
Abstract
Atomistic simulation has a broad range of applications from drug design to materials discovery. Machine learning interatomic potentials (MLIPs) have become an efficient alternative to computationally expensive ab initio simulations. For this reason, chemistry and materials science would greatly benefit from a general reactive MLIP, that is, an MLIP that is applicable to a broad range of reactive chemistry without the need for refitting. Here we develop a general reactive MLIP (ANI-1xnr) through automated sampling of condensed-phase reactions. ANI-1xnr is then applied to study five distinct systems: carbon solid-phase nucleation, graphene ring formation from acetylene, biofuel additives, combustion of methane and the spontaneous formation of glycine from early earth small molecules. In all studies, ANI-1xnr closely matches experiment (when available) and/or previous studies using traditional model chemistry methods. As such, ANI-1xnr proves to be a highly general reactive MLIP for C, H, N and O elements in the condensed phase, enabling high-throughput in silico reactive chemistry experimentation.

Similar content being viewed by others

Main
Over the past several decades, atomic-scale simulation has become an invaluable computational tool for providing microscopic explanations of experimentally observed phenomena. Many scientifically crucial chemical and materials properties can be evaluated through molecular dynamics (MD) simulation, wherein atomic motion is dictated by integrating the second law of Newtonian physics. The quantitative predictiveness of MD depends almost entirely on the accuracy of the underlying model potential energy surface (the potential) used to compute the forces acting on each atom. However, standard physics-based paradigms, such as classical force fields (FFs) and quantum mechanics (QM) methods, straddle a historical trade-off between computational cost, accuracy and generality, that is, being applicable to a broad range of systems without further specialization. This trade-off is especially pronounced in the context of modelling reactions, that is, the making and breaking of chemical bonds. Although computationally efficient, reactive FFs often need to be reparameterized to pre-determined reactions to be quantitatively accurate. By contrast, while QM methods are often quite reliable and generally applicable, their computational cost is prohibitive for many reactive MD studies. For this reason, a fast, accurate and general reactive potential is of paramount importance to many scientific applications, as it would fulfil the long-sought promise for predictive MD simulations that can provide reliable reaction rates, discover entire reaction networks and warn of dangerous conditions, all before entering the laboratory.
Recently, machine learning interatomic potentials (MLIPs) have been proposed to overcome the trade-off that has existed in physics-based computational models for many decades. MLIPs often achieve computational efficiency similar to classical FFs but with QM-level accuracy1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16. Among the many different types of MLIPs that have been proposed, neural network (NN)-based MLIPs are especially capable of describing a broad range of chemical systems without additional specialization and, thereby, represent a top candidate for developing a truly general MLIP. For example, ANAKIN-ME (or ANI) is a NN-based MLIP that has been trained to large and chemically diverse datasets of organic molecules containing the elements C, H, N, O, S, F and Cl (refs. 17,18). While previous ANI MLIPs proved to be extremely accurate for near-equilibrium conformations of organic molecules in vacuo, these potentials do not address the challenges of modelling condensed-phases (that is, periodic systems of liquids, supercritical fluids or solids) and reactive chemistry.
Several MLIPs have been developed for studying both condensed-phase and gas-phase (or in vacuo) reactive chemistry of a specific system19,20,21,22,23,24,25. However, each of these studies required considerable domain and MLIP expertise and enormous computational resources to build a non-general reactive MLIP. For this reason, a highly general reactive MLIP would be transformational towards the usage and impact of MLIPs among non-experts. While recent endeavours have yielded groundbreaking results towards a general MLIP for approximately one third of the periodic table26,27,28, these studies do not directly target reactive chemistry. Targeted, model-aware sampling strategies for dataset generation of three-dimensional atomic positions are especially essential for modelling rare events, such as chemical reactions17,29.
Active learning (AL)30 is a class of model-aware algorithms designed to automatically sample, select and label new data with the goal of efficiently generating a diverse and relevant dataset to train a more robust ML model. AL aims to ameliorate human bias through automating the decision-making process for adding new data to a training dataset. Recently, AL has been applied to develop numerous MLIPs trained to datasets of atomic positions labelled with energies and atomic forces from expensive QM calculations17,19,31,32,33,34,35,36.
To develop a general reactive MLIP with AL, existing methodologies for selecting, labelling and training are relatively straightforward to apply (Methods). However, for sampling atomic positions, adequately exploring reactive chemical space in an automated fashion is extremely challenging37 because it requires the exploration of chemical variance of molecular species in tandem with structural variance associated with non-equilibrium thermodynamic processes. The traditional approach of fitting reactive FFs38,39,40,41,42 to a limited dataset of pre-determined gas-phase reaction pathways based on chemical intuition is insufficient for developing a general reactive MLIP, as the resulting MLIP would be biased to only perform well on the assumed reaction network. Similarly, while recent work (performed simultaneous and independent to this study) presented an automated approach to sample transition states and minimum-energy-path structures for gas-phase (or in vacuo) reactions of C, H, N and O molecules43, this sampling procedure is unlikely to result in an MLIP that is robust for condensed-phase high-temperature reactive MD simulations. By contrast, training an MLIP directly to condensed-phase QM reactive data would ensure that the potential is reliable for the density ranges typically used in reactive MD simulations.
Wang and co-workers developed an elegant approach for the MD-based exploration of reaction pathways in the condensed phase, using QM methods, referred to as the ab initio nanoreactor (NR)44,45. The NR was designed to model high-velocity molecular collisions of small molecules by using a fictitious biasing force to promote chemical reactions and the formation of new molecules, thus automatically exploring reaction pathways between arbitrary reactants and products. The ab initio NR was successfully able to predict graphene ring formation from pure acetylene as well as reaction pathways to form glycine, one of the building blocks of life, from small early earth molecules.
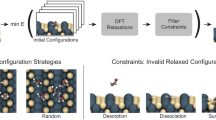
Although Wang et al. clearly demonstrated the promise of the ab initio NR to discover reactive chemistry, QM-driven MD sampling is extremely computationally intensive for generating a large training dataset within AL. In this Article, inspired by the work of Wang et al., we design an MLIP-driven NR sampling procedure that targets arbitrary reactive chemical processes and compositions of C, H, N and O elements, including near pure elemental systems and mixtures. Combined with the ANI model architecture and applying AL at scale, we aim to produce a robust and general reactive MLIP. Figure 1 shows a summary of the NR–AL workflow and the specific applications investigated in this work with the final model, referred to as ANI-1xnr.

The AL loop is an automated, iterative and efficient approach to develop a MLIP. AL generates a training dataset consisting of quantum calculations for only the high-uncertainty structures, as identified based on an ensemble of MLIPs. Structures relevant to condensed-phase reactive chemistry are sampled using NR simulations. The initial system is built by random configurations of small molecules consisting of the elements C, H, N, and O. Dynamic simulations are performed using the current MLIP with extreme fluctuations in temperature and volume to induce chemical reactions. To test the generality of the resulting model, the final MLIP is then applied to several case studies that were not directly targeted during training.
To evaluate the reliability of ANI-1xnr in practical research scenarios, we conduct several condensed-phase reactive chemistry simulations inspired by other literature with the ANI-1xnr potential, namely carbon solid-phase nucleation46,47,48, graphene ring formation from acetylene with varying O2 concentrations44,49, biodiesel ignition with different fuel additives50, methane combustion24 and the spontaneous formation of glycine from early earth molecules44,51,52. Across this wide range of applications, we show that ANI-1xnr provides results that are consistent with chemical intuition, experimental data, QM calculations (density-functional theory (DFT), Hartree–Fock and density-functional tight-binding (DFTB)) and classical reactive MD simulations (reactive force field (ReaxFF) and an application-specific MLIP) all without retraining.
This study demonstrates the capability of automated chemical exploration workflows to build a general-purpose reactive potential, resulting in ANI-1xnr, a fast, accurate and general potential capable of simulating a wide range of real-world reactive systems containing C, H, N and O elements.
Results
Nanoreactor active learning
Before assessing the performance of the ANI-1xnr model on the different case studies, we evaluated both the diversity and the completeness of the ANI-1xnr dataset. Figure 2 provides a two-dimensional visualization of a high-dimensional dataset by clustering together similar local atomic environments for the elements H (Fig. 2a), C (Fig. 2b), N (Fig. 2c) and O (Fig. 2d). Figure 2a–d compare the ANI-1xnr dataset and a non-reactive, near-equilibrium, molecule in vacuo, AL dataset (ANI-1x). Clearly, the ANI-1xnr dataset not only effectively encompasses the entire ANI-1x dataset but it also extends substantially beyond the local atomic environment space covered by ANI-1x. More importantly, the ANI-1xnr dataset provides pathways between many of the clusters in the ANI-1x dataset. These pathways probably correspond to reactions in a low-dimensional representation. Furthermore, Fig. 2e provides select examples of the over 1,000 unique molecules (consisting of ten or fewer CNO atoms; Methods) that are identified in the ANI-1xnr training dataset. Since the NR sampling simulations were initialized with only small molecules (consisting of two or fewer CNO atoms; Methods), the NR–AL procedure automatically discovered hundreds, if not thousands, of reaction pathways leading to these distinct molecular structures.

a–d, A comparison between the ANI-1xnr dataset (blue points) and a non-reactive, near-equilibrium, molecule in vacuo, AL dataset from the literature (ANI-1x; red points). Two-dimensional visualizations of the local atomic environments for the elements H (a), C (b), N (c) and O (d). The ANI-1xnr dataset not only encompasses the vast majority of the regions sampled in the ANI-1x dataset, but it also interpolates between these regions and even extends these regions substantially. For visual clarity and to manage memory loads, only a random subset of the ANI-1x dataset and ANI-1xnr dataset are depicted in a–d. e, Five examples of the over 1,000 unique molecules that formed during AL. Reaction pathways to form these molecules must, therefore, be present in the ANI-1xnr dataset.
Carbon solid-phase nucleation
Accurate simulation of amorphous carbon systems has long been one of the top interests among chemists and materials scientists, as some distinct materials (for example, graphene, diamond and carbon nanotubes) form from amorphous carbon under different conditions. Understanding this behaviour would assist in the development of functional materials by controlling the solid-phase nucleation process. Many reactive FFs have been employed to simulate amorphous carbon in MD48,53,54. With the widespread use of ML methods, researchers recently developed application-specific MLIPs to investigate amorphous carbon systems46,55. These application-specific MLIPs proved accurate at predicting pure carbon fragments and mechanical properties of the bulk system. Despite these achievements, MLIPs trained on application-specific datasets would have very poor generality to new chemistry as the model has only been fit to a limited number of structures and reactions. On the other hand, the NR–AL approach presented in this work does not sample any specific form of carbon explicitly. We rely on the NR sampler and AL algorithm to automatically select physically relevant and unbiased configurations of carbon atoms. To validate ANI-1xnr in carbon solid-phase nucleation simulations under different conditions, we perform simulations at high (3.52 g cc−1), medium (2.25 g cc−1) and low (0.50 g cc−1) densities.
Figure 3 summarizes the product of each simulation. For each of the high- (Fig. 3a), medium- (Fig. 3b) and low-density (Fig. 3c) carbon simulations, ANI-1xnr produces the expected structure of carbon for the respective density46,47,48. Specifically, for the system with the highest density (3.52 g cc−1), diamond, graphene and hexagonal diamond phase coexist after 246 ps, where 70% of carbon atoms in the simulation box forms diamond cubic crystal structure. After another 2.3 ns, the high-density system contains 86% of carbon atoms in the diamond cubic crystal structure, with very few graphene and hexagonal diamond sites. In the medium-density (2.25 g cc−1) system, 31% of atoms rapidly form graphene after 8.2 ps, and the system contains 83% graphene after another 2.3 ns. Graphene sheets tend to form a stacked and more ordered graphite-like structure, which is observed for the system slice in Fig. 3b). The low-density (0.5 g cc−1) system forms carbon atom chains after 250 ps, with 11% of atoms forming graphene sheets. After another 3 ns, the system contains 88% of atoms formed in graphene sheets. However, the graphene sheets in this low-density case are more disordered and appear to form fullerene-like closed or partially closed meshes. Extended Data Table 1 provides an analysis of the ANI-1xnr crystal lattice constants for diamond and graphite.

a–c, Specific densities at 3.52 g cc−1 (a), 2.25 g cc−1 (b) and 0.5 g cc−1 (c). Simulations are initiated with random carbon positions. The final structures agree with the expected phases of carbon for each density. Specifically, a produces diamond cubic crystal, b produces graphite-like graphene sheets and c produces fullerene-like graphene sheets.
Effect of oxygen on graphene ring formation
Wang et al.44 applied the original ab initio NR method to observe ring formation (that is, the early stages of graphene formation) from a pure acetylene (C2H2) system. Subsequently, Lei et al.49 presented DFTB NR simulations of acetylene in the presence of different amounts of oxygen, where O2/C2H2 = 0, 0.1, …, 1 is the ratio of added O2 while the number of C2H2 molecules is fixed to 40. Graphene formation is the dominant process for pure C2H2, as the generation of free radicals enables the rapid growth of hydrocarbon rings. By contrast, the addition of O2 to the system deters or, at high enough O2/C2H2 ratios, completely eliminates ring formation49. Similar to the work of Lei et al.49, we perform reactive simulations with varying ratios of C2H2 and O2.
Figure 4 shows the amount of three-, four-, five-, six- and seven-membered rings formed with respect to simulation time for eight different O2/C2H2 ratios. Increasing the oxygen ratio decreases the number of rings formed, which is in good agreement with the simulations from Lei et al. and experimental literature56. Furthermore, although the branching ratios (that is, the relative production of different ring sizes) are not completely converged for all systems, the branching ratios are clearly in qualitative agreement with Lei et al. Specifically, six-membered rings are the predominant product, followed by five-membered and seven-membered rings at noticeably lower, but nearly equal, branching ratios. However, in contrast with Lei et al., six-membered rings form even for an O2/C2H2 ratio of 0.5. In comparison, the simulations of Lei et al. predict ring formation for O2/C2H2 ratio up to 0.2, but negligible ring formation for an O2/C2H2 ratio of 0.4. The ANI-1xnr results are in much closer agreement with experimental data, which report graphene formation for O2/C2H2 ratios between 0.4 and 0.8 (no experimental measurements were reported outside of this range). A clear explanation for the improved agreement between ANI-1xnr and experiment is the longer simulation timescales and the larger system sizes achievable by ANI-1xnr compared with DFTB (Methods). Specifically, for an O2/C2H2 ratio of 0.5, six-membered rings only begin to form after 1 ns with ANI-1xnr. Considering that the DFTB simulations of Lei et al. ran for only 0.5 ns, our results suggest that six-membered rings could form under higher oxygen ratio conditions using DFTB at longer timescales. Although it is possible that even longer MD simulations could result in ring formation at even higher O2/C2H2 ratios, this case study demonstrate the value in the lower computational costs of ANI-1xnr compared with traditional methods, such as DFTB, to discover interesting phenomena that can only be observed during long timescale simulations. Further validation of the ANI-1xnr simulation results are provided in Extended Data Fig. 1.

a–h, A comparison of three-, four-, five-, six- and seven-membered ring formation for different ratios of O2/C2H2: 0.00 (a), 0.08 (b), 0.17 (c), 0.22 (d), 0.38 (e), 0.50 (f), 0.86 (g) and 1.33 (h). ANI-1xnr predicts six-membered ring formation for O2/C2H2 ratios less than 0.50, in closer agreement with experimental data than the DFTB simulation results of Lei et al.49 In comparison with these literature DFTB simulations, the computational efficiency of ANI-1xnr enables considerably longer simulation times and larger systems. Specifically, while Lei et al. performed simulations of 0.5 ns with between 160 and 270 atoms (depending on the O2/C2H2 ratio), we simulate 1,000 atoms for 10 ns (Methods).
Comparison of biofuel additives
To promote combustion processes of liquid fuel, fuel additives are utilized as detergents, oxygenates, emission depressors, corrosion inhibitors, dyes and to increase the octane number. Chen et al.50 performed high-temperature high-pressure MD simulations with ReaxFF40,42 to predict the mechanisms and kinetics of several fuel additives, including ethanol, 2-butanol and methyl tert-butyl ether (MTBE). According to their results, 2-butanol was the best fuel additive at enhancing ignition while MTBE demonstrated similar ignition enhancement to 2-butanol. By contrast, ethanol was the worst fuel additive, having a negligible effect on the O2 consumption rate and ignition delay time (IDT) compared with the clean biofuel.
To validate the reliability of ANI-1xnr for simulating biodiesel and to investigate the reported ignition enhancement of fuel additives, we reproduced four systems simulated by Chen et al.50, namely, clean biodiesel, biodiesel with ethanol as additive, biodiesel with 2-butanol as additive and biodiesel with MTBE as additive. Figure 5 shows that the main products (CO, CO2 and H2O) are produced in very similar quantities to the ReaxFF simulations of Chen et al. Despite a quantitative difference between ANI-1xnr and ReaxFF IDTs (Extended Data Table 2), the additive effect on ignition delay for ANI-1xnr agrees qualitatively with ReaxFF, namely, all three additives cause product formation to occur at earlier times compared with clean biodiesel. Furthermore, ANI-1xnr predicts that 2-butanol and MTBE both result in the enhancement of O2 consumption, similar to ReaxFF (Extended Data Table 2). The primary qualitative discrepancy with ReaxFF is that ANI-1xnr predicts that ethanol also enhances O2 consumption. However, experimental work demonstrates that ethanol can actually accelerate fuel ignition at relatively high pressures, in agreement with our simulation results57. Extended Data Fig. 3 provides further justification for the ANI-1xnr ethanol simulation results.

a–d, Tracking plots of O2 and major products (CO, CO2 and H2O) for the following biofuel simulations: biofuel+O2 (a), biofuel + O2 with ethanol additive (b), biofuel + O2 with 2-butanol additive (c) and biofuel + O2 with MTBE additive (d). IDT is defined as the average time that at least five molecules of CO, CO2 and H2O are produced (Supplementary Fig. 1). IDT is significantly decreased for each additive in comparison with the clean biofuel. For tracking plots including the entire 2 ns simulation, see Extended Data Fig. 2.
Methane combustion
Emerging research has shown the success of application-specific MLIPs on systems such as radical reactions in hydrocarbon combustion and well-known gas-phase mechanisms58,59. Zeng et al.24 trained an NN-based potential to a dataset of QM-calculated fragment clusters sampled from a ReaxFF simulation of the combustion process of a mixture of CH4 and O2. They showed that their application-specific MLIP could then simulate the combustion process of methane with a reasonable mechanism. Though our ANI-1xnr potential was trained for a more general purpose, we compare the performance of our MLIP with the application-specific MLIP of Zeng et al. for methane combustion under high temperatures and pressures. Specifically, we reproduce their MD simulation of methane combustion under the same conditions with the ANI-1xnr potential. Figure 6a shows that the ANI-1xnr potential produces very similar major products and species profiles to those of Zeng et al. However, by comparison with the CH4 and O2 consumption rates of Zeng et al., ANI-1xnr predicts an overall reaction rate that is approximately a factor of 40 times faster. Specifically, while their system required 0.5 ns of simulation time to consume half of the initial CH4, our system required only 0.012 ns. Similar to the biofuel case, the difference in the overall reaction rate is probably due to the difference in the reference DFT reaction energy barriers (Methods). Extended Data Figs. 4 and 5 provide further explanation as to the potential cause of this discrepancy.

a, A tracking plot of O2, CH4 and major products (CO, CO2 and H2O). The tracking plot for the full simulation is provided as Supplementary Fig. 3. b, Snapshots of initial reactants, intermediate species and final products. Both the tracking plot and the snapshots confirm that ANI-1xnr predicts qualitatively reasonable reactive chemistry for this system. However, ANI-1xnr is markedly more reactive than the application-specific MLIP in the literature24.
Due to the extreme simulation conditions, no experimental reference data are available for comparison. However, the similar trend for species concentration with respect to time in comparison with the work of Zeng et al. indicates that our general-purpose MLIP was able to learn the relevant physics and mechanisms as well as the application-specific MLIP of Zeng et al. Also, the CH4 and O2 consumption curves for the ANI-1xnr model are much closer to exponential decay, which is more physically reasonable than the near-linear decay plots of Zeng et al.
Miller experiment
In 1959, Stanley Miller designed a famous experiment to elucidate the origins of life on earth51. Miller applied an electric field to a gaseous system consisting of simple small-molecule species (for example, NH3, CO, H2O, H2 and CH4) and reported the formation of amino acids such as glycine (C2H5NO2). This revolutionary experiment led to the formation of the field of prebiotic chemistry, which aims to discover the reaction networks that produce molecules that are essential for the formation of life. In this spirit, computational studies have attempted to imitate the reaction conditions of the Miller experiment to predict the key reaction pathways that lead to the formation of glycine. Recently, Saitta and Saija performed relatively short (≈40 ps) near-ambient temperature (400 K) condensed-phase (≈1 g cc−1) DFT-MD simulations, wherein an electric field is applied directly to ‘spark’ chemical reactions52. As our MLIP does not contain the necessary electronic information to apply an electric field, we instead encourage reactions to occur on picosecond timescales by performing high-temperature high-density MD simulations, similar to the Miller NR simulation of Wang et al.44 Due to the low computational cost of our MLIP, we are able to run our Miller experiment simulation considerably longer (≈4 ns) than the ab initio NR simulations of Wang et al. (≈1 ns) with the same system size of 228 atoms but with periodic boundary conditions. For this reason, we use a constant condensed-phase density (with corresponding pressures around 1 GPa) rather than applying an artificial piston to periodically compress the non-periodic gas-phase system to around 10 GPa, as was the approach employed by Wang et al.
Figure 7 shows the ANI-1xnr reaction mechanism to form glycine starting from the initial reactants. During our Miller simulation, glycine is formed three times and persists for approximately 225 fs, 375 fs and 913 fs. Dissociation of glycine in less than 1 ps is expected, considering the relatively high temperature of this simulation. The final step to form glycine is hydrogen addition to C2H4NO2, similar to the mechanism of Saitta and Saija. However, hydrogen addition occurs at an oxygen atom in our mechanism, rather than at the α-carbon as in the Saitta and Saija mechanism. In one instance, our Miller simulation produced the same C2H4NO2 isomer as reported by Saitta and Saija. By contrast to the Saitta and Saija mechanism, this C2H4NO2 isomer dissociated in our simulation rather than forming glycine. The key precursor to C2H4NO2 is CH4N, which is formed through several pathways. The pathway to form CH4N that proceeds through the CH2O intermediate is very similar to the mechanism reported by Wang et al.44 The mechanisms to form the intermediates formaldehyde (CH2O) and hydrogen cyanide (CHN) from the initial reactants CO, NH3 and H2O were nearly identical to those reported by Wang et al.44 and Saitta and Saija52. Overall, there are several similarities between our mechanism and those of Wang et al. and Saitta and Saija.

The reaction pathways discovered by ANI-1xnr in a Miller experiment simulation for the formation of glycine from small-molecule species (for example, NH3, CO, H2O, H2 and CH4). The green arrows denote reactions previously identified by Wang et al. or Saitta and Saija. The orange arrows denote reactions that have a similar reaction in Wang et al. or Saitta and Saija. The majority of reactions have been previously reported in the literature, confirming the validity of the ANI-1xnr mechanism. Three-dimensional snapshots extracted from the MD simulation trajectory are reported in Extended Data Fig. 6, further confirming that the reaction pathways are physically meaningful. Note that +H does not necessarily signify a free hydrogen atom, +H is short-hand for a proton donor, for example, NH4, NH3, CHO, CHNO, H3O or H2O. Likewise, −H does not necessarily signify dissociation of a hydrogen atom. −H is short-hand for a proton acceptor, for example, NH2, CO, CNO, H2O or OH. The boxes encapsulate the key intermediates, carbon dioxide (CO2) and methylene (CH2). The novel pathways to form these key intermediates are reported in Extended Data Fig. 7. The depiction of bond orders and radical species is based simply on chemical intuition, since ANI-1xnr does not provide explicit bonding, orbital or electronic information (for an alternative interpretation of this mechanism involving ionic species, see Extended Data Fig. 8).
Conclusions
Here, we introduced a sampling procedure, dataset and MLIP (ANI-1nxr) based on the NR for organic condensed-phase MDs, including reactions. The NR-based AL process builds a reactive dataset spanning elemental compositions of C, H, N and O under a wide range of conditions starting from nine small seed molecules. The NR–AL procedure provided data with unprecedented chemical environment diversity and relevance compared with prior non-reactive AL, and uncovers more than 1,000 unique molecules in total, under condensed-phase reactive atomistic configurations. Each unique molecular species formed by MDs simulation in our NR sampler was the result of one or more reaction pathways that did not need to be known or specified before runtime.
We validated the generality of the ANI-1xnr potential on five real-world condensed-phase reactive case studies: carbon solid-phase nucleation, effect of oxygen on graphene ring formation from acetylene, ignition of biodiesel with various fuel additives, combustion of methane and the spontaneous formation of glycine in early earth conditions, all without retraining. In carbon solid-phase nucleation and graphene ring formation studies, we show that ANI-1xnr reproduces the experiment well. In other cases, in extreme simulation conditions where an experiment is not available for comparison, ANI-1xnr produces results that are generally consistent with traditional modelling approaches, such as DFT, DFTB, ReaxFF and even an application-specific MLIP. The effectiveness of the NR–AL approach demonstrates the power of coupling and automating the system exploration, data generation and model training processes to produce a robust MLIP.
Although the ANI-1xnr potential is already a broadly applicable tool for studying condensed-phase reactive chemistry, we envision continuous improvement of this MLIP. Future work could augment the condensed-phase ANI-1xnr dataset with low-density or in vacuo reactive data, for example, by sampling pathways for pre-determined reactions37,43 or for reactions identified in the NR simulations44. Future work could also extend the dataset to additional elements18. As the current dataset was computed using an affordable plane-wave DFT method, future work could also investigate the prospect for higher-accuracy QM methods (for example, double-hybrid DFT or post-Hartree–Fock) to obtain improved reaction barriers. In addition to simple retraining, any of these improvements could use more advanced ML training paradigms, such as transfer learning60, meta-learning61 and lifelong learning62. Concerning the model form, the ANI-1xnr potential is fully local, meaning long-range effects, such as London dispersion and Coulombic interactions, are not described explicitly beyond the model cutoff radius (Methods). Certain applications may require more direct treatment of long-range effects. Future work could investigate incorporating recent developments, such as explicit long-range terms63, charge equilibration schemes64 or graph NN models5,6,13,14,15,16 that can implicitly account for long-range interactions. A recent advancement in ML for natural language processing is the concept of foundational models, that is, large, general models usually trained with unlabelled data that can be specialized to specific tasks quickly with very small amounts of data65. As ANI-1xnr is trained to a large, general dataset, a clear future direction is to evaluate whether it can act as a foundational model for application-specific MLIPs when greater accuracy is required.
We are providing the ANI-1xnr dataset for future research. We are also providing the resulting ANI-1xnr potential to the community. We advise potential users to exercise strong caution if applying ANI-1xnr outside of the training domain (CHNO condensed-phase reactive chemistry). Nonetheless, considering that ANI-1xnr was developed independently of the five case study systems, the generality of ANI-1xnr is truly remarkable.
Methods
Model description and training details
The ANI-1xnr model was trained similarly to ANI models within other contexts18, including materials science32 and chemistry66. We use the ANI descriptors67, which is a modified form of the Behler and Parinello NN descriptors12. ANI-1xnr uses a local cutoff of 5.2 Å for the radial descriptors and 3.5 Å for the angular descriptors. The model is trained for the elements C, H, N and O, each of which has its own specialized NN-based potential. The NN architecture for each element and symmetry functions are reported as Supplementary Tables 2 and 3, respectively. Similar to previous ANI models, ANI-1xnr predicts energies based solely upon the atomic positions and element types. Therefore, unlike more complex MLIPs, for example, SpookyNet16 and AIMNet68, the ANI-1xnr-predicted energies do not explicitly depend on the charge or spin multiplicity. Thus, similar to ReaxFF, ANI-1xnr predicts only the ground-state energy, regardless of whether the lowest-energy electronic state corresponds to a radical or an ion.
Training
The ANI-1xnr model was trained using both energy and force terms in the loss function as described in previous work69. During training, we employ early stopping to prevent overfitting with learning rate annealing to ensure a high-fidelity fit. The model training is considered converged when the learning rate drops below 1.0 × 10−5. Model performance against the held-out test dataset is presented in Supplementary Tables 4 and 5 and Supplementary Figs. 5–8. Note that, similar to previous MLIP studies27,28, we report the per-atom energy errors. This is because energy is an extensive property and the ANI-1xnr dataset consists of systems spanning almost two orders of magnitude in the number of atoms. Therefore, the unnormalized energy spans an enormous range and, thus, the corresponding unnormalized energy error is skewed by larger systems.
Although the ANI-1xnr root mean squared errors are approximately an order of magnitude larger than an MLIP trained to near-equilibrium data (for example, ANI-1x). This is not surprising since the ANI-1xnr dataset is considerably more challenging to train due to the wider range of atomic environments and system energies present in a reactive dataset. While errors between MLIPs trained on different datasets are not precisely comparable, we note that the ANI-1xnr mean absolute errors are of similar magnitude to those for TeaNet27, which was also trained on a very challenging dataset aimed at developing a universal potential. Furthermore, the ANI-1xnr dataset is much more general than most reactive datasets that are limited to a single system of interest, for example, the Zeng et al. reactive dataset for CH4 + O2. By comparison, the force root mean squared errors of ANI-1xnr are only about 30% higher than those of the MLIP trained to the Zeng et al. single-reactive-system dataset, despite the ANI-1xnr dataset covering a substantially wider range of reactive chemistry. A validation that ANI-1xnr conserves energy is shown in Supplementary Fig. 9.
Training dataset generation
The ANI-1xnr training dataset was generated through an iterative AL process, where sampling of new atomic configurations is obtained with NR-inspired MD simulations. To bootstrap the AL process, periodic cells containing randomly placed and oriented small molecules with less than three non-H atoms and with a randomly selected composition of C, H, N and O are generated. Starting from this small initial bootstrap dataset, the AL algorithm is applied iteratively, yielding generations of datasets designed to improve upon their ancestors. Iterations of sampling, selecting, labelling and training are performed until the resulting MLIP is no longer improving. Training was described in the previous section. Details regarding sampling, selecting and labelling are provided in the following paragraphs.
Sampling
Atomic structures (that is, positions) are sampled by performing NR simulations with the current AL-generation MLIP. The MLIP-driven NR simulations are initialized with random compositions of small molecules, containing in the order of 100 atoms. Random oscillations of temperature and density (that is, simulation box volume) promote reactions and the formation of new products during the allotted simulation time (less than 1 ns).
Selecting
From all the atomic structures sampled along the NR trajectories, only high-uncertainty structures are selected for the ever-growing dataset, as these structures are deemed to be poorly described by the current AL-generation MLIP. Similar to previous ANI studies, we utilize a query-by-committee70 uncertainty metric, that is, the normalized ensemble standard deviation in energy and atomic forces32,60. To achieve a balance between exploration of chemical space and exploitation of the most important regions of the potential energy surface, the uncertainty thresholds vary between AL iterations, where the latter AL iterations generally have larger thresholds than earlier iterations. The final uncertainty threshold values for the normalized energy and forces were 1.85 \({{{\rm{kcal}}}}\; {{{{\rm{mol}}}}}^{-1}\; {{\mathrm{N}}}^{-\frac{1}{2}}\) and 6.92 kcal mol−1 Å−1, respectively.
Labelling
Each selected structure is then labelled with QM system energy and atomic forces. These QM calculations are computed with the open-source CP2K software71 using unrestricted Kohn–Sham DFT72, Becke, Lee, Yang and Parr (BLYP) functional73,74, triple-zeta valence basis set with two sets of polarisation functions (TZV2P)75, Goedecker, Teter and Hutter (GTH) pseudopotentials76, Grimme third-generation dispersion (D3) correction with zero damping77 and energy cutoffs of 600 and 60 Ry, respectively, for the plane-wave and Gaussian contributions to the basis set, as recommended in previous work78. Ensuring that each DFT calculation converges to the global-minimum energy is challenging for complex condensed-phase systems with a large number of molecules and partially broken bonds. Indeed, it is likely that the few large outliers observed in Supplementary Fig. 5 are DFT calculations that converged to a meta-stable electronic state. Fortunately, the fraction of these outliers is relatively low. Thus, these presumed meta-stable calculations do not meaningfully impede the MLIP from learning the dominant branch of convergence for the DFT calculations.
The overall spin multiplicity for each DFT calculation is constrained to a singlet state. Note that this is the spin for the entire box, not just a single molecule or radical. The assumption that a condensed-phase system does not accumulate an impactful amount of spin is effectively an infinite-system size approximation. This choice of spin multiplicity is consistent with previous studies that perform CP2K simulations of bulk systems containing radical species79. However, a singlet spin multiplicity for low-density gas phase or in vacuo calculations would not always be appropriate (for example, for radicals or molecules with partially formed bonds). The use of a singlet spin multiplicity may explain, in part, why ANI-1xnr performs poorly for in vacuo bond-breaking calculations (Extended Data Fig. 4).
Below is a detailed step-by-step description of the AL workflow (for a high-level overview, see Fig. 1):
-
1.
Generate a bootstrap dataset (labelled with energies and forces) of 100 randomly generated periodic cells containing randomly placed and oriented small molecules including C2, H2, N2, O2, NH3, CH4, CO2, H2O and C2H2 with random composition
-
2.
Train ensemble of ANI potentials to the current training dataset using eightfold (16 blocks) cross validation (14/1/1: train/validation/test split) scheme
-
3.
Prepare for NR–AL sampling: build a new random box of small molecules with random size, density, placements orientations. Define a random schedule function for oscillating temperature (T) and density (ρ). Oscillating functional form is the same for temperature and density (see equations below), where t is time, \({t}_{\max }\) is a hyperparameter for the max time the simulation will run, and Tstart, Tend, Tamp, ρstart, ρend, ρamp and tper are randomly selected values within a pre-determined range (Supplementary Table 6):
$$\begin{array}{rcl}T(t)&=&{T}_{{{{\rm{start}}}}}+\frac{t}{{t}_{\max }}({T}_{{{{\rm{end}}}}}-{T}_{{{{\rm{start}}}}})+{T}_{{{{\rm{amp}}}}}{\sin }^{2}\left(\frac{t}{{t}_{{{{\rm{per}}}}}}\right)\\ \rho (t)&=&{\rho }_{{{{\rm{start}}}}}+\frac{t}{{t}_{\max }}({\rho }_{{{{\rm{end}}}}}-{\rho }_{{{{\rm{start}}}}})+{\rho }_{{{{\rm{amp}}}}}{\sin }^{2}\left(\frac{t}{{t}_{{{{\rm{per}}}}}}\right)\end{array}$$ -
4.
Run the NR MD simulation using forces from current AL-generation MLIP
-
5.
Monitor energy and force uncertainty metrics every 5–50 MD steps (with an MD time step of 0.5 fs). If the uncertainty values exceed a pre-selected threshold value, end the simulation and add configuration to a new batch of structures. Otherwise, continue running MD for a maximum of 1 ns
-
6.
Run DFT single-point calculations on the new batch of structures to obtain energy and force labels
-
7.
Add new labelled data to the training dataset
-
8.
Go back to step 2 and repeat until the MLIP converges. We define convergence as when MLIP-driven MD sampling simulations run for on the order of 50 ps on average. In other words, convergence is achieved when the MLIP is confident in all new MD simulations
Resulting training dataset
After more than 50 iterations of AL, the resulting training dataset includes 26,650 simulation cells of atomic positions with corresponding DFT system energy and atomic forces. Two-dimensional visualizations of the local atomic environments present in the dataset (Fig. 2a–d) are generated using t-distributed stochastic neighbour embeddings80. Distributions of the system sizes, compositions and densities can be found in Supplementary Figs. 10–12, respectively. The average system size is 139 atoms. The vast majority (≈95%) of configurations in the training dataset have a density between 0.5 and 2.0 g cc−1. While the minimum density in the dataset is ≈0.03 g cc−1, less than 1% of the configurations in the dataset have a density less than 0.1 g cc−1, suggesting that ANI-1xnr should not be trusted for low-density gas-phase simulations or in vacuo calculations.
By cross-referencing the ANI-1xnr training dataset with the existing PubChem database81 for only CHNO molecules with ten or fewer CNO atoms, we conclude that the ANI-1xnr dataset contains 1,212 unique known PubChem molecules, or approximately 0.2% of the ≈555k PubChem CHNO molecules with ten or fewer CNO atoms. Supplementary Fig. 13 shows a histogram of the sizes of all molecules that are found in the ANI-1xnr dataset, which includes one molecule up to 145 atoms. The majority are small molecules of similar size or slightly larger than those from which the systems were initialized. There are many occurrences of molecules in the range of 10–90 atoms. The largest structures, ascertained by visual inspection, are graphene sheets. Furthermore, the 1,212 unique PubChem molecules only represent the simulation frames that were selected by the uncertainty estimate. Therefore, 1,212 should be considered a lower bound of molecules discovered during AL. There are probably many more molecules formed over all NR–AL sampling, which is estimated to be hundreds of nanoseconds of MD simulation time in aggregate.
To automate the extraction of common molecular entities that formed during the AL process, we developed a NetworkX-based package called MolFind. This Python software tool employs user prescribed cutoff distances for defining when two atoms are bonded or not and discovers clusters of atoms connected via bonds. The three-dimensional molecular architecture is partially captured through a graphical representation (that is, nodes and edges) of the bonding topology where atoms are nodes and bonds are edges. Graphs are encoded according to the open-source Python package called NetworkX82. The graphical representation and the NetworkX package enables (1) the counting of the number of topologically distinct molecular species in a simulation via a graph isomorphism check and (2) a comparison to known molecular entities with a specified topology. Previously, we tabulated a large database of known molecules and associated topologies by scraping the entirety of the PubChem database up to ten non-hydrogen atoms. The existence of a species in the database is not required for MolFind to extract a bonded atomic cluster but if found, it can affix a chemical/species name with the entity.
Simulation details
All MD simulations in this study are performed with the NeuroChem package67 and the Atomic Simulation Environment83. The average computational speed of our Atomic Simulation Environment–NeuroChem MD simulations was approximately 50k atomic gradients per second on a single NVIDIA Titan V graphics processing unit (GPU). We acknowledge that a more optimized code, such as Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS)84, would be noticeably more computationally efficient. For example, recent studies have demonstrated that highly optimized MLIPs within LAMMPS can obtain 1–10 million atomic gradients per second on a single NVIDIA A100 GPU15,85. However, because of the relatively small system sizes of our AL simulations (less than 500 atoms) and our case study simulations (no greater than 5,000 atoms), it was not necessary to fully optimize our computational performance by utilizing a code such as LAMMPS. To demonstrate that there is an opportunity to greatly improve our performance, we achieved 90k atomic gradients per second simply by increasing our simulation size to 25k atoms and, thereby, more efficiently utilizing the GPU.
Carbon solid-phase nucleation
To investigate the formation process of diamond and graphene, MD simulations were performed for amorphous carbon under different densities. Three initial system structures with three different densities (0.5 g cc−1, 2.25 g cc−1 and 3.52 g cc−1) were generated by varying the simulation box length for a constant total number of carbon atoms of 5,000. The initial system structure was built with in-house code. First, the initial position for the first carbon atom in the simulation box was randomly selected. Then, random positions were proposed for each additional carbon atom. A proposed position was accepted only if the distance to all previous positions was larger than twice the van der Waals radius for carbon atoms (1.7 Å). This process was repeated until all 5,000 carbon atoms were inserted. Langevin dynamics were performed at a temperature of 2,500 K for 5 ns with step length of 0.5 fs. Coordinates and properties were recorded every 50 fs (100 time steps). Eight independent trajectories were run for each density to verify that the correct phase was identified from different starting structures. Different phases (diamond cubic, hexagonal diamond or graphene) in each snapshot were distinguished with the Open Visualization Tool86.
Effect of oxygen on graphene ring formation
To investigate ring formation from acetylene, MD simulations were performed for eight different systems with varying O2/C2H2 ratios: (0.00, 0.08, 0.17, 0.22, 0.38, 0.50, 0.86 and 1.33). All systems contained 1,000 atoms, resulting in a range of 150–250 C2H2 molecules and 0–200 O2 molecules, depending on the O2/C2H2 ratio. To have a nearly identical density of 0.2 g cc−1 for each system, the box lengths ranged between 37 Å and 44 Å. The initial structures were generated with PackMol87. Next, the minimum-energy structure was obtained with the Limited-memory Broyden–Fletcher–Goldfarb–Shanno optimizer88. Then, Langevin dynamics simulations were run at 2,000 K for 10 ns with a 0.5 fs time step and a friction constant of 0.01. Snapshots and properties were recorded every 0.5 ps (1,000 time steps).
Ring structures of varying sizes were identified and counted with our in-house code MolFind. Considering that the distance between bonded atoms can fluctuate, a 0.02 Å buffer was utilized when scanning C–C bonds so that any pair of carbon atoms that has distance smaller than 1.72 Å (two times the covalent radius of carbon atom plus the buffer) were considered bonded. Similar buffers were also added when analysing other simulations.
Comparison of biofuel additives
To investigate the effect of different fuel additives on ignition performance, MD simulations were performed for clean biofuel and biofuel with three different additives: ethanol, 2-butanol and MTBE. The biofuel composition, the number of additive molecules and the number of O2 molecules were the same as presented in Table 2 of the ReaxFF reference paper50. Each system consists of approximately 2,000 atoms. Initial structures were generated using Packmol such that the initial separation of all molecules was at least 2 Å. The initial density was 0.2 g cc−1 in all four cases, consistent with Chen et al. Langevin dynamics were run at a temperature of 100 K for 1 ps for relaxation. Then, the system temperature was gradually increased to 3,000 K at a 50 K ps−1 heating rate. After reaching the desired temperature of 3,000 K, the simulation was ran for an additional 10 ns. A fixed time step of 0.1 fs was utilized. The temperature, time step and heating profile were the same as those utilized by Chen et al.50 During the whole process (including relaxation and temperature ramping) snapshots and properties were recorded every 1 ps (10,000 time steps). Five independent trajectories were performed for each system to reduce uncertainty in species profiles.
ANI-1xnr was trained to BLYP reference calculations, whereas ReaxFF was primarily developed based on B3LYP calculations (supplemented with high-accuracy bond dissociation energy data). Since reaction rates are extremely sensitive to energy barriers, this difference in the DFT functional can lead to a substantial difference in overall reaction rates.
Methane combustion
The methane combustion system was initialized with 100 CH4 molecules and 200 O2 molecules. All molecules were inserted using Packmol and ensuring that all molecules were separated by at least 2.0 Å. The cubic simulation box length was 37.60 Å, resulting in a density of 0.25 g cc−1. The temperature was initialized to 3,000 K by Maxwell–Boltzmann distribution. Langevin dynamics were run for 1 ns with a time step of 0.1 fs and with a friction constant of 0.01. The initial density, number of molecules, temperature and time step were consistent with Zeng et al.24. Snapshots and properties were recorded every 0.1 ps (1,000 time steps).
ANI-1xnr was trained to reference calculations computed with BLYP functional and TZV2P basis set, whereas Zeng et al. utilized the MN15 functional and 6-31G** basis set24. Since reaction rates are extremely sensitive to energy barriers, this difference in the DFT functional and basis set can lead to a substantial difference in overall reaction rates.
Miller experiment
To investigate the ability to simulate complex organic system that involve biologically relevant molecules, an MD simulation was performed with a similar species composition to the Miller experiment. Packmol was utilized to randomly place 16 H2, 14 H2O, 14 CO, 14 NH3 and 14 CH4 in a cubic simulation box with edge lengths of 12.1 Å, resulting in a density of 1.067 g cc−1. The simulation was run with Langevin dynamics for over 4 ns with a time step of 0.25 fs. The temperature was linearly increased from 0 K to 300 K in the first 100 ps. Then, the temperature was linearly increased from 300 K to 2,500 K in the next 100 ps. The temperature was then maintained at 2,500 K for 4,000 ps. The system was then cooled from 2,500 K to 300 K over the final 200 ps. Snapshots and properties were recorded every 12.5 fs (50 time steps).
Although some differences exist between our mechanism and those reported in previous simulation studies, this is not surprising considering not only the difference in levels of theory (that is, Hartree–Fock versus DFT versus MLIP), but also the difference in the simulation methodologies (that is, our Miller simulation did not utilize a ‘piston’ nor induce an electric field). For this reason, we further validate our Miller experiment results by comparing the ANI-1xnr energies and forces directly with DFT calculations along the MD trajectory. These validation results are provided in Supplementary Fig. 4.
Data availability
The ANI-1xnr training dataset is publicly available at https://doi.org/10.6084/m9.figshare.22814579. Initial and final structures for case study simulations are provided as Supplementary Data files. Details are provided in the corresponding section in Methods. Source data are provided with this paper.
Code availability
The ANI-1xnr model can be found at https://github.com/atomistic-ml/ani-1xnr/.
References
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Behler, J. First principles neural network potentials for reactive simulations of large molecular and condensed systems. Angew. Chem. Int. Ed. 56, 12828–12840 (2017).
Kulichenko, M. et al. The rise of neural networks for materials and chemical dynamics. J. Phys. Chem. Lett. 12, 6227–6243 (2021).
Bartók, A. P. & Csányi, G. Gaussian approximation potentials: a brief tutorial introduction. Int. J. Quantum Chem. 115, 1051–1057 (2015).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Thölke, P. & Fabritiis, G. D. Equivariant transformers for neural network based molecular potentials. In International Conference on Learning Representations https://openreview.net/forum?id=zNHzqZ9wrRB (2022).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Comm. 14, 579 (2023).
Khorshidi, A. & Peterson, A. A. Amp: a modular approach to machine learning in atomistic simulations. Comput. Phys. Commun. 207, 310–324 (2016).
Yao, K., Herr, J. E., Toth, D. W., Mckintyre, R. & Parkhill, J. The TensorMol-0.1 model chemistry: a neural network augmented with long-range physics. Chem. Sci.9, 2261–2269 (2018).
Singraber, A., Morawietz, T., Behler, J. & Dellago, C. Parallel multistream training of high-dimensional neural network potentials. J. Chem. Theory Comput. 15, 3075–3092 (2019).
Kang, P.-L. & Liu, Z.-P. Reaction prediction via atomistic simulation: from quantum mechanics to machine learning. iScience 24, 102013 (2021).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Haghighatlari, M. et al. NewtonNet: a Newtonian message passing network for deep learning of interatomic potentials and forces. Digital Discov. 1, 333–343 (2022).
Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csanyi, G. in Advances in Neural Information Processing Systems (eds Koyejo, S. et al.) 35, 11423–11436 (Curran Associates, 2022).
Chigaev, M. et al. Lightweight and effective tensor sensitivity for atomistic neural networks. J. Chem. Phys. 158, 184108 (2023).
Unke, O. T. et al. SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 12, 7273 (2021).
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018).
Devereux, C. et al. Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens. J. Chem. Theory Comput. 16, 4192–4202 (2020).
Young, T. A., Johnston-Wood, T., Zhang, H. & Duarte, F. Reaction dynamics of Diels–Alder reactions from machine learned potentials. Phys. Chem. Chem. Phys. 24, 20820–20827 (2022).
Jiang, B., Li, J. & Guo, H. High-fidelity potential energy surfaces for gas-phase and gas-surface scattering processes from machine learning. J. Phys. Chem. Lett. 11, 5120–5131 (2020).
Kolb, B., Zhao, B., Li, J., Jiang, B. & Guo, H. Permutation invariant potential energy surfaces for polyatomic reactions using atomistic neural networks. J. Chem. Phys. 144, 224103 (2016).
Cooper, A. M., Hallmen, P. P. & Kästner, J. Potential energy surface interpolation with neural networks for instanton rate calculations. J. Chem. Phys. 148, 094106 (2018).
Li, J., Song, K. & Behler, J. A critical comparison of neural network potentials for molecular reaction dynamics with exact permutation symmetry. Phys. Chem. Chem. Phys. 21, 9672–9682 (2019).
Zeng, J., Cao, L., Xu, M., Zhu, T. & Zhang, J. Z. Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation. Nat. Commun. 11, 5713 (2020).
Chen, R., Shao, K., Fu, B. & Zhang, D. H. Fitting potential energy surfaces with fundamental invariant neural network. II. Generating fundamental invariants for molecular systems with up to ten atoms. J. Chem. Phys. 152, 204307 (2020).
Takamoto, S. et al. Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements. Nat. Commun. 13, 2991 (2022).
Takamoto, S., Izumi, S. & Li, J. TeaNet: universal neural network interatomic potential inspired by iterative electronic relaxations. Comput. Mater. Sci. 207, 111280 (2022).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Behler, J. Constructing high-dimensional neural network potentials: a tutorial review. Int. J. Quantum Chem. 115, 1032–1050 (2015).
Ren, P. et al. A survey of deep active learning. ACM Comput. Surv. 54, 1–40 (2021).
Sivaraman, G. et al. Machine-learned interatomic potentials by active learning: amorphous and liquid hafnium dioxide. NPJ Comput. Mater. 6, 104 (2020).
Smith, J. S. et al. Automated discovery of a robust interatomic potential for aluminum. Nat. Commun. 12, 1257 (2021).
Yoo, P. et al. Neural network reactive force field for C, H, N, and O systems. NPJ Comput. Mater. 7, 9 (2021).
Zaverkin, V., Holzmüller, D., Steinwart, I. & Kästner, J. Exploring chemical and conformational spaces by batch mode deep active learning. Digit. Discov. 1, 605–620 (2022).
Young, T. A., Johnston-Wood, T., Deringer, V. L. & Duarte, F. A transferable active-learning strategy for reactive molecular force fields. Chem. Sci. 12, 10944–10955 (2021).
Ang, S. J., Wang, W., Schwalbe-Koda, D., Axelrod, S. & Gómez-Bombarelli, R. Active learning accelerates ab initio molecular dynamics on reactive energy surfaces. Chem 7, 738–751 (2021).
Guan, X. et al. A benchmark dataset for hydrogen combustion. Sci. Data 9, 215 (2022).
Warshel, A. & Weiss, R. M. An empirical valence bond approach for comparing reactions in solutions and in enzymes. J. Am. Chem. Soc. 102, 6218–6226 (1980).
Baskes, M. Determination of modified embedded atom method parameters for nickel. Mater. Chem. Phys. 50, 152–158 (1997).
van Duin, A. C. T., Dasgupta, S., Lorant, F. & Goddard, W. A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 105, 9396–9409 (2001).
Brenner, D. W. et al. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 14, 783–802 (2002).
Senftle, T. P. et al. The ReaxFF reactive force-field: development, applications and future directions. NPJ Comput. Mater. 2, 15011 (2016).
Schreiner, M., Bhowmik, A., Vegge, T., Busk, J. & Winther, O. Transition1x—a dataset for building generalizable reactive machine learning potentials. Sci. Data 9, 779 (2022).
Wang, L.-P. et al. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 6, 1044–1048 (2014).
Wang, L.-P. in Computational Approaches for Chemistry Under Extreme Conditions 127–159 (Springer, 2019).
Deringer, V. L. & Csányi, G. Machine learning based interatomic potential for amorphous carbon. Phys. Rev. B 95, 094203 (2017).
Powles, R. C., Marks, N. A. & Lau, D. W. M. Self-assembly of sp2-bonded carbon nanostructures from amorphous precursors. Phys. Rev. B 79, 075430 (2009).
Tomas, C. D., Suarez-Martinez, I. & Marks, N. A. Graphitization of amorphous carbons: a comparative study of interatomic potentials. Carbon 109, 681–693 (2016).
Lei, T. et al. Mechanism of graphene formation via detonation synthesis: a DFTB nanoreactor approach. J. Chem. Theory Comput. 15, 3654–3665 (2019).
Chen, Z., Sun, W. & Zhao, L. Combustion mechanisms and kinetics of fuel additives: a ReaxFF molecular simulation. Energy Fuels 32, 11852–11863 (2018).
Miller, S. L. & Urey, H. C. Organic compound synthesis on the primitive earth. Science 130, 245–251 (1959).
Saitta, A. M. & Saija, F. Miller experiments in atomistic computer simulations. Proc. Natl Acad. Sci. USA 111, 13768–13773 (2014).
Los, J. H., Ghiringhelli, L. M., Meijer, E. J. & Fasolino, A. Improved long-range reactive bond-order potential for carbon I construction. Phys. Rev. B 72, 214102 (2005).
Srinivasan, S. G., Van Duin, A. C. & Ganesh, P. Development of a ReaxFF potential for carbon condensed phases and its application to the thermal fragmentation of a large fullerene. J. Phys. Chem. A 119, 571–580 (2015).
Wang, J. et al. A deep learning interatomic potential developed for atomistic simulation of carbon materials. Carbon 186, 1–8 (2022).
Sorensen, C., Nepal, A. & Singh, G. P. Process for high-yield production of graphene via detonation of carbon-containing material. US patent 9,440,857 (2016).
Cooper, S. P., Mathieu, O., Schoegl, I. & Petersen, E. L. High-pressure ignition delay time measurements of a four-component gasoline surrogate and its high-level blends with ethanol and methyl acetate. Fuel 275, 118016 (2020).
Brickel, S., Das, A. K., Unke, O. T., Turan, H. T. & Meuwly, M. Reactive molecular dynamics for the [Cl–CH3–Br]− reaction in the gas phase and in solution: a comparative study using empirical and neural network force fields. Electron. Struct. 1, 024002 (2019).
Li, J., Chen, J., Zhang, D. H. & Guo, H. Quantum and quasi-classical dynamics of the OH + CO → H + CO2 reaction on a new permutationally invariant neural network potential energy surface. J. Chem. Phys. 140, 044327 (2014).
Smith, J. S. et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 10, 2903 (2019).
Allen, A. E. A. et al. Learning together: towards foundational models for machine learning interatomic potentials with meta-learning. Preprint at arXiv https://doi.org/10.48550/arXiv.2307.04012 (2023).
Eckhoff, M. & Reiher, M. Lifelong machine learning potentials. J. Chem. Theory Comput. 19, 3509–3525 (2023).
Rezajooei, N., Thien Phuc, T. N., Johnson, E. & Rowley, C. A neural network potential with rigorous treatment of long-range dispersion. Digit. Discov. 2, 718–727 (2023).
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 12, 398 (2021).
Bommasani, R. et al. On the opportunities and risks of foundation models. Preprint at arXiv https://doi.org/10.48550/arXiv.2108.07258 (2022).
Smith, J. S. et al. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 7, 134 (2020).
Smith, J. S., Isayev, O. & Roitberg, A. E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. J. 8, 3192–3203 (2017).
Zubatyuk, R., Smith, J. S., Leszczynski, J. & Isayev, O. Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network. Sci. Adv. 5, eaav6490 (2019).
Smith, J. S., Lubbers, N., Thompson, A. P. & Barros, K. Simple and efficient algorithms for training machine learning potentials to force data. Preprint at arXiv https://doi.org/10.48550/arXiv.2006.05475 (2020).
Seung, H. S., Opper, M. & Sompolinsky, H. Query by Committee. In Proc. Association for Computing Machinery (1992). https://doi.org/10.1145/130385.130417
Kühne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package—Quickstep: efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
VandeVondele, J. & Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (2007).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54, 1703–1710 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Jadrich, R. B., Ticknor, C. & Leiding, J. A. First principles reactive simulation for equation of state prediction. J. Chem. Phys. 154, 244307 (2021).
Fetisov, E. O. et al. First-principles Monte Carlo simulations of reaction equilibria in compressed vapors. ACS Cent. Sci. 2, 409–415 (2016).
van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380 (2022).
Hagberg, A. A., Schult, D. A. & Swart, P. J. Exploring network structure, dynamics, and function using NetworkX. In Proc. 7th Python in Science Conference (eds Varoquaux, G., Vaught, T. & Millman, J.) 11–15 (2008).
Larsen, A. H. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 271, 108171 (2022).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 14, 579 (2023).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model. Simul. Mat. Sci. Eng. 18, 015012 (2009).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Liu, D. C. & Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 45, 503–528 (1989).
Acknowledgements
The authors thank A. E. Roitberg for useful discussions on validating MLIPs for reactive chemistry. S.Z., K.B., B.T.N., S.T., N.L. and R.A.M. acknowledge support from the US Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under Triad National Security, LLC (‘Triad’) contract grant 89233218CNA000001 (FWP: LANLE3F2). M.Z.M. gratefully acknowledges the resources of the Los Alamos National Laboratory (LANL) Applied Machine Learning summer student programme. The work at LANL was supported by the LANL Directed Research and Development Funds 20210087DR. Work at LANL was performed in part at the Center for Nonlinear Studies and the Center for Integrated Nanotechnologies, a US Department of Energy Office of Science user facility at LANL. This research used resources provided by the LANL Institutional Computing Program. O.I. acknowledges support from Office of Naval Research through Energetic Materials Program (MURI grant number N00014-21-1-2476). M.Z.M. and E.K. acknowledge funding from National Science Foundation, grant CHE 2102461.
Author information
Authors and Affiliations
Contributions
J.S.S. and N.L. conceptualized the approach. R.B.J., J.S.S, S.T. and B.T.N. designed the QM methods. The NR sampler was designed by J.S.S. and N.L. The validation cases were designed by J.S.S., M.Z.M., S.Z. and R.A.M. Software was written by J.S.S., N.L., M.Z.M., R.B.J. and S.Z. J.S.S. performed the AL procedure. The validation case studies were performed by the following authors: carbon solid-phase nucleation by J.S.S.; effect of oxygen on graphene ring formation by J.S.S., S.Z., M.Z.M. and N.L.; comparison of biofuel additives by S.Z., M.Z.M. and R.A.M.; methane combustion by S.Z. and R.A.M.; Miller experiment by J.S.S., M.Z.M., R.A.M. and N.L. with reaction network analysis by M.Z.M. and R.A.M. Figure 2 was designed by S.Z., J.S.S., N.L., R.A.M. and K.B. Figure 2 was generated by M.Z.M. and J.S.S. Figure 7 was generated by R.A.M. Other figures were rendered by S.Z., J.S.S., M.Z.M., N.L. and R.A.M. Supervision was provided by E.K., O.I., S.T., K.B., R.A.M., J.S.S. and N.L. Funding acquisition was provided by B.T.N., J.S.S., S.T., K.B., O.I. and E.K. All authors discussed the results. S.Z., J.S.S. and R.A.M. wrote the original draft of the manuscript. Editing and revision of the manuscript were performed by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ANI-1xnr uncertainty for all eight O2/C2H2 ratios in graphene ring formation simulations.
Uncertainty (ε) is computed as ensemble standard deviation in energy normalized by the square-root of number atoms. Dashed black line corresponds to AL energy threshold of 1.85 \({\rm{kcal}}\,\,{\rm{mol}}^{-1}\,\,{N}^{-\frac{1}{2}}\). The MLIP uncertainty is fairly constant and approximately equal to the AL energy threshold through nearly the entire simulation (with only a few snapshots as exceptions). The relatively low and constant uncertainty confirms that each system is well-modeled by our MLIP. It is also interesting that the MLIP uncertainty decreases with increasing O2/C2H2 ratio, suggesting that ANI-1xnr is most confident under a higher O2/C2H2 ratio. The reason for such a tendency is that more oxidation reactions happen in the system with a larger O2/C2H2 ratio, which is a common reaction in our training dataset, although typically involving species other than acetylene. By contrast, the system with a smaller O2/C2H2 ratio has more ring formation, large carbon sheet formation and even phase change process, which are less common in the training set. Although C2H2 and O2 are common species in the ANI-1xnr dataset (see Methods for details), the fact that the uncertainties remain slightly larger than those for any structure in the entire ANI-1xnr training set demonstrates that these specific systems were not studied directly in the NR AL sampling. Therefore, this case study serves as an assessment of the generality of the ANI-1xnr potential.
Extended Data Fig. 2 Biofuel additive simulation results for ANI-1xnr over the entire simulation.
Tracking plot of O2 and major products (CO, CO2, and H2O) for biofuel simulations: (a) biofuel+O2 (b) biofuel+O2 with ethanol additive (c) biofuel+O2 with 2-butanol additive (d) biofuel+O2 with MTBE additive.
Extended Data Fig. 3 Comparison of normalized production rate of OH for biofuel additive simulations.
Number of OH species (NOH) is normalized by the initial number of O2 molecules \(({N}_{{{{{\rm{O}}}}}_{2}}^{t = 0})\). In comparison to the pure biofuel, all three systems with additives have a higher and earlier peak in OH radical, when normalized by the initial amount of O2. The enhancement in OH production for ethanol is intuitive since ethanol contains a hydroxyl group with a similar bond dissociation energy to 2-butanol. Considering the important role that the OH radical plays in ignition and combustion chemistry, the accelerated rate of OH production is consistent with a lower IDT for all three additive systems. Thus, although the ANI-1xnr results for ethanol are in conflict with ReaxFF, the enhancement in OH production provides understanding as to how ethanol accelerates the ignition process, similar to 2-butanol and MTBE.
Extended Data Fig. 4 Bond dissociation diagram for C-H bond in methane.
Data are presented as mean (center line) ± ensemble standard deviation (shaded region). Comparison between ANI-1xnr and ANI-1x with the DFT and experimental bond dissociation energies (BDE). The C-H bond dissociation energy from ANI-1xnr (116.6 kcal mol−1) is similar to that from our reference DFT (110.0 kcal mol−1), the reference DFT of Zeng et al. (115.0 kcal mol−1), and experiment (103.3 kcal mol−1). Although an in vacuo energy barrier may not correspond to a condensed-phase reaction rate, this analysis demonstrates that the accelerated consumption of CH4 is not due to a vastly under predicted C-H bond dissociation energy by ANI-1xnr or our reference DFT. However, the relatively large uncertainties in ANI-1xnr along the bond dissociation path suggest that ANI-1xnr should not be utilized for studying low-density gas-phase (or in vacuo) bond-breaking reactions. Although ANI-1xnr exhibits markedly greater uncertainty between 2-3 Å, the MLIP remains smooth and the forces are continuous. The poor performance in this high-uncertainty region is possibly due, in part, to the fact that the spin multiplicity of the minimum-energy electronic state changes from a singlet to a triplet in the gas-phase. Whereas, ANI-1xnr does not explicitly learn the relationship between energy and spin multiplicity and was trained on condensed-phase singlet data, where the notion of spin is much less pronounced. Although this may seem surprising that ANI-1xnr performs worse for in vacuo calculations where the physics are definitively simpler than for the condensed-phase, it is important to recall that the performance of MLIPs is highly dependent on having relevant training data. Specifically, since ANI-1xnr was trained solely to condensed-phase NR simulations (with densities between 0.5 and 2.5 g cc-1), we do not recommend using ANI-1xnr for in vacuo calculations.
Extended Data Fig. 5 Uncertainty analysis of ANI-1xnr on reactive literature datasets.
Distribution of ANI-1xnr ensemble uncertainties in (a/c) energy and (b/d) force for the (a/b) ANI-1xnr dataset, (a/b) Zeng et al. dataset for methane, and (c/d) Transition-1x reactive dataset. Energy uncertainty is the ensemble standard deviation for energy normalized by the square-root of number atoms. Force uncertainty is the ensemble standard deviation for force averaged over all atoms and Cartesian coordinates. The energy and force uncertainties in (a/b) for the Zeng et al. dataset are smaller than the AL thresholds of 1.85 \({\rm{kcal}}\,\,{\rm{mol}}^{-1}\,\,{N}^{-\frac{1}{2}}\) and 6.92 kcal mol−1 A−1, respectively, for approximately 77% of the ≈ 567000 structures. Note that the AL selection criterion of Zeng et al. was based solely on the force uncertainty. The ANI-1xnr force uncertainty is larger than the Zeng et al. AL threshold of 11.53 kcal mol−1 A−1 for only 5% of the structures in the Zeng et al. dataset, suggesting that ANI-1xnr has a similar confidence on this dataset as the application-specific MLIP of Zeng et al. Note that with such a high AL threshold, it is quite likely that there are some non-physical structures in the Zeng et al. dataset. Furthermore, the Zeng et al. dataset also consists of structures sampled with ReaxFF without curation. It is also important to recall that the Zeng et al. training dataset consists of small clusters of molecules extracted from a condensed-phase MD simulation. Therefore, atoms near the center of the cluster are in condensed-phase environments while atoms on the border of the cluster are effectively in a gas-phase environment. By contrast, the unilateral high uncertainties for the Transition-1x dataset in (c/d) demonstrate that ANI-1xnr is not intended for in vacuo reactive chemistry. Note that the distribution of Transition-1x uncertainties is nearly the same for reactants, transition states, and products, demonstrating that the issue is the vacuum environment rather than unsampled transition states.
Extended Data Fig. 6 Three-dimensional structures of reactions discovered by ANI-1xnr in Miller experiment simulation.
Structures are snapshots extracted directly from the MD simulation trajectory, providing visual confirmation that the reaction pathways are physically meaningful.
Extended Data Fig. 7 Reaction mechanisms for formation of key intermediates (CH2 and CO2) from initial reactants (NH3, CO, CH4, H2O) in Miller simulation.
Green arrows denote reactions previously identified by Wang et al. or Saitta and Saija. Orange arrows denote reactions that have closely-related reactions in Wang et al. or Saitta and Saija.
Extended Data Fig. 8 Alternative mechanism for the formation of glycine in the Miller experiment simulation.
In this pathway, the final step to form glycine involves H-abstraction from H3O, which chemical intuition would label as a cationic species (H3O+). The penultimate species (\({{{{\rm{C}}}}}_{2}{{{{\rm{H}}}}}_{4}{{{{\rm{NO}}}}}_{2}^{-}\)) formed prior to glycine, therefore, cannot be unambiguously labeled as an anion or a radical. The uncertainty regarding the ionic nature of this mechanism illustrates an issue with electron-agnostic MLIPs, like ANI-1xnr. The depiction of bond orders, charges on ions, and radical species is based simply on chemical intuition, since ANI-1xnr does not provide explicit bonding, orbital, or electronic information.
Supplementary information
Supplementary Information
Supplementary Tables 1–6 and Figs. 1–13.
Supplementary Data 1
Initial and final Cartesian coordinates for simulations performed in each case study.
Source data
Source Data Fig. 4
Effect of oxygen on graphene ring formation simulation results for ANI-1xnr.
Source Data Fig. 5
Biofuel additive simulation results for ANI-1xnr.
Source Data Fig. 6
Methane combustion simulation results for ANI-1xnr.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Makoś, M.Z., Jadrich, R.B. et al. Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential. Nat. Chem. 16, 727–734 (2024). https://doi.org/10.1038/s41557-023-01427-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-023-01427-3
