in the common vampire bat core involved in the reverse Krebs cycle (Supplementary Information 10), which is used by some bacteria to produce carbon compounds from CO2 and water (abundant blood components). We speculate that the presence of such metabolic pathways indicates the growth of specific microbes in the gut’s environmental conditions resulting from blood consumption.
The holobiont has also provided solutions to the lack of important nutrients in blood (Fig. 4c and Supplementary Information 8). For example, the PDZD11 gene in the common vampire bat genome, involved in vitamin B5 metabolism, evolved faster in the common vampire bat genome relative to the other examined bats (branch test P = 1.97 × 10−10). We further postulate that the microbiome also contributes in tackling the low nutritional challenge by providing necessary nutrients. For example, compared to the other bat microbiomes, the common vampire bat gut microbiome had the highest number of enriched enzymes related to the biosynthesis of cofactors and vitamins, such as carotenoid (Supplementary Fig. 8B and Supplementary Information 10 and 11). Furthermore, we identified enzymes involved in the metabolism of butyrate, an important nutrient for cells lining the mammalian colon derived from bacterial fermentation40, enriched in the common vampire bat gut microbiome as well as in the vampire bat gut microbiome core (Supplementary Information 10 and 11).
Lipid and glucose assimilation
Besides vitamins and other nutrients, lipids and glucose may not be readily available in blood. Furthermore, vampire bats have a reduced capacity to store energy reserves. In agreement with this, we identified GO enrichment of lipid metabolism on genes with dN/dS values statistically higher or lower compared to other bats (Fig. 4a,c and Supplementary Information 8). For example, we identified the gene FFAR1, which plays an important role in glucose homeostasis, as evolving faster in the common vampire bat compared to the other bats (branch test P = 3.68 × 10−5) and containing amino-acid substitutions with a possible functional impact on its binding ability (Supplementary Table 4). This may enable the common vampire bat to better utilize the available glucose. The common vampire gut microbiome also exhibits unique solutions to the challenge (Fig. 4b,d and Supplementary Information 10 and 11). Differences in the carbohydrate and glycan metabolism functional profile were identified in the principal component analysis (PCA) comparing the different microbiomes (Supplementary Fig. 11), which place the vampire bat gut microbiome profile within a cluster separate from those of the non-sanguivorous bats. Importantly, we identified enrichment in the microbial gene glycerol kinase in the common vampire bat (Fisher’s P = 0.0027), which plays a key role in the formation of triacylglycerol and in fat storage, and its deficiency causes symptoms such as hypoglycaemia and lethargy in a mouse model41.
Hologenomic solutions to non-nutritional challenges
Immunity
Due to its sanguivorous lifestyle, the common vampire bat risks direct contact with blood-borne pathogens from prey. Consequently, we observed > 280 bacterial species known to be pathogenic to some mammalian species present exclusively in the common vampire bat gut microbiome (Supplementary Information 12). For example, we identified enrichment of genes from Borrelia (Supplementary Information 11) and Bartonella as one of the most abundant genera in the common vampire bat compared to the other bats (Supplementary Information 13). These bacteria are known to be transmitted by sanguivorous invertebrates (ticks, fleas, mosquitoes and lice). This suggests that the abundance of this genus could be a shared pattern of sanguivorous species. While several studies have elucidated part of the expected genomic immunity-related adaptations6, analysis of the full genome enabled us to identify more elements related to immunity, such as defence response to virus and antigen processing and presentation (Fig. 4a,c and Supplementary Information 8). For example, we identified the antimicrobial gene RNASE7 to be positively selected (branch-site test P = 0.004) and containing amino-acid substitutions that may increase its bactericidal capacity (Supplementary Table 4). In addition, when compared to the gut microbiomes of non-sanguivorous bats, that of the common vampire bat contains a large abundance of potentially protective bacteria, such as Amycolatopsis mediterranei (P < 0.05), which has been shown to produce antiviral compounds against bacteriophages and poxviruses42 (Supplementary Information 13).
Iron assimilation
Iron concentration represents a significant challenge to sanguivory. Although the concentration of free iron ion is not high in the blood, severe haemolysis (for example, during digestion of blood) could result in high levels of iron that, if absorbed in excess, could accumulate and disrupt the normal function in organs such as the liver, heart and pancreas. Interestingly, we identified the light and heavy chains of the iron-storing protein ferritin (encoded by FTL, FTH1) under gene family expansion in the common vampire bat genome (Viterbi P = 0 and Viterbi P = 0.0012, respectively, Supplementary Information 5). In addition, we identified an enrichment of the iron-storing protein ferritin (Fisher’s P = 0.0014), suggesting that the gut microbiome also contributes to solving this challenge (Fig. 4d and Supplementary Information 11).
Nitrogen waste and blood/osmotic pressure
The high abundance of protein in the blood and its rapid ingestion could lead to accumulation of nitrogenous waste products, primarily urea, which could lead to renal disease-like symptoms (for example, high blood pressure and fluid retention). This challenge is exacerbated by the abundance of salts in blood, which pose additional osmotic and blood pressure challenges. We see at the genome level that this is addressed by a higher rate of evolution in the common vampire bat genes compared to the other bats involved in disposal of excess nitrogen (Fig. 4a,c and Supplementary Information 8), such as PSMA3 (branch test P = 2.08 × 10−7). This challenge seems to also be addressed by the gut microbiome. The PCA of the copy number of genes involved in amino-acid metabolism distinguishes the common vampire bat in a single cluster separated from the other bat species analysed (Supplementary Fig. 11), suggesting a specialized microbial amino-acid metabolism capacity. We also identified enrichment in the common vampire bat microbial gene urease subunit alpha (ureA, Fisher’s P = 0.016) involved in urea degradation (Supplementary Information 11).
Conclusions
It is clear from our results that the common vampire bat has adapted to sanguivory through a close relationship between its genome and gut microbiome. We identified a phylogenetic and dietary impact on the common vampire bat gut microbiome and uncovered an unexpected genomic viral and repetitive element genomic make-up. We showed that extreme dietary specializations, such as that of the common vampire bat, provide a comparative framework with which to tease apart the relative roles of genomes and microbiomes in adaptation. In conclusion, our study illustrates the benefits of studying the evolution of complex adaptations under a holobiome framework, and suggests that vertebrate adaptation studies that do not account for the action of the hologenome may fail to recover the full complexity of adaptation.
Methods
Genome sequencing and raw-read processing
We shotgun-sequenced the D. rotundus genome using a wing biopsy from a sample collected by the NIH through the Catoctin Wildlife and Zoo in Thurmont, Maryland, USA. The capturing method and dead preservation procedure of the specimen are unknown. The age and sex of the dead individual are unknown. Sampling permits were given to BGI for the sequencing of the specimen, originally as part of the BGI 10K genome project. Genomic DNA was extracted at the Laboratory of Genomic Diversity and was fragmented to 2–10 kilobases (kb). Sequencing libraries were constructed with insert sizes 170 bp−10 kb, according to the Illumina protocol for sequencing on Illumina HiSeq2000 following the manufacturer’s instructions. We sequenced reads of 49 bp for the long-insert-size libraries (2 kb, 5 kb and 10 kb) and 100 bp for the short-insert-size libraries (170 bp, 500 bp and 800 bp). Sequencing errors were corrected on the basis of the frequency of nucleotide strings of a given length and low-quality reads were filtered out using SOAPfilter43 as follows. (1) Remove reads with >10% uncalled nucleotides (Ns). (2) For short-insert-size libraries (<2 kb), reads were removed if the quality score of >60% bases was <7. For large-insert-size libraries (≥2 kb), reads were removed if the quality score of >80% bases was <7. (3) Adapter sequences, and duplicate or identical reads were removed. (4) Read pairs were removed if Read1 and Read2 were completely identical. (5) For short-insert-size paired-end sequences, reads with overlapping length ≥10 bp between the Read1 and Read2 were removed.
Genome assembly
We estimated the genome size of D. rotundus using Kmerfreq44 by dividing the total number of seven decamers by the peak of the seven decamer Poisson distribution. High-quality reads were assembled using SOAPdenovo43 as follows. (1) Short-insert library reads were assembled as initial contigs ignoring the sequence pair information. (2) Reads were aligned to the previously generated contig sequences. Scaffolds were constructed from short-insert-size libraries to large-insert-size libraries by weighting the paired-end relationships between pairs of contigs, with at least three read pairs required forming a connection between any two contigs. (3) Gaps in the scaffolds were closed using GapCloser43. Genome quality assessment was performed by downloading a publicly available D. rotundus transcriptome45 and aligning the transcripts to the genome using BLAT46.
Genome contiguity improvement
We prepared two Chicago libraries22 using 5 μg of high-molecular-weight DNA obtained from D. rotundus cultured cells from the San Diego Zoo collection, which were originally derived from a skin sample taken from between the shoulder blades of a D. rotundus individual. Permits for this were obtained from the San Diego Zoo Global. The capturing method and dead preservation procedure of the specimen are unknown. The age and sex of the dead individual are unknown. DNA was extracted with Qiagen Blood and Cell Midi kits according to the manufacturer’s instructions. The steps required for building the Chicago libraries were performed as described in ref. 22. The libraries were sequenced using Illumina HiSeq 2500 2 × 100 bp rapid run. Our initial D. rotundus assembly, shotgun sequence data and Chicago library sequences were used by Dovetail Genomics as input data for HiRise, as described in ref. 22. Genome assembly contiguity statistics were obtained using a minimum N track length of 1 to delimit the contig blocks within the scaffolds.
Protein-coding gene and functional annotation
Homology-based gene prediction was performed using as a reference the Ensembl gene sets of Myotis lucifugus, Pteropus alecto, Myotis davidii, horse and human. We aligned the protein sequences of the reference gene sets to the D. rotundus assembly using tblastn47 and linked the blast hits into candidate gene loci with genBlastA48. We filtered out those candidate loci with homologous block length <90% of the query length. We extracted genomic sequences of candidate gene loci, including the intronic regions and 3 kb upstream/downstream sequences. The sequences were passed to GeneWise49 to search for accurately spliced alignments. We filtered out pseudogenes containing more than one frame error for single-exon genes. Potentially pseudogenized single exons were removed if they were part of a multi-exon gene. We then aligned protein sequences of these genes against UniProt using blastp and filtered out genes without matches. We also filtered out genes that had >80% repeat regions. De novo gene prediction was performed with AUGUSTUS50 using a published common vampire bat transcriptome45 as a training data set and with masked transposable-element-related repeats. We filtered out partial and <150 bp predicted genes. Genes that aligned over 50% of their length to annotated transposable elements were filtered out. Finally, we built a non-redundant gene set with the homology-based evidence prioritized over the de novo evidence. If de novo genes were chosen in the reference gene set, we retained only those with > 30% of their length aligning against UniProt51 and that contained at least 3 exons. The integrated gene set was translated into amino-acid sequences, which were used to search the InterPro database with iprscan_4.852. We used BLAST to search the metabolic pathway database in KEGG53 and homologues in the SwissProt and TrEMBL databases in UniProt. The quality and annotation of the D. rotundus genome was assessed and compared to that of E. europaeus, R. ferrumequinum, M. brandtii, M. davidii, M. lucifugus and P. parnellii with BUSCO54.
Repeat annotation
Repeat annotation was performed on the genomes of D. rotundus, P. parnelli, M. lyra and P. vampyrus. Transposable elements were identified using RepeatMasker55 and RepeatProteinMask against the Repbase transposable element library56. We used RepeatScount, PILER-DF and RepeatModeler-1.0.555,57 to construct a de novo transposable element library, which was then used by RepeatMasker to predict repeats. We predicted tandem repeats using TRF58. LTR_Finder59 was used to detect long terminal repeats (LTRs). The Repbase-based annotations and the de novo annotations were merged.
Non-retroviral EVEs
We constructed a comprehensive library of all non-retroviral virus protein sequences available in GenBank and EMBL. We used DIAMOND60 to search these sequences against the D. rotundus genome. We extracted the matching amino-acid sequences and performed reciprocal blastp-like searches61 using DIAMOND with the selected subset of D. rotundus amino-acid sequences and the set of non-redundant protein sequences. D. rotundus genome sequences were considered of viral origin if they unambiguously matched viral proteins in the reciprocal best hits. Putative viral open reading frames were inferred from automated alignments, using exonerate62. For each putative D. rotundus viral peptide, we retrieved the function and predicted the taxonomic assignation by comparison to the best reciprocal blastp-like hit viral proteins. Phylogenetic analyses were performed by aligning the sequences using MAFFT63 and curated using AliView64. Maximum-likelihood inferences were performed on each multiple amino-acid alignment using RAxML65. Support for nodes was obtained from 100 non-parametric bootstrap iterations, and the root of maximum-likelihood trees was determined by midpoint rooting. We confirmed the presence of selected viruses by constructing double-indexed Illumina libraries66 on DNA derived from the spleen tissue of four different D. rotundus. Libraries were pooled and sequenced using 150-bp paired-end Illumina platform NextSeq 500. The raw paired-end reads were quality assessed, merged and filtered by mapping against the bacterial, human and chiropteran reference genome database with SMALT (https://www.sanger.ac.uk/resources/software/smalt/). Reads >150 bp were run through a viral assignment pipeline67 using blastx68 to search against the viral non-redundant protein database. Viral-matching reads were assigned a taxonomy with MEGAN569. Reads matching viral sequences were manually verified by reciprocal blastx analysis. Positive viral hits were defined as those with at least two different reads matching two different proteins or two different regions within the same viral protein.
ERVs
We identified flanking LTR regions using RepeatMasker70 and RMBLAST (http://www.repeatmasker.org/RMBlast.html), Tandem Repeats Finder58 and RepBase71 to the D. rotundus genome. We used blastn and tblastx with the retro-viral reference genome sequences in the RefSeq database of NCBI against the D. rotundus genome. Reads matching each of the RefSeq sequences were extracted and sorted by GI, collapsed by ID and manually verified by reciprocal blastn. Tblastx was re-run and the kept reads were mapped to the D. rotundus genome with SMALT and manually verified by reciprocal blastp. ERV sequences validation was carried out with tblastx against the Retroviridae. The retrieved ERV sequences were mapped using SMALT v0.7.6 against the genomes of D. rotundus, Mastomys natalensis, Eptesicus fuscus, M. lucifugus, M. brandtii, M. davidii, P. alecto, R. aegyptiacus and P. parnellii. Tblastx of the full genomic sequences of DrERV and DrgERV was also performed against Chiroptera. Selected genomic contigs were further analysed by reciprocal blastx against the non-redundant GenBank coding DNA sequence (CDS) translations + PDB + SwissProt + PIR + PRF database restricting the search to the Retroviridae, or against the DrgERV sequences. Retrieved sequences were aligned using MUSCLE in SeaView72. The best-fit amino-acid substitution model for each alignment was identified using jModelTest273 and trees were inferred under maximum-likelihood criteria using RAxML65.
Orthologous gene families
Using the D. rotundus genome against a range of other mammalian species, we performed clustering of orthologous genes using two strategies. (1) Identifying single-copy orthologues in the species by using the TreeFam method74. (2) Identifying 1:1 orthologues by building pair-wise orthologues between D. rotundus and the other species and using a reciprocal best hit (RBH) plus synteny approach as described in ref. 75.
dN/dS analyses
We used PAML codeml76 on the cleaned CDS alignments from the two sets of orthologous families and the corresponding phylogenetic tree as reported in ref. 77. To identify genes with accelerated evolution in the D. rotundus lineage we ran the two-ratio branch model. As a null model, we used the one-ratio model. Using these results, we performed likelihood ratio tests (LRTs) to identify genes with significant P values. To adjust for multiple testing, we used the FDR method. To identify genes with positively selected sites in D. rotundus, we also used a branch site with PAML codeml76. LRT and FDR were computed as carried out for the branch model tests.
Gene family expansion/contraction
We used CAFE78 with the results from the single-copy orthologous gene families. To obtain the dated tree required as input for CAFE, we obtained divergence times from the fossil dating records from TimeTree79. We concatenated the CDSs aligned with MAFFT and used PAML mcmctree to determine split times with the approximate likelihood calculation method. Genes with >200 copies in 1 of the species were filtered out.
Putative gene loss
We identified genes putatively lost in D. rotundus as previously described in ref. 80. The human gene set was downloaded from Ensembl and we obtained a non-redundant gene catalogue to be used as a reference by collapsing redundant genes and keeping the longest open reading frame. We used Bos taurus, Equus caballus and E. europaeus as outgroups. Genes identified as missing in D. rotundus were searched with blastp against the protein catalogues of the other available bats. Further validation on selected genes was performed by: evaluating the conservation of their syntenic regions compared to other species, and the GC content of the syntenic regions compared to the D. rotundus genome average GC content; searching them against the published D. rotundus transcriptome; and through PCR assays.
Functional characterization
We performed GO analysis using GOrilla81 as well as manual characterization using the GO annotations of the human genes downloaded from Ensembl and literature mining. We used PROVEAN82 to characterize the functional impact of the non-synonymous substitutions present only on the proteins in D. rotundus.
Species-specific PSS identification and protein modelling
We tested for positive selection and positive selected sites (PSSs) using the complete CDS alignments for the proteins FFAR1, PLXNA4, REG4, RNAS7 and TA2R3 from various species downloaded from the OrthoMaM database83. Sequences were realigned using MUSCLE84 in SeaView72 and phylogenetic analysis was performed using PhyML85. We used PAML codeml76 to test: the M1/M2 branch model constraining the D. rotundus node76; the M8a/M8 site model; and the branch-site model A constraining the D. rotundus node. The branch-site model A was evaluated under a LRT against the null hypothesis, while PSSs were scored under naïve empirical Bayes (NEB) and Bayes empirical Bayes (BEB). We performed modelling of the mentioned proteins from D. rotundus using the PyMOL Molecular Graphics System (Schrödinger, LLC). The three-dimensional protein models of the D. rotundus sequences were constructed using Phyre286 based on profile hidden Markov model analysis using the structures reported in refs 87,88,89,90.
Metagenomic data
We used faecal and anal swab samples from D. rotundus, R. aegyptiacus, R. ferrumequinum and M. gigas. The D. rotundus samples were obtained from wild D. rotundus individuals captured using mist nets and cloth bags and preserved in RNAlater according to the manufacturer’s instructions. The R. ferrumequinum samples were collected in Woodchester Mansion, Gloucestershire, UK. The M. gigas samples from Australia were collected by the consulting company Biologic in Pilbarra, Australia. The R. aegyptiacus faecal samples were collected from the Copenhagen Zoo. We have complied with all relevant ethical regulations for the collection of these samples. DNA from faecal samples was extracted using the PowerFecal DNA Isolation Kit (MoBio) with modifications to the manufacturer’s instructions. DNA from the D. rotundus milk sample was extracted using a standard commercial kit (Qiagen), following the manufacturer’s instructions. Anal swab samples were extracted using the BiOstic Bacteremia DNA Isolation Kit (MoBio) following the manufacturer’s protocol. All samples were kept at −20 °C. Libraries were built using the NEBnext DNA Library Prep Mast Mix Set for 454 (New England BioLabs) following the manufacturer’s instructions. Samples were 100-bp paired-end sequenced on the Illumina 2500 HiSeq platform.
Taxonomic and functional identification
The reads were cleaned with Trimmomatic91 and prinseq-lite92. The data sets were mapped against the closest available bat genome and only the non-mapping reads were kept. MGmapper93 was used for the taxonomic identification of invertebrates, protozoa, fungi, virus and bacteria. We kept the species identified with a coverage value higher than the first-quartile from the coverage distribution of the corresponding database and filtered out those found on the corresponding extraction blanks. Rarefaction curves from each data set were obtained using an in-house script with the MGmapper results. We then performed de novo assembly using IDBA_UD94, predicted genes using Prodigal95 and generated a non-redundant gene catalogue with usearch96. The non-redundant catalogue was searched with ublast96 against the UniProt database51 for functional and taxonomic annotation. We used DIAMOND60 to search the unmapped reads against the UniProt database keeping only the best hit for functional and taxonomic annotation. We annotated the UniProt protein identifications using KEGG orthology (KO) and eggNOG IDs.
Taxonomic and functional metagenomics comparison
We filtered on the basis of the breadth of coverage and the identifications of the extraction blanks. We removed any non-microbial hit and any taxa in which the paired reads matched different genera or only one of the reads had a hit. The counts were normalized by percentage. We identified a microbial taxonomic and functional sanguivorous core by comparing the filtered sets of the bats and keeping as core those taxa and genes identified only in the D. rotundus samples. We manually examined the taxa from the filtered taxonomic identifications, and the KO and COG annotations from the filtered non-redundant gene set catalogue. We compared the normalized abundance of taxa and functions between D. rotundus and the non-sanguivorous bats as follows. (1) Using the distributions of the different functional categories from D. rotundus and each non-sanguivorous bat species with a Wilcoxon rank-sum test. (2) Using the entire taxonomic and functional data sets, as well as down-sampling the normalized count values. Sampling values were the minimum, median and third-quartile values of the count distributions. With the resulting data sets, we calculated the Euclidean, Bray–Curtis and Jaccard distance metrics with the R package vegan97, and used the Ward hierarchical clustering method using UPGMA and Ward, and performed PCAs with prcomp and the GPA method. (3) We identified taxa and functions significantly contributing to the variation between the D. rotundus and the non-sanguivorous bat species. We examined the rotation matrix from the PCA of the normalized counts, excluding the four deepest sequenced samples, of the species and genus microbial taxonomic levels and the KEGG functional pathways. We identified the most significantly abundant D. rotundus microbial taxa as those with a significantly higher median normalized count value (P < 0.05) in D. rotundus and a median and mean normalized count value of 0 in the other 3 bat species for the first 3 principal components. We also identified significantly more abundant genes in D. rotundus by generating and annotating with KEGG a non-redundant gene set with all of the predicted genes from all of the bat samples. The reads of the samples were mapped against this bat non-redundant gene set, and a normalized count matrix was generated and used for Fisher tests on each of the functional pathways.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Code availability
In-house scripts used for the processing of the data are available from the corresponding authors upon request.
Data availability
The NCBI BioProject accession code for the genome assembly is PRJNA414273, and the sequence reads are available at the NCBI sequence read archive (SRA) under the accession SRA619672. The BioProject code for the metagenomic sequencing data is PRJNA415003, and the reads can be accessed at SRA with the accession SRA620977.
References
Breidenstein, C. P. Digestion and assimilation of bovine blood by a vampire bat (Desmodus rotundus). J. Mammal. 63, 482–484 (1982).
Edwards, M. A., Kaufman, M. L. & Storvick, C. A. Microbiologic assay for the thiamine content of blood of various species of animals and man. Am. J. Clin. Nutr. 5, 51–55 (1957).
Gracheva, E. O. et al. Molecular basis of infrared detection by snakes. Nature 464, 1006–1011 (2010).
Kishida, R., Goris, R. C., Terashima, S. & Dubbeldam, J. L. A suspected infrared-recipient nucleus in the brainstem of the vampire bat, Desmodus rotundus. Brain. Res. 322, 351–355 (1984).
Singer, M. A. Vampire bat, shrew, and bear: comparative physiology and chronic renal failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R1583–R1592 (2002).
Escalera-Zamudio, M. et al. The evolution of bat nucleic acid sensing Toll-like receptors. Mol. Ecol. 24, 5899–5909 (2015).
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R. & Gordon, J. I. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 (2008).
Graf, J., Kikuchi, Y. & Rio, R. V. M. Leeches and their microbiota: naturally simple symbiosis models. Trends Microbiol. 14, 365–371 (2006).
Hornborstel, H. Ueber die bakteriologischen Eigenschaften des Darmsymbionten beim medizinischen Blutegel (Hirudo officinalis) nebst Bemerkungen zur Symbiosefrage. Zbl. Bakteriol. 148, 36–47 (1942).
Graf, J. The effect of symbionts on the physiology of Hirudo medicinalis, the medicinal leech. Invertebr. Reprod. Dev. 41, 269–275 (2002).
Indergand, S. & Graf, J. Ingested blood contributes to the specificity of the symbiosis of Aeromonas veronii Biovar Sobria and Hirudo medicinalis, the medicinal leech. Appl. Environ. Microbiol. 66, 4735–4741 (2000).
Semova, I. et al. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12, 277–288 (2012).
Hajela, N. et al. Gut microbiome, gut function, and probiotics: implications for health. Indian J. Gastroenterol. 34, 93–107 (2015).
Cryan, J. F. & Dinan, T. G. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13, 701–712 (2012).
Qin, J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 (2012).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Sokol, H. et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl Acad. Sci. USA 105, 16731–16736 (2008).
Jefferson, R. The Hologenome. Agriculture, Environment and the Developing World: A Future of PCR. VHS Recording, Cold Spring Harbor Laboratory Press, Part 4: The Hologenome Plenary Lecture (Cold Spring Harbor Laboratory Press, New York, 1994).
Bordenstein, S. R. & Theis, K. R. Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 13, e1002226 (2015).
Ballal, S. A., Gallini, C. A., Segata, N., Huttenhower, C. & Garrett, W. S. Host and gut microbiota symbiotic factors: lessons from inflammatory bowel disease and successful symbionts. Cell. Microbiol. 13, 508–517 (2011).
Smith, J. D. L. & Gregory, T. R. The genome sizes of megabats (Chiroptera: Pteropodidae) are remarkably constrained. Biol. Lett. 5, 347–351 (2009).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2015).
Kojima, K. K. & Jurka, J. Crypton transposons: identification of new diverse families and ancient domestication events. Mob. DNA 2, 12 (2011).
Robertson, H. M. Members of the pogo superfamily of DNA-mediated transposons in the human genome. Mol. Gen. Genet. 252, 761–766 (1996).
Aiewsakun, P. & Katzourakis, A. Endogenous viruses: Connecting recent and ancient viral evolution. Virology 479–480, 26–37 (2015).
Hayward, J. A. et al. Identification of diverse full-length endogenous betaretroviruses in megabats and microbats. Retrovirology 10, 35 (2013).
Escalera-Zamudio, M. et al. A novel endogenous betaretrovirus in the common vampire bat (Desmodus rotundus) suggests multiple independent infection and cross-species transmission events. J. Virol. 89, 5180–5184 (2015).
Hong, W. & Zhao, H. Vampire bats exhibit evolutionary reduction of bitter taste receptor genes common to other bats. Proc. R. Soc. B 281, 20141079 (2014).
Dotson, C. D. et al. Bitter taste receptors influence glucose homeostasis. PLoS ONE 3, e3974 (2008).
Phillips, C. D. & Baker, R. J. Secretory gene recruitments in vampire bat salivary adaptation and potential convergences with sanguivorous leeches. Front. Ecol. Evol. 3, 122 (2015).
Gracheva, E. O. et al. Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature 476, 88–91 (2011).
Wang, Y. The functional regulation of TRPV1 and its role in pain sensitization. Neurochem. Res. 33, 2008–2012 (2008).
Phillips, C. D. et al. Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627 (2012).
Apitz-Castro, R. et al. Purification and partial characterization of draculin, the anticoagulant factor present in the saliva of vampire bats (Desmodus rotundus). Thromb. Haemost. 73, 94–100 (1995).
Truelove, E., Fielding, A. K. & Hunt, B. J. The coagulopathy and thrombotic risk associated with L-asparaginase treatment in adults with acute lymphoblastic leukaemia. Leukemia 27, 553–559 (2013).
Kikuchi, Y. & Graf, J. Spatial and temporal population dynamics of a naturally occurring two-species microbial community inside the digestive tract of the medicinal leech. Appl. Environ. Microbiol. 73, 1984–1991 (2007).
Worthen, P. L., Gode, C. J. & Graf, J. Culture-independent characterization of the digestive-tract microbiota of the medicinal leech reveals a tripartite symbiosis. Appl. Environ. Microbiol. 72, 4775–4781 (2006).
Guyton, A. C., Coleman, T. G., Young, D. B., Lohmeier, T. E. & DeClue, J. W. Salt balance and long-term blood pressure control. Annu. Rev. Med. 31, 15–27 (1980).
Bar-Peled, L., Schweitzer, L. D., Zoncu, R. & Sabatini, D. M. Ragulator is a GEF for the Rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012).
Donohoe, D. R. et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell. Metab. 13, 517–526 (2011).
Huq, A. H., Lovell, R. S., Ou, C. N., Beaudet, A. L. & Craigen, W. J. X-linked glycerol kinase deficiency in the mouse leads to growth retardation, altered fat metabolism, autonomous glucocorticoid secretion and neonatal death. Hum. Mol. Genet. 6, 1803–1809 (1997).
August, P. R. et al. Biosynthesis of the ansamycin antibiotic rifamycin: deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei S699. Chem. Biol. 5, 69–79 (1998).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Preprint at https://arxiv.org/abs/1308.2012 (2013).
Francischetti, I. M. B. et al. The ‘Vampirome’: transcriptome and proteome analysis of the principal and accessory submaxillary glands of the vampire bat Desmodus rotundus, a vector of human rabies. J. Proteom. 82, 288–319 (2013).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
She, R., Chu, J. S.-C., Wang, K., Pei, J. & Chen, N. GenBlastA: enabling BLAST to identify homologous gene sequences. Genome Res. 19, 143–149 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
UniProt Consortium. The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195 (2008).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-3.0; http://www.repeatmasker.org.
Jurka, J. Repeats in genomic DNA: mining and meaning. Curr. Opin. Struct. Biol. 8, 333–337 (1998).
Smit, A. & Hubley, R. RepeatModeler Open-1.0; http://www.repeatmasker.org/RepeatModeler.
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2014).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Larsson, A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 (2010).
Taboada, B. et al. Is there still room for novel viral pathogens in pediatric respiratory tract infections?. PLoS ONE 9, e113570 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Huson, D. H. & Weber, N. Microbial community analysis using MEGAN. Methods Enzymol. 531, 465–485 (2013).
Tarailo-Graovac, M. & Chen, N. UNIT 4.10 Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14 (2009).
Jurka, J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420 (2000).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 (2010).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012).
Schreiber, F., Patricio, M., Muffato, M., Pignatelli, M. & Bateman, A. TreeFamv9: a new website, more species and orthology-on-the-fly. Nucleic Acids Res. 42, D922–D925 (2014).
Jarvis, E. D. et al. Whole genome analyses resolve early branches in the tree of life of modern birds. Science 346, 1320–1331 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Teeling, E. C. et al. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 307, 580–584 (2005).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Hedges, S. B., Marin, J., Suleski, M., Paymer, M. & Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 32, 835–845 (2015).
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320 (2014).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 (2009).
Choi, Y. & Chan, P. A. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Ranwez, V. et al. OrthoMaM: a database of orthologous genomic markers for placental mammal phylogenetics. BMC Evol. Biol. 7, 241 (2007).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Kang, Y. et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015).
He, H., Yang, T., Terman, J. R. & Zhang, X. Crystal structure of the plexin A3 intracellular region reveals an autoinhibited conformation through active site sequestration. Proc. Natl Acad. Sci. USA 106, 15610–15615 (2009).
Ho, M.-R. et al. Human RegIV protein adopts a typical C-type lectin fold but binds Mannan with two calcium-independent sites. J. Mol. Biol. 402, 682–695 (2010).
Huang, Y.-C. et al. The flexible and clustered lysine residues of human ribonuclease 7 are critical for membrane permeability and antimicrobial activity. J. Biol. Chem. 282, 4626–4633 (2007).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Hasman, H. et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 52, 3136–3136 (2014).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.3-5. (R Foundation for Statistical Computing, Vienna, 2016).
Acknowledgements
We thank M. L. Méndez Ojeda, G. Czirjak, L. Caballero, O. Ríos, A. Patraca, C. Tello, D. Becker and M. E. Roelke-Parker for the collection and exportation of vampire bat samples, as well as P. Perelman and M. Korody for the preparation of genomic DNA. We thank Dovetail Genomics for the improvement of the genome contiguity of D. rotundus, in particular R. E. Green, M. Hartley and B. Rice. We also thank G. Jones for his support in the capture of the R. ferrumequinum bat samples and for useful comments on the manuscript, and R. Dunn and L. Nichols for their support at the early stages of the project. Likewise, we thank A. Alberdi and O. Aizpurua for assistance regarding samples from Rhinolophus euryale, Miniopterus schreibersii, Nyctalus leisleri and Nyctalus lasiopterus. We thank the Danish National High-Throughput DNA Sequencing Centre for assistance in generating the metagenome and Dovetail Illumina sequence data. We also thank the Instituto de Biotecnologia-UNAM for giving us access to its computer cluster and J. Verleyenfor his computer support. M.T.P.G. and M.L.Z.M. recognize Lundbeck Foundation R52-A5062, ERC Consolidator Grant (681396 – Extinction Genomics) and Danish National Research Foundation DNRF94 grants as the principal funding behind this work. M.E.-Z. and A.D.G. acknowledge the Deutsche Forschungsgemeinschaft (DFG) (GR 3924/9-1 to A.D.G) and an international doctoral scholarship provided by the Consejo Nacional de Ciencia y Tecnología (CONACyT) and the German Academic Exchange Service (DAAD) (311664). O.G.P. and J.T. acknowledge funding from the European Research Council under FP7/2007-2013/ERC grant agreement no. 614725-PATHPHYLODYN. A.K. recognizes funding from the Royal Society. H.K.F. acknowledges funding from a Bing-Mooney Fellowship in Environmental Science and Conservation. Field work in Peru was funded by the US National Science Foundation (grant DEB-1020966) and a Sir Henry Dale Fellowship to D.S., jointly funded by the Wellcome Trust and the Royal Society (grant 102507/Z/13/Z). Funding for Costa Rican fieldwork came from the Stanford Woods Institute for the Environment Environmental Venture Project.
Author information
Authors and Affiliations
Contributions
M.L.Z.M. and Z.X. contributed equally to the work. M.L.Z.M. and M.T.P.G. devised the study and supervised all parts of the project. M.L.Z.M., Z.X., M.E.-Z., S.L. and G.Z. designed the genomic and comparative genomic methods. M.L.Z.M. and T.S.-P. designed the metagenomics methods. M.L.Z.M. led the data integration, bioinformatic analyses and carried out the primary interpretation of analytical outcomes in close collaboration with M.E.-Z., A.D.G., D.S and M.T.P.G. The design of the viral analyses was carried out by J.T., A.K., O.G.P., M.E.-Z., U.L. and B.T., as was the performance of the viral bioinformatic analyses. Z.X., Y.L., S.L. and M.L.Z.M. performed the genomic methods. A.C.B. and J.A.S.C. provided support on a variety of bioinformatic analyses. S.L. performed the laboratory protocols for the genomic analysis. G.P. performed validation of the chosen putatively lost genes. O.A.R., M.B., T.S.-P., M.T.P.G. and G.Z. aided in the generation of the common vampire bat genome. A.K.R. and M.E.-Z. performed the laboratory metagenomic protocols. H.K.F., E.L.-R., E.R.-A., K.B., M.F.B. and N.E.W. carried out the collection and export of samples for the metagenomic data sets. C.F.A., E.R.-A. and E.L.-R. carried out the collection of the samples used for the viral analyses. M.L.Z.M. drafted the manuscript. M.E.-Z., J.T., D.S., H.K.F., K.B., A.D.G., M.F.B., M.B., G.Z., T.S.-P. and M.T.P.G. provided critical and substantial contribution to the various draft versions of the paper. All authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary Notes 1–11, Supplementary Figures 1–21, Supplementary Tables 1–16, Supplementary References.
Supplementary Information 1
This excel file contains ten sheets with the information on the genome assembly and annotation. Sheet 1: statistics of reads for D. rotundus genome sequencing. Sheet 2: statistics of seven decamers in reads from D. rotundus genome. Sheet 3: D. rotundus genome assembly statistics. Sheet 4: coverage of D. rotundus transcriptome by genome assembly. We used blat to map the transcriptome against genome assembly. Coverage percentages were calculated through dividing the total bases of transcripts that anchored the genome assembly by the total bases of transcripts, and we classed the data into three levels, >1kb, >2kb and >5kb. Sheet 5: statistics of TE content in D. rotundus genome. Sheet 6: prediction of genes in D. rotundus genome. Sheet 7: proteomic prediction and functional annotation of D. rotundus genome. Sheet 8: genomic assembly comparison using BUSCO of D. rotundus and the other used bat genomes. Sheet 9: genomic assembly comparisons of the scaffold and contig lengths of the used bat genomes. Sheet 10: statistics of high-quality reads mapping to the common vampire bat final genome assembly.
Supplementary Information 2
Gene ontology enrichment (for process, function and component) of the genes surrounding TEs in the D. rotundus genome. Sheet 1: De novo predicted MuDR elements in a single list comparison. Sheet 2: De novo predicted MuDR elements using as background list for comparison all the D. rotundus annotated genes. Sheet 3: RepeatMasker predicted MuDR elements in a single list comparison. Sheet 4: RepeatMasker predicted MuDR elements using as background list for comparison all the D. rotundus annotated genes. Sheet 5: all the de novo predicted TEs using as background list for comparison of all the D. rotundus annotated genes. Sheet 6: all the ProteinMask predicted TEs using as background list for comparison all the D. rotundus annotated genes. Sheet 7: all the RepeatMasker predicted TEs using as background list for comparison all the D. rotundus annotated genes. Sheet 8: all the de novo predicted TEs in a single list comparison. Sheet 9: all the ProteinMask predicted TEs in a single list comparison. Sheet 10: all the RepeatMasker predicted TEs in a single list comparison.
Supplementary Information 3
Viral elements in the D. rotundus genome. Sheet 1: confirmed viral hits within each vampire bat sample. Sheet 2: all bioinformatically identified significant D. rotundus non-retroviral EVEs.
Supplementary Information 4
Sheet 1: identified putative retroviral elements from the repeat elements characterization. Sheet 2: sequences used to confirm the presence of the putative retroviral elements identified in the repeat elements characterization. Sheet 3: RepeatMasker results of retroviral elements. Sheet 4: table taken from Table S1 from Cui et al. 2012 of previously reported ERV abundance. Sheet 5: BLASTN results for DrERV. Sheet 6: TBLASTX results for DrERV.
Supplementary Information 5
Comparative genomic analyses. Sheet 1: summary of gene families contraction in D. rotundus. Sheet 2: summary of gene families expansion in D. rotundus. Sheet 3: summary of gene families expansion in P. parnelli – D. rotundus. Sheet 4: summary of gene families contraction and expansion in Phyllostomidae-Vespertilioniada. Sheet 5: summary of gene families expansion in Microchiroptera-Pteropodidae. Sheet 6: statistics for functional annotation.
Supplementary Information 6
Extended discussion on some of the most relevant identifications from the comparative genomic and metagenomics analyses.
Supplementary Information 7
Sheet 1: putative gene loss results. These results contain those sequences identified as pseudogenes or as absent in the common vampire bat genome. Sheet 2: summary of the results of the supportive analyses on the selected putatively lost genes. Sheet 3: sequences of the designed PCR primers.
Supplementary Information 8
Statistically significant gene from the orthologous clusters from the single copy pipeline using six species. Sheet 2: GO enrichment of the significant genes from the orthologous clusters from the single copy pipeline using six species. Sheet 3: statistically significant gene from the orthologous clusters from the single copy pipeline using ten species. Sheet 4: GO enrichment of the significant genes from the orthologous clusters from the single copy pipeline using ten species. Sheet 5: statistically significant gene from the orthologous clusters from the 1:1 pipeline using ten species. Sheet 6: GO enrichment of the significant genes from the orthologous clusters from the 1:1 pipeline using ten species.
Supplementary Information 9
Metagenomic samples metadata and sequencing statistics.
Supplementary Information 10
Metagenomic vampire core. Sheet 1: bacterial core. Sheet 2: plasmid core. Sheet 3: fungal core. Sheet 4: protozoan core. Sheet 5: viral core. Sheet 6: gene core.
Supplementary Information 11
Microbial functional enrichment. Sheet 1: amino-acid metabolism. Sheet 2: biosynthesis of other secondary metabolites. Sheet 3: carbohydrate metabolism. Sheet 4: energy metabolism. Sheet 5: glycan biosynthesis and metabolism. Sheet 6: lipid metabolism. Sheet 7: metabolism of cofactors and vitamins. Sheet 8: other amino-acids metabolism. Sheet 9: terpenoids and polyketides metabolism. Sheet 10: nucleotide metabolism. Sheet 11: xenobiotics degradation and metabolism.
Supplementary Information 12
Potential microbial pathogenic species
Supplementary Information 13
Bacterial (sheet 1) and plasmid (sheet 2) microbial abundance matrix.
Supplementary Information 14
Gammaretrovirus-like ERV sequences identified in D. rotundus genome.
Supplementary Information 15
Statistically significant antiviral genes deeper selection analyses. Sheet 1: M1vsM2 test. Sheet 2: M8avsM8. Sheet 3: positive selected sites detected in D. rotundus under M8. Sheet 4: BSAn vs BSA test. Sheet 5: positive selected sites detected in the D. rotundus under BSA. Sheet 6: protein annotation functional features.
Supplementary Information 16
Microbial functional core pathway coverage compared to the total microbial functions.
Supplementary Information 17
MGmapper metagenomics identifications. Sheet 1: greengenes database. Sheet 2: human microbiome database. Sheet 3: MetaHit Assembly database. Sheet 4: plasmid database. Sheet 5: Silva database.
Supplementary Information 18
Physiological information of the sampled D. rotundus individuals. Sheet 1: individuals from Peru. Sheet 2: individuals from Costa Rica.
Supplementary Information 19
Microbiome functional (sheet 1) and taxonomic (sheets 2 and 3) profiles.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zepeda Mendoza, M.L., Xiong, Z., Escalera-Zamudio, M. et al. Hologenomic adaptations underlying the evolution of sanguivory in the common vampire bat. Nat Ecol Evol 2, 659–668 (2018). https://doi.org/10.1038/s41559-018-0476-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-018-0476-8
This article is cited by
-
Exploring the potential effects of forest urbanization on the interplay between small mammal communities and their gut microbiota
Animal Microbiome (2024)
-
Tracking the hologenome dynamics in aquatic invertebrates by the holo-2bRAD approach
Communications Biology (2024)
-
Sex differences and individual variability in the captive Jamaican fruit bat (Artibeus jamaicensis) intestinal microbiome and metabolome
Scientific Reports (2024)
-
Sugar assimilation underlying dietary evolution of Neotropical bats
Nature Ecology & Evolution (2024)
-
Co-diversification of an intestinal Mycoplasma and its salmonid host
The ISME Journal (2023)
