
Abstract
Under climate change, species unable to track their niche via range shifts are largely reliant on genetic variation to adapt and persist. Genomic vulnerability predictions are used to identify populations that lack the necessary variation, particularly at climate-relevant genes. However, hybridization as a source of novel adaptive variation is typically ignored in genomic vulnerability studies. We estimated environmental niche models and genomic vulnerability for closely related species of rainbowfish (Melanotaenia spp.) across an elevational gradient in the Australian Wet Tropics. Hybrid populations between a widespread generalist and several narrow range endemic species exhibited reduced vulnerability to projected climates compared to pure narrow endemics. Overlaps between introgressed and adaptive genomic regions were consistent with a signal of adaptive introgression. Our findings highlight the often-underappreciated conservation value of hybrid populations and indicate that adaptive introgression may contribute to evolutionary rescue of species with narrow environmental ranges.
Similar content being viewed by others


Main
The environmental conditions experienced by species throughout their evolutionary history contribute to determine their present-day niche and constrain their distributional limits1,2. Ancestral environments also shape contemporary patterns of standing genetic variation that are a key component of evolutionary potential. On the one hand, species that evolved in narrow environmental ranges may lack variation at genomic regions important for adaptation to a changing environment. On the other hand, generalist species that tolerate a much wider range of conditions may be better placed to respond to rapid climate change.
In predicting species’ vulnerability to rapid climate change, three evolutionary responses are typically considered: genetic adaptation, dispersal to a more suitable environment or acclimation to the altered environment through phenotypic plasticity3. An alternative and perhaps complementary evolutionary mechanism that is less often assessed is interspecific introgression following hybridization. That is the transfer of genetic material from one species into another by repeated backcrossing. Through this process, vulnerable species may adopt and exploit aspects of the evolutionary history of species more suited to the changed environmental conditions4,5.
The role of hybrid populations in conservation is controversial due to concerns about diluting the genetic integrity of parental species, as well as policy and legislative uncertainty6. Hybridization can potentially increase the risk of extinction via outbreeding depression, through demographic swamping due to infertile or maladapted hybrids, or by genetic swamping leading to complete replacement of the local gene pool7,8. The threat posed by these issues, however, is likely to be case-specific and may be less of a concern if hybridization is natural and has occurred over an extended period. Introgression as a source of novel genetic variation that can increase evolutionary potential is only recently gaining widespread appreciation, particularly in animals9. This has led to a call for hybrid populations to be given greater conservation value in policy and management decisions10. Hybrid zones could potentially facilitate evolutionary rescue of many species threatened by climate change5. This concept is also the basis of proposals for human-mediated evolutionary rescue of threatened species via translocations4,11,12.
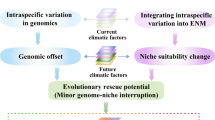
Genomic vulnerability assessments are increasingly used to identify populations that lack the genetic variation likely to be important for adaptation to climate change13,14,15. A range of statistical methods has been used16,17,18,19, although the basic framework is mostly similar regardless of the approach (but see ref. 20). The first of two steps is to build a statistical model of the relationship between putatively adaptive genetic variation and the current environment. Secondly, this model is applied to projections of future environmental conditions to predict the change in allele frequencies required to maintain present patterns of local adaptation (also termed genomic offset). In addition to estimating the amount of evolutionary change required, it is equally important to understand the capacity for that change to occur naturally. This second component, rarely assessed in studies of genomic vulnerability, infers whether the adaptive alleles are present in a population. Most studies have focused on abiotic factors influencing vulnerability, although species’ interactions and other evolutionary processes can also impact species’ responses to climate change21,22,23.
Here we explored an ideal biogeographic scenario involving rainbowfishes endemic to the Wet Tropics bioregion of northeastern Australia to assess whether natural hybridization can influence vulnerability to climate change. Rainbowfishes (genus Melanotaenia) are a suitable ectotherm system to investigate climatic-driven adaptive evolution24,25,26,27,28,29,30,31. They are a species-rich group of small fish found across the full spectrum of freshwater habitats in the Australian continent32. Their adaptive capacity to respond to projected climates appears to be biogeographically determined27 and their patterns of local adaptation are linked to hydroclimatic gradients and divergent thermal environments26,28,29,30. When experimentally exposed to future climates, rainbowfishes show associations between upper thermal tolerance and gene expression responses influenced by the biogeographic context where species evolved25,27,31. They also display range-wide differences in genotype and environment associations linked to seasonal variation in stream flow and temperature28,29, as well as to phenotypic traits that affect fitness26,30. The combined evidence from experimental and wild populations of Melanotaenia species24,25,26,27,28,29,30,31 supports the hypothesis that historical climatic variation, regional climatic differences and population connectivity influence variation in rainbowfish traits that determine regional patterns of adaptive resilience to climate change.
In this study we targeted five closely related species of tropical rainbowfishes that differ in predicted sensitivity to climate change. These include a widespread lowland generalist, Melanotaenia splendida splendida, and four narrow range specialists: the upland species M. eachamensis, Malanda rainbowfish and Tully rainbowfish (the latter two are undescribed), and the lowland species, M. utcheensis. We henceforth refer to the four narrow range species as narrow endemic rainbowfishes (NERs). Melanotaenia splendida are widely distributed and abundant across northeastern Australia, whereas the NERs exhibit restricted distributions confined to short river valleys within and below the Atherton Tablelands (Fig. 1a)33. The study area is centred at a well-described contact zone between lineages of many species expanding from two major Quaternary refugia34,35,36,37,38,39. The Australian Wet Tropics bioregion is listed as a World Heritage Area and as a biodiversity hotspot. It is severely threatened by climate change, with the extinction of many endemic species predicted as temperatures increase and the cooler upland rainforest habitat disappears40. Since cessation of volcanic activity in the early Holocene, major drainage patterns have largely resembled the present-day arrangement41 and it is expected that M. splendida and the NERs have intermittently been in contact during most of that time. In the absence of large waterfalls separating populations, species boundaries (and potentially hybrid zones) have probably been maintained by local hydroclimatic conditions associated with each species’ climatic niche. In the last few decades, climate change has resulted in M. splendida encroaching further into higher elevation habitat occupied by the NERs, and hybrids have been found where the species meet33,42. This has raised concerns over the potential for NER populations to become threatened with extinction due to hybridization with M. splendida42.

a, Sampling sites in the Wet Tropics of Queensland, Australia. b, Admixture plots for K = five ancestral species. c, Treemix maximum likelihood tree showing introgression among species. d, Topographic relief profile indicating the difference in elevation between upland and lowland habitat along a transect between sites 13 and 35.
We constructed environmental niche models (ENMs) for all species to track range size variation throughout the Holocene and into the future. We predict that NERs, but not M. splendida, will lose large areas of suitable habitat under projected climates. We then used genotype–environment association (GEA) analyses to identify candidate adaptive variation to estimate genomic vulnerability. We predict that hybrid NER populations will retain more adaptive alleles in the future and will require less evolutionary change to maintain patterns of local adaptation than pure NER populations. We also extended the genomic vulnerability framework to infer historical evolutionary responses to variation in climate throughout the Holocene. That extension enables the interpretation of future vulnerability estimates in the context of the rate and magnitude of past environmental changes. By examining patterns of hybridization and introgression between M. splendida and the NERs, we hypothesize that introgressive hybridization could be a source of novel adaptive variation that would be likely to facilitate evolutionary rescue of species threatened by climate change.
Genomic variation, hybrid detection and introgression
We examined 13,734 single nucleotide polymorphism (SNP) loci to reveal extensive patterns of hybridization and introgression among the 344 individuals representing the five species (Fig. 1 and Supplementary Table 1). More than 98% of SNPs mapped to one of 24 pseudo-chromosomes and were evenly distributed (mean = 565, s.d. = 68.7 SNPs per chromosome; Supplementary Table 2). Estimates of genetic diversity varied across species and among sampling sites but were significantly elevated (expected heterozygosity (He); pairwise Wilcoxon rank sum, Bonferroni corrected P < 0.001) for hybrid populations compared with pure NER populations (Supplementary Tables 3 and 4).
Individual ancestry proportions43 for K = five ancestral species (Fig. 1b) were used to classify 167 individuals as pure (Q > 0.95) and 85 individuals as M. splendida–NER hybrids (M. splendida and one NER Q > 0.1, all other species Q < 0.05). Pure individuals included 41 M. splendida, 22 M. eachamensis, 36 Malanda rainbowfish, 57 M. utcheensis and 11 Tully rainbowfish. We found hybrids between M. splendida and M. eachamensis (31), Malanda rainbowfish (49) and M. utcheensis (5), while no hybrid Tully rainbowfish were identified. The pure and hybrid classifications were well supported by the hybrid index44 estimates among M. splendida and each NER (Supplementary Table 5 and Extended Data Fig. 1). Parental interspecific heterozygosity was marginally greater than expected based on hybrid indexes, suggesting the presence of ancestral polymorphisms in both parental species. Hybrid individuals showed reduced levels of interspecific heterozygosity (Extended Data Fig. 2), providing evidence for advanced generation hybrids. Our maximum likelihood (ML) tree demonstrated that each lineage is monophyletic, although the best model supported nine migration events from M. splendida into NERs and one between NER species (Fig. 1c). NewHybrids45 simulations demonstrated the high power of our data to resolve hybrid classes providing an overall posterior probability of >0.995 across all simulated genealogical classes for each species. Empirical results supported the other analyses in identifying advanced generation hybrids for all species (Supplementary Tables 5 and 6 and Extended Data Figs. 1 and 2). Overall, our findings support the hypothesis that contact between the narrow endemics and M. splendida has probably been recurrent over long periods of time.
To assess historical admixture and genome-wide introgression, we calculated D, f4-ratios and fdM statistics for all M. splendida–NER trios with Dsuite. We found strong evidence for introgression between M. splendida and M. eachamensis, M. utcheensis and Malanda rainbowfish with a significant excess of ABBA (positive D) for all trios (Table 1). Positive f4-ratios were also observed for all trios, indicating substantial introgression from M. splendida into the three NERs (Table 1). Results for the sliding window statistic fdM identified 28 introgressed regions for the hybrid individuals from M. eachamensis, 26 for the hybrid Malanda rainbowfish and 18 introgressed regions for M. utcheensis. The introgressed regions were well distributed across 23 of 24 pseudo-chromosomes (Supplementary Table 2) and contained 153, 89 and 61 SNPs mapping to the main dataset for M. eachamensis, Malanda rainbowfish and M. utcheensis, respectively.
Environmental niche models
The ENMs based on maximum temperature of the warmest month (Bio05) and precipitation of the coldest quarter (Bio19) for M. splendida, M. eachamensis, M. utcheensis and Malanda rainbowfish (Extended Data Fig. 3) provided a good fit for the current species distributions (Supplementary Table 7). Throughout the Holocene, suitable habitat area for the NERs remained stable and similar to their recent historical known range. However, Malanda rainbowfish, for example, are predicted to lose as much as 92% of their current range by 2070 under the intermediate (RCP4.5) emissions scenario and more than 95% under the high (RCP8.5) emissions scenario (Fig. 2). A large area of high habitat suitability was confirmed for M. splendida throughout the Holocene, and ENMs based on projected 2070 climate suggested that most of that area is likely to remain suitable for the species (Fig. 2). Due to the low number of occurrence records for Tully rainbowfish (they are only known from four locations), ENMs could not be reliably estimated for this species.

Narrow endemic species, Malanda rainbowfish, are predicted to lose up to 95% of their current distribution by 2070 (RCP8.5 emissions scenario). Habitat area is expressed as the log number of suitable pixels on a binary representation of ensemble environmental niche models using a probability of occurrence threshold of 70% to estimate range sizes at each time period.
Climate adaptation and genomic vulnerability
The GEA analysis, temporal climatic change models and genomic vulnerability analyses together demonstrate the threat climate change poses to NERs (Fig. 3). Maximum temperature of the warmest month (Bio05) and precipitation of the coldest quarter (Bio19) were retained for the GEA analysis after accounting for correlated variables. The redundancy analysis (RDA) model was significant (R2 = 0.15, P < 0.001) and identified 211 candidate adaptive SNPs associated with the two climatic variables, with the first two axes explaining 15.99% and 12.84% of the variation constrained by the environment (Fig. 3a and Supplementary Tables 8 and 9). Four candidate adaptive SNPs occurred in introgressed regions identified using the fdM sliding window statistic. These included one for Malanda–M. splendida hybrids and three for M. eachamensis–M. splendida hybrids. In total 163/211 GEA candidate SNPs were annotated to 158 genes, and 294/301 candidate introgressed SNPs were annotated to 201 genes (Supplementary Table 10). Gene ontology enrichment analyses revealed several enriched categories for the GEA candidates (q < 0.05), including mitogen-activated protein kinase signalling pathway genes that have regulatory functions on heat shock proteins and cellular responses to thermal stress46 (Supplementary Table 11). Annotation of genomic positions and predicted functional effects indicated that a substantial number of candidate SNPs were in exons, with expected moderate to high impact on gene function (Extended Data Fig. 4).

a, Biplot summarizing the redundancy analysis. b, Current climate PCA indicating the relative size and position of each species niche. c–f, Temporal PCA plots demonstrating direction and magnitude of climate change between time periods: (c) early Holocene to mid Holocene, (d) mid Holocene to late Holocene, (e) late Holocene to current and (f) current to 2070 (RCP8.5 projections).
The environmental niche PCA highlights the much larger environmental envelope occupied by M. splendida compared with NERs. The lowland specialist M. utcheensis inhabits a warmer and wetter environment than the upland species that are restricted to much cooler conditions (Fig. 3b). The climate change PCAs reveal relatively little variation in climate over approximately 10 ky from the early Holocene until the present. However, for 2070 the climate is expected to become much hotter and slightly drier (RCP8.5 projections; Fig. 3c–f).
Following model calibration based on the current environment and observed candidate adaptive allele frequencies, eight populations for which the AlleleShift model performed poorly (R2 < 0.5) were not considered in the final vulnerability estimates47. Allele frequencies were then predicted for the remaining 28 sampling sites for historic (Fig. 4a) and projected future environments (Fig. 4b). Melanotaenia splendida ancestry was a reasonable predictor of genomic vulnerability (R2 = 0.11, P < 0.04) supporting the hypothesis that introgression with M. splendida may provide an evolutionary rescue effect for NER species most vulnerable to climate change (Fig. 4c). Allele frequencies of the candidate loci were estimated for each species and the percentage of loci missing the adaptive allele (based on the future climate models) were aggregated for pure and hybrid populations (Fig. 4d). Populations of pure Malanda rainbowfish lacked 30% of adaptive alleles, whereas hybrid Malanda populations had variation at >99% of candidate loci. For M. utcheensis, 26% of adaptive alleles were missing from pure populations with just 2% of adaptive alleles absent from hybrid populations. Pure populations of M. eachamensis appeared less depauperate than the other NERs with 13% of adaptive alleles absent, while only 7% of adaptive variation was missing from hybrid populations. In contrast, pure M. splendida populations were missing just 0.4% of adaptive alleles.

a–b, Estimates of the allele frequency change required to keep pace with climate change for Melanotaenia eachamensis, Malanda rainbowfish, Tully rainbowfish and M. utcheensis based on the candidate adaptive SNPs for (a) late Holocene to current and (b) current to 2070 (RCP8.5). c, Required change in allele frequency as a function of M. splendida ancestry (R2 = 0.11, P < 0.04). d, Percentage of adaptive alleles (relative to 2070 RCP8.5 climate predictions) absent from pure and hybrid populations. Hollow circles indicate pure populations and crosses show hybrid populations.
Discussion
Our genomic vulnerability assessments reveal that populations of narrow endemic rainbowfishes which demonstrate introgressive hybridization with a warm-adapted widespread generalist also exhibit reduced genomic vulnerability to climate change. This supports the hypothesis that natural evolutionary rescue may moderate the effects of climate change for these populations. Examining the genomic vulnerability estimates in the context of historical ENMs indicated that the evolutionary change required in the next 50 years far exceeds that which is likely to have occurred since the early Holocene. These findings are consistent with evidence from experiments of adaptive resilience to projected climates, and from range-wide surveys of adaptation which indicated that physiological performance limits and adaptive capacity in rainbowfishes are closely linked to local climatic conditions and range sizes25,26,27,28,29,30,31. Our approach expands on assessments of genomic vulnerability15,17,48,49,50 by considering the potential for adaptive introgression to enhance longer-term genomic responses to rapid environmental changes. Hybrid populations were shown to be less vulnerable, based on both the amount of evolutionary change required (that is, adaptive allele frequency change) and the capacity for that change to occur naturally (that is, whether the adaptive alleles are present). The latter is an often-overlooked component of genomic vulnerability and one that highlights the importance of standing genetic diversity for evolutionary potential.
Increasing empirical work demonstrates that introgression can accelerate adaptive shifts in response to environmental change51,52,53,54 which might help lineages threatened by climate warming55,56,57. For example, there is strong evidence for adaptive introgression between archaic humans58, and also among more modern human populations59 that probably facilitated rapid adaptation to new environments. Nolte et al.60 identified a hybrid population of sculpins (Cottus gobio) that were able to invade habitat unsuitable for either of the two parental lineages. One striking aspect of this example is the speed at which the hybrid population was able to adapt, with evidence that divergent phenotypic and life history traits arose along with the new habitat preferences in just a few decades. Our findings are consistent with a signal of adaptive introgression that could promote evolutionary rescue of cool-adapted species. This interpretation, however, requires further validation via common garden experiments (for example, ref. 61) to provide fitness estimates for the parental and hybrid lineages in current and predicted temperatures.
Contact between the cool-adapted rainforest species and warm-adapted lowland species has probably occurred recurrently throughout the Quaternary, consistent with findings for many lineages in this region62,63. Rainbowfish ecotypes are known to be constrained by climate26,27,28,29,30, and previous experiments of upper thermal tolerance and adaptive resilience to projected temperatures indicate that biogeographic factors might strongly influence climate change vulnerability25,27,31. The hybrid zones examined here have also probably been maintained by the respective climatic niches of the parental species, although the ENMs predict that by 2070 Malanda rainbowfish may lose >95% of suitable habitat, and M. eachamensis face >92% reduction (RCP8.5 projections). As the cooler upland climatic niche retreats, it is unclear to what extent pure and hybrid NER populations might either persist or be replaced completely by M. splendida. Translocating pure populations of upland species outside their current range is unlikely to provide a long-term solution as they currently occupy the only remaining cool tropical montane rainforest region on mainland Australia. Their impending niche loss coupled with high genomic vulnerability potentially provides few options for conservation managers in the future. We suggest that low levels of introgression (for example sites 10 and 16) should not be cause for concern and these populations should be afforded the equivalent conservation status as pure populations. We also argue that more advanced hybrid populations should be conferred greater value for their potentially crucial role in retaining unique diversity from the NER lineages in the future. These populations may also buffer species-level vulnerability in the short term, either directly via introgressed adaptive alleles or through indirect effects such as increased local effective population sizes64. Interestingly, although M. utcheensis is a lowland species and has evolved in a warmer environment, populations of this species exhibit some of the highest genomic vulnerability and lowest adaptive capacity to future conditions. While the coastal lowland environment of M. utcheensis appears to have remained relatively stable throughout the Holocene, populations may have been isolated by their unique climatic niche and we found little evidence for gene flow via hybridization. This highlights that the evolutionary history of specialist warm-adapted species could render them more vulnerable to climate change than might be expected.
In 1985, Soule65 described the (then) emerging field of conservation biology as a crisis discipline, and this has never been more true than now. The rate of anthropogenic climate change is challenging many species to mount evolutionary responses to environmental changes occurring on an ecological time scale. Conservation biologists and managers are also increasingly obliged to make difficult decisions without the time or resources necessary to fully understand the potential implications of these decisions. The genetic and demographic consequences of hybridization are difficult to predict but should be carefully considered when assessing whether possible negative effects are offset by potential gains in adaptive resilience. Here we identified long-established hybrid rainbowfish populations harbouring potentially important and novel genetic variation for responding to climate change. These populations would typically be ignored in management plans that focus on maintaining pure lineages66,67. The high resolution of modern genomic techniques can reveal subtle signals of admixture and managers must be cautious in how they interpret invasiveness and the threat of hybridization68. Nonetheless, the ability to more precisely characterize ancestry means that patterns of hybridization can be well defined. Our work highlights the conservation value of hybrid populations and exemplifies how adaptive introgression may contribute to natural evolutionary rescue of species threatened by climate change.
Methods
Study system
Thought to have a Gondwanan origin, rainbowfishes (Melanotaeniidae) are the most speciose freshwater fish family endemic to Australia and New Guinea69. Multiple lineages exist sympatrically which occasionally hybridize32,69, and many species readily do so in captivity70. This tendency may have helped facilitate their rapid adaptive radiation (for example, ref. 71) across the diverse range of climatic ecoregions they now inhabit within Australia32. Previous work, including for the generalist M. splendida splendida, has identified genomic signatures of local adaptation and adaptive plasticity associated with biogeographic history, hydroclimatic variation and projected climates25,26,27,28,29,30,31. Additionally, rainbowfishes are renowned for their high morphological diversity72,73, and strong links between morphological variation and local adaptation have also been established for several Australian species, including for the NER, M. eachamensis24,30,74.
Sampling and genomic data
Samples for 344 individuals from five Australian rainbowfish species69 were collected from 38 sites in the Australian Wet Tropics (Supplementary Table 1). In addition to the five focal species, two samples from a sixth rainbowfish species, M. trifasciata, were also included as an outgroup69. Fish were either sampled live and returned to the water with caudal fin clips stored in 100% ethanol, or euthanized in an overdose of AQUI-S solution (50% isoeugenol), frozen in liquid nitrogen and stored at −70 °C in the Australian Biological Tissues Collection at the South Australian Museum, Adelaide.
DNA was extracted following a modified salting-out protocol75 with DNA assessed for integrity using gel electrophoresis and for purity with a NanoDrop 1000 spectrophotometer (Thermo Scientific). Double digest restriction-site-associated DNA sequencing libraries76 were prepared using the restriction enzymes SbfI and MseI (New England Biolabs). Using custom individual barcodes to multiplex samples (96 per lane for all ingroup samples and 48 per lane for the outgroup samples), libraries were randomly assigned to each of seven Illumina HiSeq2500 lanes and sequenced as single-end, 100 base pair (bp) reads. Raw sequencing data were demultiplexed using the process_radtags module from STACKS 2.477. Individual fastq files were trimmed using Trimmomatic v.0.3978 and aligned to a M. duboulayi reference genome using Bowtie2 v.2.3.5.179. The SAM files were converted to BAM files and duplicate reads were marked and removed using Picard v.2.21.7 (https://github.com/broadinstitute/picard), before using GATK v.3.8-1-0 (ref. 80) for indel realignment. BCFtools v.1.9 (ref. 81) (bcftools call -m) was used to call SNPs. Raw genotypes were filtered for missing data, mapping quality (>30), HWE, MAF (>0.01) before pruning to reduce the effect of linkage disequilibrium. We first estimated linkage disequilibrium decay across the genome and plotted pairwise R2 among 66,762 raw SNPs before fitting a spline of exponential decay to estimate the distance in base pairs at which decay is no longer significant (P > 0.05) based on Tukey’s criteria for anomalies. We found that average R2 did not change significantly after 605 bp and pruning SNPs <300 bp apart resulted in 99.3% of SNP pairs separated by >100 Kbp, with <0.005% separated by less than 600 bp (Supplementary Table 12).
Genomic variation, hybrid detection and introgression
Genetic diversity summary statistics of expected heterozygosity (He), observed heterozygosity (Ho) and percentage of polymorphic loci were estimated for each sampling site and for aggregated pure and hybrid populations per species using the hierfstat R package82. We used a Wilcoxon rank sum test implemented in the stats R package83 (pairwise.wilcox.test) to assess differences in heterozygosity among pure and hybrid populations of the NERs.
To identify individuals with hybrid ancestry we first used ADMIXTURE v.1.3.0 to estimate individual ancestry proportions (Q) assuming a K value of five ancestral species43. We determined pure individuals as those with a Q value of >0.95. Hybrid status was assigned to individuals with both M. splendida and one other species with ancestry of >0.1, with the remaining species’ Q values <0.05. This allowed the evaluation of introgression between M. splendida and each of the narrow endemic species while reducing noise associated with individuals with multiple species ancestry. To additionally assess patterns of hybrid ancestry we estimated hybrid indices using the method implemented in the gghybrid R package84 and generated triangle plots to visualize the relationship between interspecific heterozygosity and hybrid index. We also performed simulations using NewHybrids v.1.1 (ref. 45) and the Hybriddetective R package85 to test the power of our data for detecting hybrids and to assign individuals to hybrid classes. We selected panels of around 200 informative SNPs and generated three replicates of three simulations with pure parents, F1, F2, and backcrosses between F1 and pure parents. Samples were then assigned to hybrid classes, based on the posterior probability thresholds estimated with the simulations. We used a Jeffreys-like prior and default genotype proportions with a burn-in of 20,000 iterations followed by 200,000 MCMC sweeps.
We used Treemix86 to examine introgression between branches of the rainbowfish phylogeny. This method models both topology and gene flow by first using allele frequencies and a Gaussian approximation for genetic drift to estimate a ML tree. The residual fit of the ML tree is used to identify populations that are a poor fit to the tree, before migration edges are fitted between branches in stepwise iterations to maximize the likelihood. We ran Treemix testing 1–20 migration events, using blocks of 500 SNPs (-k 500), no sample size correction (-noss) and two M. trifasciata samples as an outgroup. The final model was selected as the number of migration events at the asymptote of the log-likelihood estimations for all models.
Based on the ADMIXTURE results we used Dsuite87 to assess gene flow between M. splendida and the other species and to identify introgressed loci. For these analyses we refiltered the original raw genotypes, based on the 249 pure and hybrid individuals (as described above). The data were again filtered for missing data (<20%), MAF (>0.01), although the HWE filter was not applied as divergent allele frequencies are expected among species. PLINK v.1.9 (ref. 88) was used to prune the SNPs for linkage disequilibrium (–indep 50 5 2). The resulting 27,009 SNP dataset was used to calculated Patterson’s D89,90, also known as the ABBA–BABA statistic based on the tree (((P1,P2),P3),O). The D and f4-ratio statistics were calculated using the Dtrios function in Dsuite with default parameters. Trios were assessed to test the hypothesis of introgression between M. splendida and each of the NERs for which hybrids fitting the above criteria existed. In this case we tested trios where P1 represented the pure narrow endemic samples, P2 the hybrid samples, P3 the pure M. splendida samples and O the outgroup, M. trifasciata. In addition to assessing evidence for gene flow between M. splendida and the narrow endemics, we also estimated the sliding window statistic fdM91 to identify specific introgressed genomic regions. Implemented with the Dinvestigate function in Dsuite, a sliding window of 50 SNPs with a step of 10 SNPs was used (-w 50,10). Windows in the top 5% of the fdM distribution were considered as candidate introgressed loci. Overlapping candidate windows were merged using BEDtools v.2.29.1 (ref. 92) to provide a minimum set of candidate regions for each trio. We then used BCFtools view to map the 13,734 SNP dataset to the candidate introgressed regions to identify any overlap with the candidate climate-adapted SNPs.
Ecological niche models
Bioclimatic (BIOCLIM) variables were extracted from CHELSA v.1.2 (refs. 93,94). Projections for 2070 under intermediate (RCP4.5) and high (RCP8.5) emissions scenarios were also obtained from CHELSA based on the Australian Community Climate and Earth System Simulator (ACCESS1.0) global circulation model95. Historic climate models (also derived from CHELSA) were downloaded from PaleoClim96 for the early Holocene (11.7–8.326 ka), mid Holocene (8.326–4.2 ka) and late Holocene (4.2–0.3 ka)97. These data were resampled from 2.5 arc-minutes to match the current and future datasets’ 30 arc-second resolution. All rasters were cropped to an area encompassing the catchments from which samples were obtained. Coastlines for all time periods were also cropped to the current coastline to control for the effect of sea level changes throughout the Holocene. This was to enable direct comparison of habitat suitability across time periods for the specific extent of potential habitat available now and in the future (2070).
To predict species vulnerability to climate change, ecological niche models were generated for each species and each time period using biomod2 v.3.4.6 (ref. 98). In addition to locations for the genomic samples, occurrence data for a further 420 locations within the study extent were obtained from the Atlas of Living Australia (ALA; http://www.ala.org.au). These data were filtered for duplicate entries, geographic accuracy and to remove outliers based on known distributional limits. To avoid collinearity among variables, and to reduce the likelihood of overfitting the RDA and environmental niche models, we initially conducted a PCA on raster data from all 19 BIOCLIM variables across the study area using the raster_pca function from the synoptReg R package99. We then selected one temperature and one precipitation variable that most highly correlated with the first two axes of the initial climate PCA to ensure that spatial variation in climate was well captured. The ecological importance of the retained variables, maximum temperature of the warmest month (Bio05) and precipitation of the coldest quarter (Bio19), has also previously been demonstrated in studies of rainbowfish adaptation26,27,28. Ensemble models were built using four commonly used algorithms: maximum entropy (Maxent), generalized linear model (GLM), generalized boosting model (GBM) and random forest (RF)100. Five hundred pseudo-absences were randomly selected from the model extent. Each model was replicated three times and those with a relative operating characteristic curve statistic of >0.8 were retained. The weighted mean of probabilities ensemble models for each species were converted to binary representation using a probability threshold of 70% and used to estimate relative range sizes at each time period. A more sophisticated method of determining the binary threshold (minimum suitability of the top 90% of training sites) was trialled initially, which was found to bias the narrow endemic species range estimates downwards due to limited occurrence records. After exploring a range of parameters, we found that using the 70% suitability threshold provided a good balance between model accuracy and precision across all species.
Climate adaptation and genomic vulnerability
To identify a candidate set of climate-adapted loci for tropical Australian rainbowfish we used a GEA analysis using RDA to detect associations between population allele frequencies and the same two climatic variables used for the ENMs (Bio05 and Bio19). To control for the nonlinear spatial phylogenetic structure in the RDA, we estimated Moran’s eigenvector maps (MEM)101 using the mgQuick function from the MEMGENE R package102, before using a forward selection procedure to identify significant MEM eigenvectors to use as conditioning variables. We used the rda function in the vegan R package103 and tested significance of the final model using the anova.cca function and 1,000 permutations. The mean locus score across all SNPs was calculated for each of the first two RDA axes, and those scoring greater than three standard deviations from the mean were considered candidates for hydroclimatic selection104. Overlap between the GEA and introgressed candidate loci identified were considered as potential signals of adaptive introgression. We used SnpEff105 to perform gene, genomic position and functional effect annotations for the candidate loci based on the M. duboulayi genome. Gene ontology terms and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were explored using the STRING web server106.
To assess changes in the environment since the early Holocene, and how climate is predicted to change over the next 50 years, principal components analyses (PCA) were performed on climate data for each time period based on the retained bioclim variables from the RDA. The population.shift function from the AlleleShift R package19 was then used to visualize and compare the magnitude and direction of environmental changes between periods. An additional PCA was also performed using the retained current environmental data and plotting convex hulls surrounding the sampling sites for each species to highlight the relative size and any overlap of the environmental niche space occupied by each species.
AlleleShift was also used to model the rate of past evolutionary change and to predict future genomic vulnerability based on the candidate adapted loci. An initial two-step calibration first used RDA to build a model (AlleleShift::count.model) and predict the relationship between allele counts and the environmental data (AlleleShift::pred.model). Secondly, the predicted allele counts were used as independent variables in a generalized additive model with observed allele frequencies as the response (AlleleShift::freq.model). This step constrains the final allele frequency predictions to fall between 0 and 1. Model fit was then evaluated for each population and those for which the model performed poorly (R2 < 0.5) were omitted from the final analyses as suggested by Blumstein et al.47. Based on the calibrated allele frequency–environment model, allele counts were predicted for the 2070 projected environmental data and then converted to allele frequencies to enable direct comparison. Genomic vulnerability was then expressed simply as the difference between median values of the observed and predicted allele frequencies among the current and projected environmental models (referred to as delta allele frequency). Allele frequency shifts were also estimated using the historic environmental models to help interpret the genomic vulnerability assessments in the context of inferred rates of evolutionary responses to climate change throughout the Holocene. To test the hypothesis that hybrid populations show reduced genomic vulnerability to climate change, we constructed a linear model examining the relationship between genomic vulnerability and the proportion of M. splendida ancestry for each population. Finally, to assess the capacity for the pure NER populations to adapt in situ assuming no gene flow from M. splendida or the hybrid populations, we identified how many loci were missing the adaptive allele (as predicted by the AlleleShift model).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Melanotaenia duboulayi reference genome assembly is available at NCBI GenBank accession number JAPDEC000000000. Genotypes for the 13,734 and 27,009 SNP datasets and environmental data files can be accessed on Figshare: https://doi.org/10.6084/m9.figshare.21692918 (ref. 107).
Code availability
Code can be accessed on GitHub at https://github.com/pygmyperch/NER (ref. 108).
References
Ackerly, D. D. Community assembly, niche conservatism, and adaptive evolution in changing environments. Int. J. Plant Sci. 164, S165–S184 (2003).
Kellermann, V., Van Heerwaarden, B., Sgrò, C. M. & Hoffmann, A. A. Fundamental evolutionary limits in ecological traits drive Drosophila species distributions. Science 325, 1244–1246 (2009).
Hansen, M. M., Olivieri, I., Waller, D. M. & Nielsen, E. E. Monitoring adaptive genetic responses to environmental change. Mol. Ecol. 21, 1311–1329 (2012).
Aitken, S. N. & Whitlock, M. C. Assisted gene flow to facilitate local adaptation to climate change. Annu. Rev. Ecol. Evol. Syst. 44, 367–388 (2013).
Becker, M. et al. Hybridization may facilitate in situ survival of endemic species through periods of climate change. Nat. Clim. Change 3, 1039–1043 (2013).
Allendorf, F. W., Leary, R. F., Spruell, P. & Wenburg, J. K. The problems with hybrids: setting conservation guidelines. Trends Ecol. Evol. 16, 613–622 (2001).
Todesco, M. et al. Hybridization and extinction. Evol. Appl. 9, 892–908 (2016).
Rhymer, J. M. & Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 27, 83–109 (1996).
Taylor, S. A. & Larson, E. L. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3, 170–177 (2019).
vonHoldt, B. M., Brzeski, K. E., Wilcove, D. S. & Rutledge, L. Y. Redefining the role of admixture and genomics in species conservation. Conserv. Lett. 11, e12371 (2018).
Hamilton, J. A. & Miller, J. M. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv. Biol. 30, 33–41 (2016).
Ralls, K., Sunnucks, P., Lacy, R. C. & Frankham, R. Genetic rescue: a critique of the evidence supports maximizing genetic diversity rather than minimizing the introduction of putatively harmful genetic variation. Biol. Conserv. 251, 108784 (2020).
Capblancq, T., Fitzpatrick, M. C., Bay, R. A., Exposito-Alonso, M. & Keller, S. R. Genomic prediction of (mal) adaptation across current and future climatic landscapes. Annu. Rev. Ecol. Evol. Syst. 51, 245–269 (2020).
Rellstab, C., Dauphin, B. & Exposito‐Alonso, M. Prospects and limitations of genomic offset in conservation management. Evol. Appl. 14, 1202–1212 (2021).
Bay, R. A. et al. Genomic signals of selection predict climate-driven population declines in a migratory bird. Science 359, 83–86 (2018).
Rellstab, C. et al. Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol. Ecol. 25, 5907–5924 (2016).
Fitzpatrick, M. C. & Keller, S. R. Ecological genomics meets community-level modelling of biodiversity: mapping the genomic landscape of current and future environmental adaptation. Ecol. Lett. 18, 1–16 (2015).
Exposito-Alonso, M. et al. Genomic basis and evolutionary potential for extreme drought adaptation in Arabidopsis thaliana. Nat. Ecol. Evol. 2, 352–358 (2018).
Kindt, R. AlleleShift: an R package to predict and visualize population-level changes in allele frequencies in response to climate change. PeerJ 9, e11534 (2021).
Gain, C. & François, O. LEA 3: factor models in population genetics and ecological genomics with R. Mol. Ecol. Resour. 21, 2738–2748 (2020).
Aguirre-Liguori, J. A., Ramírez-Barahona, S. & Gaut, B. S. The evolutionary genomics of species’ responses to climate change. Nat. Ecol. Evol. 5, 1350–1360 (2021).
Taylor, S. A., Larson, E. L. & Harrison, R. G. Hybrid zones: windows on climate change. Trends Ecol. Evol. 30, 398–406 (2015).
Hoffmann, A. A. & Sgro, C. M. Climate change and evolutionary adaptation. Nature 470, 479–485 (2011).
McGuigan, K., Franklin, C. E., Moritz, C. & Blows, M. W. Adaptation of rainbow fish to lake and stream habitats. Evolution 57, 104–118 (2003).
Smith, S., Bernatchez, L. & Beheregaray, L. RNA-seq analysis reveals extensive transcriptional plasticity to temperature stress in a freshwater fish species. BMC Genomics 14, 375 (2013).
Smith, S. et al. Latitudinal variation in climate‐associated genes imperils range edge populations. Mol. Ecol. 29, 4337–4349 (2020).
Sandoval-Castillo, J. et al. Adaptation of plasticity to projected maximum temperatures and across climatically defined bioregions. Proc. Natl Acad. Sci. USA 117, 17112–17121 (2020).
Brauer, C., Unmack, P. J., Smith, S., Bernatchez, L. & Beheregaray, L. B. On the roles of landscape heterogeneity and environmental variation in determining population genomic structure in a dendritic system. Mol. Ecol. 27, 3484–3497 (2018).
Attard, C. R. et al. Fish out of water: genomic insights into persistence of rainbowfish populations in the desert. Evolution 76, 171–183 (2022).
Gates, K. et al. Environmental selection, rather than neutral processes, best explain patterns of diversity in a tropical rainforest fish. Preprint at bioRxiv https://doi.org/10.1101/2022.1105.1113.491913 (2022).
McCairns, R. J. S., Smith, S., Sasaki, M., Bernatchez, L. & Beheregaray, L. B. The adaptive potential of subtropical rainbowfish in the face of climate change: heritability and heritable plasticity for the expression of candidate genes. Evol. Appl. 9, 531–545 (2016).
McGuigan, K., Zhu, D., Allen, G. & Moritz, C. Phylogenetic relationships and historical biogeography of melanotaeniid fishes in Australia and New Guinea. Mar. Freshwat. Res. 51, 713–723 (2000).
Unmack, P. J. et al. Malanda Gold: the tale of a unique rainbowfish from the Atherton Tablelands, now on the verge of extinction. Fish. Sahul. 30, 1039–1054 (2016).
Moritz, C. Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst. Biol. 51, 238–254 (2002).
Pope, L., Estoup, A. & Moritz, C. Phylogeography and population structure of an ecotonal marsupial, Bettongia tropica, determined using mtDNA and microsatellites. Mol. Ecol. 9, 2041–2053 (2000).
Hugall, A., Moritz, C., Moussalli, A. & Stanisic, J. Reconciling paleodistribution models and comparative phylogeography in the Wet Tropics rainforest land snail Gnarosophia bellendenkerensis (Brazier 1875). Proc. Natl Acad. Sci. USA 99, 6112–6117 (2002).
Moritz, C. et al. Identification and dynamics of a cryptic suture zone in tropical rainforest. Proc. R. Soc. B. 276, 1235–1244 (2009).
Phillips, B. L., Baird, S. J. & Moritz, C. When vicars meet: a narrow contact zone between morphologically cryptic phylogeographic lineages of the rainforest skink, Carlia rubrigularis. Evolution 58, 1536–1548 (2004).
Krosch, M. N., Baker, A. M., Mckie, B. G., Mather, P. B. & Cranston, P. S. Deeply divergent mitochondrial lineages reveal patterns of local endemism in chironomids of the Australian Wet Tropics. Austral Ecol. 34, 317–328 (2009).
Williams, S. E., Bolitho, E. E. & Fox, S. Climate change in Australian tropical rainforests: an impending environmental catastrophe. Proc. R. Soc. B. 270, 1887–1892 (2003).
Whitehead, P. et al. Temporal development of the Atherton Basalt Province, north Queensland. Aust. J. Earth Sci. 54, 691–709 (2007).
Moy, K. G., Unmack, P. J., Lintermans, M., Duncan, R. P. & Brown, C. Barriers to hybridisation and their conservation implications for a highly threatened Australian fish species. Ethology 125, 142–152 (2019).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Buerkle, C. A. Maximum‐likelihood estimation of a hybrid index based on molecular markers. Mol. Ecol. Notes 5, 684–687 (2005).
Anderson, E. & Thompson, E. A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160, 1217–1229 (2002).
Dorion, S. & Landry, J. Activation of the mitogen-activated protein kinase pathways by heat shock. Cell Stress Chaperones 7, 200 (2002).
Blumstein, M. et al. Protocol for projecting allele frequency change under future climate change at adaptive-associated loci. STAR Protoc. 1, 100061 (2020).
Gougherty, A. V., Keller, S. R. & Fitzpatrick, M. C. Maladaptation, migration and extirpation fuel climate change risk in a forest tree species. Nat. Clim. Change 11, 166–171 (2021).
Blumstein, M. et al. A new perspective on ecological prediction reveals limits to climate adaptation in a temperate tree species. Curr. Biol. 30, 1447–1453. e1444 (2020).
Razgour, O. et al. Considering adaptive genetic variation in climate change vulnerability assessment reduces species range loss projections. Proc. Natl Acad. Sci. USA 116, 10418–10423 (2019).
Goicoechea, P. G. et al. Adaptive introgression promotes fast adaptation in oaks marginal populations. Preprint available at bioRxiv https://doi.org/10.1101/731919 (2019).
Lavergne, S. & Molofsky, J. Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc. Natl Acad. Sci. USA 104, 3883–3888 (2007).
De Carvalho, D. et al. Admixture facilitates adaptation from standing variation in the European aspen (Populus tremula L.), a widespread forest tree. Mol. Ecol. 19, 1638–1650 (2010).
De-Kayne, R. et al. Genomic architecture of adaptive radiation and hybridization in Alpine whitefish. Nat. Commun. 13, 4479 (2022).
Baskett, M. L. & Gomulkiewicz, R. Introgressive hybridization as a mechanism for species rescue. Theor. Ecol. 4, 223–239 (2011).
Meier, J. I. et al. The coincidence of ecological opportunity with hybridization explains rapid adaptive radiation in Lake Mweru cichlid fishes. Nat. Commun. 10, 1–11 (2019).
Svardal, H. et al. Ancestral hybridization facilitated species diversification in the Lake Malawi cichlid fish adaptive radiation. Mol. Biol. Evol. 37, 1100–1113 (2020).
Racimo, F., Sankararaman, S., Nielsen, R. & Huerta-Sánchez, E. Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 16, 359–371 (2015).
Jeong, C. et al. Admixture facilitates genetic adaptations to high altitude in Tibet. Nat. Commun. 5, 1–7 (2014).
Nolte, A. W., Freyhof, J., Stemshorn, K. C. & Tautz, D. An invasive lineage of sculpins, Cottus sp. (Pisces, Teleostei) in the Rhine with new habitat adaptations has originated from hybridization between old phylogeographic groups. Proc. R. Soc. B. 272, 2379–2387 (2005).
Fitzpatrick, M. C., Chhatre, V. E., Soolanayakanahally, R. Y. & Keller, S. R. Experimental support for genomic prediction of climate maladaptation using the machine learning approach Gradient Forests. Mol. Ecol. Resour. 21, 2749–2765 (2021).
Schneider, C., Cunningham, M. & Moritz, C. Comparative phylogeography and the history of endemic vertebrates in the Wet Tropics rainforests of Australia. Mol. Ecol. 7, 487–498 (1998).
Hewitt, G. M. Quaternary phylogeography: the roots of hybrid zones. Genetica 139, 617–638 (2011).
Pfennig, K. S., Kelly, A. L. & Pierce, A. A. Hybridization as a facilitator of species range expansion. Proc. R. Soc. B. 283, 20161329 (2016).
Soulé, M. E. What is conservation biology? A new synthetic discipline addresses the dynamics and problems of perturbed species, communities, and ecosystems. Bioscience 35, 727–734 (1985).
Biermann, C. & Havlick, D. Genetics and the question of purity in cutthroat trout restoration. Restor. Ecol. 29, e13516 (2021).
Fredrickson, R. J. & Hedrick, P. W. Dynamics of hybridization and introgression in red wolves and coyotes. Conserv. Biol. 20, 1272–1283 (2006).
Hirashiki, C., Kareiva, P. & Marvier, M. Concern over hybridization risks should not preclude conservation interventions. Conserv. Sci. Pract. 3, e424 (2021).
Unmack, P. J., Allen, G. R. & Johnson, J. B. Phylogeny and biogeography of rainbowfishes (Melanotaeniidae) from Australia and New Guinea. Mol. Phylogenet. Evol. 67, 15–27 (2013).
Allen, G. Rainbowfishes in Nature and the Aquarium (Tetra Publications, 1995).
Seehausen, O. Hybridization and adaptive radiation. Trends Ecol. Evol. 19, 198–207 (2004).
Pusey, B., Kennard, M. J. & Arthington, A. H. Freshwater Fishes of North-eastern Australia (CSIRO Publishing, 2004).
Zhu, D., Degnan, S. & Moritz, C. Evolutionary distinctiveness and status of the endangered Lake Eacham rainbowfish (Melanotaenia eachamensis). Conserv. Biol. 12, 80–93 (1998).
McGuigan, K., Chenoweth, S. F. & Blows, M. W. Phenotypic divergence along lines of genetic variance. Am. Nat. 165, 32–43 (2005).
Sunnucks, P. & Hales, D. F. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 13, 510–524 (1996).
Peterson, B., Weber, J., Kay, E., Fisher, H. & Hoekstra, H. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7, e37135 (2012).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes Genomes Genet. 1, 171–182 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357 (2012).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Goudet, J. Hierfstat, a package for R to compute and test hierarchical F‐statistics. Mol. Ecol. Notes 5, 184–186 (2005).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2021).
Bailey, R. ribailey/gghybrid: gghybrid R package for Bayesian hybrid index and genomic cline estimation. v2.0.0 https://doi.org/10.5281/zenodo.3676498 (2020).
Wringe, B. hybriddetective: automates the process of detecting hybrids from genetic data. R package version 0.1.0.9000 https://github.com/bwringe/hybriddetective (2016).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Malinsky, M., Matschiner, M. & Svardal, H. Dsuite‐Fast D‐statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 21, 584–595 (2021).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Durand, E. Y., Patterson, N., Reich, D. & Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011).
Malinsky, M. et al. Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science 350, 1493–1498 (2015).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Karger, D. N. et al. Climatologies at high resolution for the earth’s land surface areas. Sci. Data 4, 1–20 (2017).
Karger, D. N. et al. CHELSA climatologies at high resolution for the Earth’s land surface areas (v.1.0). https://doi.org/10.1594/WDCC/CHELSA_v1 (2016).
Ackerley, D. & Dommenget, D. Atmosphere-only GCM (ACCESS1.0) simulations with prescribed land surface temperatures. Geosci. Model Dev. 9, 2077–2098 (2016).
Brown, J. L., Hill, D. J., Dolan, A. M., Carnaval, A. C. & Haywood, A. M. PaleoClim: high spatial resolution paleoclimate surfaces for global land areas. Sci. Data 5, 1–9 (2018).
Fordham, D. A. et al. PaleoView: a tool for generating continuous climate projections spanning the last 21,000 years at regional and global scales. Ecography 40, 1348–1358 (2017).
Thuiller, W., Lafourcade, B., Engler, R. & Araújo, M. B. BIOMOD–a platform for ensemble forecasting of species distributions. Ecography 32, 369–373 (2009).
Lemus-Canovas, M., Lopez-Bustins, J. A., Martin-Vide, J. & Royé, D. synoptReg: an R package for computing a synoptic climate classification and a spatial regionalization of environmental data. Environ. Model. Softw. 118, 114–119 (2019).
Hao, T., Elith, J., Guillera‐Arroita, G. & Lahoz‐Monfort, J. J. A review of evidence about use and performance of species distribution modelling ensembles like BIOMOD. Divers. Distrib. 25, 839–852 (2019).
Galpern, P., Peres‐Neto, P. R., Polfus, J. & Manseau, M. MEMGENE: spatial pattern detection in genetic distance data. Methods Ecol. Evol. 5, 1116–1120 (2014).
Peres‐Neto, P. R. & Galpern, P. memgene: spatial pattern detection in genetic distance data using Moran’s eigenvector maps. R package version 1.0.1 https://cran.r-project.org/web/packages/memgene/ (2019).
Oksanen, J. et al. vegan: community ecology package. R package version 2.3–0 https://cran.r-project.org/web/packages/vegan/ (2015).
Forester, B. R., Jones, M. R., Joost, S., Landguth, E. L. & Lasky, J. R. Detecting spatial genetic signatures of local adaptation in heterogeneous landscapes. Mol. Ecol. 25, 104–120 (2015).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Szklarczyk, D. et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612 (2021).
Brauer, C. J. et al. Data for ‘Natural hybridisation reduces vulnerability to climate change’. figshare https://doi.org/10.6084/m9.figshare.21692918 (2022).
Brauer, C. J. et al. Code for ‘Natural hybridisation reduces vulnerability to climate change’. GitHub https://github.com/pygmyperch/NER (2022).
Acknowledgements
We acknowledge the Traditional Owners of Country covering the Wet Tropics, recognize their continuing connection to land, water and fishes, and pay respects to Elders past, present and emerging. Collections were obtained under permits from various state fisheries’ agencies and research was carried out under Flinders University Animal Welfare Committee approval E342. We thank M. Sasaki for laboratory assistance. Financial support was provided by the Australian Research Council via grants to L.B.B. (grant nos. FT130101068; DP150102903). K. Martin provided samples, assistance in the field and valuable discussion about rainbowfish species distributions and habitat preferences.
Author information
Authors and Affiliations
Contributions
C.J.B., P.J.U. and L.B.B. conceived, designed and refined the project. C.J.B. and J.S.-C. performed the data analysis with assistance from K.G. and P.J.U. All authors contributed to interpreting the results. C.J.B. and L.B.B. led the writing of the manuscript, with input from J.S.-C., K.G., M.P.H., P.J.U. and L.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Climate Change thanks Jorge Avaria-Llautureo, Michael M. Hansen, Kristina Sefc and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Hybrid index estimations among Melanotaenia splendida and narrow endemic species (NERs).
a) M.eachamensis, b) Malanda rainbowfish and c) M. utcheensis populations. Pure reference populations are coded as S0 (blue, M. splendida), S1 (red, NERs). The point estimates are based on the mode from the posterior distribution, and error bars indicate 95% credibility intervals based on an initial burn-in of 1000 MCMC iterations followed by 5000 iterations. Dashed blue and red lines define the 95% credible intervals for classifying a sample as parental or hybrid.
Extended Data Fig. 2 Triangle plots contrasting hybrid index with interspecific heterozygosity among Melanotaenia splendida and narrow endemic species (NERs).
a) M.eachamensis, b) Malanda rainbowfish and c) M. utcheensis populations. Parental interspecific heterozygosity is marginally greater than expected for all species, suggesting the presence of ancestral polymorphisms in both parental species. Hybrid individuals show reduced levels of interspecific heterozygosity, providing evidence for advanced-generation hybrids.
Extended Data Fig. 3 Weighted mean of probabilities ensemble environmental niche models built from individual Maximum Entropy, Generalised Linear Model, Generalised Boosting Model, and Random Forest models.
a) Melanotaenia splendida, b) Malanda rainbowfish, c) M. eachamensis, and D) M. utcheensis climate models for the early-Holocene (11.7–8.326 ka), mid-Holocene (8.326–4.2 ka) and late-Holocene (4.2–0.3 ka) and projected models for 2070 under intermediate (RCP4.5) and high (RCP8.5) emissions scenarios were obtained from CHELSA v1.1 and based on the Australian Community Climate and Earth System Simulator (ACCESS1.0) global circulation model. Plot axes are (°E, °S).
Extended Data Fig. 4 Genomic and functional annotation of candidate SNP loci.
Annotation by a) genomic region, b) functional effect, and c) impact of 211 genome environment association (GEA) candidates, and 301 introgression regions (INTRO) candidates, based on the crimson spotted rainbowfish (Melanotaenia duboulayi) genome annotation.
Supplementary information
Supplementary Information
Supplementary Tables 1–4 and 6–9.
Supplementary Tables
Supplementary Tables 5 and 10–12.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brauer, C.J., Sandoval-Castillo, J., Gates, K. et al. Natural hybridization reduces vulnerability to climate change. Nat. Clim. Chang. 13, 282–289 (2023). https://doi.org/10.1038/s41558-022-01585-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41558-022-01585-1
This article is cited by
-
Sea of opportunities: marine genomics in an era of global environmental change
BMC Genomics (2023)
-
Three systems of molecular markers reveal genetic differences between varieties sabina and balkanensis in the Juniperus sabina L. range
Annals of Forest Science (2023)
-
Real-time mode of operation data analysis to catch the thread-tip denotes the failure cause of the grid-tie PV central inverter
Scientific Reports (2023)
-
Population genomic diversity and structure in the golden bandicoot: a history of isolation, extirpation, and conservation
Heredity (2023)
-
Hybridization provides climate resilience
Nature Climate Change (2023)
