
Abstract
Mechanically interlocked structures, such as catenanes and rotaxanes, are fascinating synthetic targets and some are used for molecular switches and machines. Today, the vast majority of catenated structures are built upon macrocycles and only a very few examples of three-dimensional shape-persistent organic cages forming such structures have been reported. However, the catenation in all these cases was based on a thermodynamically favoured π–π-stacking under certain reaction conditions. Here, we show that catenane formation can be induced by adding methoxy or thiomethyl groups to one of the precursors during the synthesis of chiral [8 + 12] imine cubes, giving dimeric and trimeric catenated organic cages. To elucidate the underlying driving forces, we reacted 11 differently 1,4-disubstituted terephthaldehydes with a chiral triamino tribenzotriquinacene under various conditions to study whether monomeric cages or catenated cage dimers are the preferred products. We find that catenation is mainly directed by weak interactions derived from the substituents rather than by π-stacking.

Similar content being viewed by others
Main
Since the first report by Wasserman in the early 1960s of a catenane as a statistically occurring by-product during a macrocyclization via acyloin condensation1, interest in interlocked molecular structures has developed rapidly in recent decades2,3, especially because such compounds provide fundamental knowledge for supramolecular switches and machines4,5,6. Although Schill and Lüttringhaus introduced rational synthetic approaches towards a number of interlocked structures as early as the 1960s7, the real spark for this research field was the work by Sauvage and co-workers to create a high-yielding catenane synthesis by exploiting the templated coordination of two molecular strands by a metal ion before closing these to two interlocked macrocycles via Williamson ether synthesis8,9. This concept of using a template was and still is the most frequently applied strategy for the synthesis of more complex interlocked structures such as Borromean rings10, various knots11,12,13,14, a Star of David catenane15, poly[n]catenanes16 or interlocked coordination cages17,18,19,20,21. In addition to ligand-to-metal ion coordination, weaker and less-directing supramolecular interactions—such as hydrogen-bonding or π–π-stacking—have been used to arrange molecular precursors in the right fashion to synthesize interlocked structures22.
In contrast to the relatively large number and diversity of interlocked coordination cages18, only a few examples of purely organic cage catenanes have been reported to date. The first example was reported by Beer et al.23 They exploited a template effect of sulfate anions interacting with carbamate units to prearrange two tripodal precursor molecules in such a way that the end-capping of these via a copper-mediated 1,3-dipolar cycloaddition resulted in the formation of a triply interlocked cage dimer in 21% yield. One year later, in 2010, Cooper and co-workers described that by changing conditions for the synthesis of a [4 + 6] imine cage by adding catalytic amounts of trifluoroacetic acid to the reaction solution in acetonitrile or dichloromethane (DCM), these [4 + 6] imine cages form triply interlocked dimers24, which was proven by single-crystal X-ray diffraction. It was suggested that π–π-stacking is most probably the driving force for the catenane formation, and if a competing aromatic solvent was present in certain amounts, this indeed suppressed the catenane formation. In 2014, the formation of a quadruply interlocked dimer of a giant [12 + 8] boronic ester cage was described25, which was clearly characterized by single-crystal X-ray diffraction. The only difference between the interlocked cage dimer and a corresponding monomeric [12 + 8] boronic ester cage26 published before is the position and length of solubilizing alkyl chains in the molecular precursors, which led to the hypothesis that weak dispersion interactions may additionally be responsible for the catenane formation by overcoming any entropic penalty. In similar fashion, albeit more distinct, this entropic penalty was balanced by dispersion interactions in the formation of a hydrocarbon cage and its catenated dimer made by alkyne metathesis27. Depending on the concentration of reacting monomers, the equilibrium between monomeric and interlocked cages can be shifted towards one or the other metathesis product. The authors assumed that a triply interlocked structure is energetically more favoured than a singly interlocked one due to a maximization of filled space. In 2015, Li et al. exploited the hydrophobic effect to achieve an interlocked cage dimer via a hydrazone bond formation in water28. Very recently, the group of Shaodong Zhang presented the formation of a triply interlocked catenane of a [2 + 3] imine cage29,30. Again, it was concluded that the energetic benefits of additional π–π-stacking provide the driving force. In contrast to the aforementioned examples, in the present work dimer formation has been studied in more detail by kinetic NMR measurements and time-dependent mass spectrometry; however, no thermodynamic assumptions were corroborated experimentally. It is worth mentioning that Greenaway et al. described the unexpected formation of a bridged cage catenane during large high-throughput screening31.
During our ongoing work on condensing chiral triamino-tribenzotriquinacenes (TBTQs) with aromatic aldehydes to study self-sorting of cages32,33, we serendipitously found a substituent-driven formation of dimeric and trimeric cage catenanes, which is described herein.
Results and discussion
Synthesis and characterization of cage and catenanes
Inspired by Xu and Warmuth’s chiral cube34, based on the condensation of eight molecules of cyclotriveratrylene trisaldehyde and para-phenylene diamine, we intended to use a chiral TBTQ precursor instead, which, in contrast to the cyclotriveratrylene, is structurally fixed and cannot racemize during cage formation. Indeed, the condensation of enantiopure triamino-TBTQ (P)-1 (ref. 35) with 2,4-dihydroxy-terephthalaldehyde 2 under typical conditions we have used before for similar systems (TFA catalyst, CDCl3, r.t)34,36 gave the clean chiral [8 + 12] cage OH-cube in 86% isolated yield (Fig. 1) which was identified by NMR and mass spectrometry.

a, Schematic representation of the acid-catalysed 24-fold imine condensation of chiral triamino-TBTQ 1 and 2,5-dihydroxy-terephthalaldehyde 2 to OH-cube. Note that the alkyl substituents of TBTQ are omitted from the cubic structure of OH-cube for clarity. Reactants and cube are also drawn as cartoons. Red balls represent the TBTQ units and blue struts the aldehyde or imine linker units. b, 1H NMR spectrum (500 MHz, CD2Cl2, r.t.) of pure OH-cube. For assignment, see atom labels in the molecular structure of OH-cube in a and Supplementary Information, section 2. c, MALDI–TOF mass spectrum (DCTB) of pure OH-cube. Inset: comparison of measured and calculated isotopic patterns for OH-cube.
Originally we were interested in post-stabilizing the OH-cube by Pinnick oxidation to turn imine bonds into amide bonds37. As reported before, this does not work with the phenolic hydroxy groups present. To avoid a 24-fold post-synthetic Williamson etherification on OH-cube (ref. 38), we instead condensed TBTQ (P)-1 with dimethoxy-terephthalaldehyde 3 under the same conditions (Fig. 2a). In contrast to the reaction with aldehyde 2, here the 1H NMR spectrum of the crude product was very complex with a large number of peaks in the aromatic as well as in the aliphatic region (Fig. 2b). The corresponding matrix-assisted laser desorption/ionization–time of flight mass spectrometry (MALDI–TOF-MS) revealed that in addition to the [8 + 12] OMe-cube (m/z 5,623.21), a [16 + 24] condensation product (m/z 11,245.46) was generated and even a small peak with m/z 16,868.53 was detected (Fig. 2c), suggesting that a larger [24 + 36] species may have formed. Taking into consideration the complex 1H NMR spectra reported previously for triply interlocked cages24, it was assumed that these species are most probably catenated dimer (OMe-cube)2 and trimer (OMe-cube)3 rather than larger more symmetric and non-interlocked species. By applying recycling gel-permeation chromatography (r-GPC) with DCM as solvent, it was possible to separate the three compounds after multiple cycles (Fig. 2d and Supplementary Information, section 8).

a, Schematic representation of the acid-catalysed 24-fold imine condensation of 1 and 3 to OMe-cubes. b,c, 1H NMR (600 MHz, CDCl3) (b) and MALDI–TOF mass spectrum (DCTB) (c) of the crude reaction mixture of OMe-cubes. The 1H NMR spectrum of the crude product was very complex but that the MALDI–TOF mass spectrum was relatively clear with three distinct peaks. Peaks in the MALDI–TOF mass spectra are labelled with the structures of the products; the small peak for the trimeric catenane is highlighted and shown in the inset. d, r-GPC traces (solvent, DCM) of the crude reaction mixture of OMe-cube, (OMe-cube)2 and (OMe-cube)3 clearly show three peaks.
As described previously in the literature, the equilibrium between monomeric and catenated cages shifts towards the latter by increasing the concentration of reactants and conversely to the monomeric cage by decreasing it. Therefore, the reaction was performed at different concentrations (between 0.42 and 42.8 mM) and analysed mainly by MALDI–TOF-MS (Supplementary Table 1). As expected, with higher concentrations more catenated compounds (OMe-cube)2 and (OMe-cube)3 were found and the concentration needs to be 0.42 mM or below to avoid the formation of those and to form monomeric cage OMe-cube exclusively. For comparison, reactions with dihydroxy terephthaldehyde 2 at various concentrations (up to 42.8 mM) did not give any catenated species and in each experiment only monomeric cage OH-cube was detected by 1H NMR spectroscopy (Supplementary Fig. 398). It is worth mentioning that as soon as a CHCl3 or DCM solution of monomeric cage OMe-cube was concentrated by rotary evaporation (50 °C, reduced pressure), the equilibrium immediately shifted towards the catenated products (OMe-cube)2 and (OMe-cube)3 as found by NMR and r-GPC analysis. On one hand, this clearly demonstrated the dynamic covalent chemistry character and thus thermodynamically driven formation of the catenane39. On the other hand, it made the separation and characterization of monomeric cage OMe-cube more challenging.
Despite these findings, we were able to develop a synthetic protocol to isolate OMe-cube in 85% yield by using shorter reaction times, certain concentration and temperature thresholds, and exploiting the low solubility of the cage in acetonitrile (Supplementary Information, section 2). To our delight, by changing the solvent to 1,1,2,2-tetrachloroethane (TCE), monomeric cage OMe-cube could be synthesized even at higher concentration (5.4 mM), without the necessity of GPC separation, in 76% yield (Fig. 3a). On the other hand, running the reaction of 1 and 3 in CD2Cl2 instead of CHCl3 at 10.7 mM concentration (w.r.t. 1) and 80 °C for 3 days allowed us to push the equilibrium towards the tricatenane (OMe-cube)3, which was isolated in 80% yield (Fig. 3a). The best results for the dimeric cage (OMe-cube)2 were achieved when 1 and 3 were reacted at 10.7 mM scale. However, (OMe-cube)2 still needed to be separated by r-GPC from OMe-cube and (OMe-cube)3 at 30°C to be obtained in 46% isolated yield (Fig. 3a).

a, Schematic representation of the acid-catalysed 24-fold imine condensation of 1 and 3 in different solvents for the selective formation of OMe-cube, (OMe-cube)2 and (OMe-cube)3. For reaction details, see Supplementary Information, section 2. b–g, r-GPC traces and MALDI–TOF mass spectra of OMe-cube, (OMe-cube)2 and (OMe-cube)3. r-GPC traces (solvent, DCM) of pure OMe-cube (b), (OMe-cube)2 (d) and (OMe-cube)3 (f) show retention times of 27.7, 25.9 and 25.4 min, respectively, which are consistent with their sizes. Depicted is the first cycle for each. The corresponding MALDI–TOF mass spectra of pure OMe-cube (c), (OMe-cube)2 (e) and (OMe-cube)3 (g) show exclusively a single peak for each species. h,i, 1H NMR spectra (600 MHz, 295 K, CD2Cl2) of pure OMe-cube (h) and (OMe-cube)2 (i). j, 1H NMR spectra (700 MHz, 375 K, toluene-d8) of pure (OMe-cube)3.
Mechanistic investigation of the monomeric cage to dimeric catenane reaction
To obtain some more mechanistic information on catenane formation, the transformation of OMe-cube to (OMe-cube)2 was studied in more detail. Kinetic NMR experiments indicated that full equilibrium between OMe-cube (c0 = 1.35 mM, CDCl3) and (OMe-cube)2 was achieved after ∼800 min (k1 = 10.5 ± 0.4 M−1 s−1) if catalytic amounts of TFA are present (Supplementary Fig. 506). In the absence of acid, no conversion at all was detected even after 24 h (Supplementary Fig. 507). Mixing a 15N-labelled cage *OMe-cube in a 1:1 stoichiometry with non-labelled OMe-cube under reaction conditions (TFA catalyst, CDCl3) and analysing the mixture after 3 days by MALDI–TOF-MS revealed that all units (TBTQ and linkers) are fully scrambling to a statistical mixture, suggesting that during catenane formation all TBTQ units must be disconnected from the cage scaffolds at a certain stage, opening the cages for catenation (Supplementary Figs. 508 and 509).
Structural analysis of cage and dimeric and trimeric catenanes
The GPC-purified fractions were again separately injected into r-GPC, resulting in the detection of three single distinct peaks, each of nearly Gaussian shape with retention times of 25.4 min (first fraction), 25.9 min (second fraction) and 27.7 min (third fraction) (Fig. 3b,d,f). MALDI–TOF-MS analysis of each fraction (Fig. 3c,e,g) now show single peaks exclusively at m/z 16,868.69 (first fraction), m/z 11,245.57 (second fraction) and m/z 5,623.24 (third fraction), which exactly fit to a [24 + 36], a [16 + 24] and a [8 + 12] species, respectively. The 1H NMR spectrum (Fig. 3h) of the third fraction was very simple, showing signals comparable to OH-cube, and in combination with the mass spectrum (Fig. 3c) this compound was clearly identified as the monomeric chiral [8 + 12] OMe-cube. Diffusion-ordered NMR spectroscopy (DOSY) in deuterated DCM at 295 K showed only one trace with a diffusion coefficient of D = 3.09 × 10−10 m2 s−1, which according to the uncorrected Stokes–Einstein equation corresponds to a solvodynamic radius of rS = 16.9 Å (Supplementary Fig. 308). In contrast to the relatively simple 1H NMR spectrum of monomeric OMe-cube (Fig. 3h), that of the [16 + 24] species was much more complex (Fig. 3i). Nevertheless, despite the large number of signals, most of them were sharp and did not superimpose, allowing a more detailed analysis of the structure (Fig. 4a; for detailed structural analysis, see Supplementary Information, section 13). Two-dimensional NMR experiments identified eight different types of imine protons and eight different methoxy protons (Fig. 4b,c). This is exactly the number expected for a triply interlocked cage dimer (see cartoon in Fig. 4h) Other possible catenanes, such as a singly interlocked dimer (Fig. 4g, 12 imine peaks) or quadruply interlocked dimer (Fig. 4i, six imine peaks), can clearly be ruled out. DOSY NMR in CD2Cl2 at 295 K showed only one trace of signals for (OMe-cube)2, confirming that this is a single species. The diffusion coefficient D = 2.67 × 10−10 m2 s−1 corresponds to a solvodynamic radius of 19.6 Å (Supplementary Fig. 309). This is slightly larger than for OMe-cube (16.9 Å) which is consistent with its slightly larger size.

a, Full 1H NMR (600 MHz, CD2Cl2) spectrum of (OMe-cube)2. b,c, Partial 1H NMR (600 MHz, CD2Cl2) showing eight different types of imine peaks (b) and methoxy peaks (c) of (OMe-cube)2. d, NOESY spectrum showing cross-peaks between imine proton B and methoxy proton d. e, NOESY spectrum showing cross-peaks between methoxy protons b and d. f, NOESY spectrum showing cross-peaks between highly shielded aliphatic protons of propyl chains and methoxy protons e and f. g, Cartoon of the singly catenated cubes highlighting the 12 different magnetically equivalent imine protons. h, Cartoon of the triply interlocked catenated cube, highlighting the eight different magnetically equivalent imine protons. The colour code and assignment are the same as in b and c. i, Cartoons of the quadruply interlocked catenated cubes, highlighting the six different magnetically equivalent imine protons.
The trimeric interlocked cage (OMe-cube)3 shows much less resolved multiple broad peaks at room temperature in the 1H NMR spectrum (Supplementary Fig. 72) in contrast to dimer (OMe-cube)2. However, in toluene-d8 at 375 K a much better resolved spectrum was obtained, showing sets of 12 magnetically different peaks, such as 12 imine protons and 12 signals of the terminal CH3 group of the propyl chains (Fig. 5a–c and Supplementary Figs. 88 and 89). This excludes a syn-distal connectivity (36 imine peaks; Fig. 5f, for models, see Supplementary Fig. 522) and a syn-proximal connectivity at two adjacent corners at the central cube (here 72 imine peaks are expected; Fig. 5d, for models, see Supplementary Fig. 523). For both a chain-like anti-connected catenane (OMe-cube)@(OMe-cube)@(OMe-cube) (Fig. 5e) and for an interwoven catenane [(OMe-cube)@(OMe-cube)]@(OMe-cube) (Fig. 5d) the same number—12—of magnetically different peaks are expected, as has been found (for models, see Supplementary Figs. 520 and 521). However, since the trimeric catenane (OMe-cube)3 is still very soluble under reaction conditions and no traces of larger oligomers such as tetrameric and pentameric cages (OMe-cube)4 or (OMe-cube)5 are found by mass spectrometry, it seems to be more likely that the interwoven catenane [(OMe-cube)@(OMe-cube)]@(OMe-cube) and not the chain-like anti-conformed catenane (OMe-cube)@(OMe-cube)@(OMe-cube) has formed. If it were the latter motif, we would expect at least some formation of longer oligomers, which is not the case. On the other hand, an interwoven tetrameric catenane is simply not possible for steric reasons, which once more would explain the absence of larger species and thus favours this motif for the trimeric catenane [(OMe-cube)@(OMe-cube)]@(OMe-cube). DOSY NMR of (OMe-cube)3 again shows a single trace with a diffusion coefficient D = 2.64 × 10−10 m2 s−1. The calculated solvodynamic radius of 19.8 Å (Supplementary Fig. 310) was found to be almost similar to that of the dicatenane (OMe-cube)2 (19.6 Å), once more suggesting a tightly packed, interlocked structure. In the DOSY NMR spectra of a 1:1 stoichiometric mixture of pure (OMe-cube)2 and (OMe-cube)3 (Supplementary Fig. 512) the difference between the diffusion coefficient values is very small, further supporting the more dense interwoven catenane [(OMe-cube)@(OMe-cube)]@(OMe-cube) model.

a, Full 1H NMR (700 MHz, 375 K, toluene-d8) spectrum of (OMe-cube)3. b,c, Partial 1H NMR (700 MHz, 375 K, toluene-d8) showing 12 different types of imine peaks (b) and methyl peaks (c) of (OMe-cube)3. d–g, Cartoons of the interwoven [(OMe-cube)@(OMe-cube)]@(OMe-cube) (d), anti (OMe-cube)@(OMe-cube)@(OMe-cube) (e), syn-distal (f) and syn-proximal (g) trimeric catenanes, highlighting the 12, 12, 36 and 72 different magnetically equivalent imine protons in different colours, respectively.
It is worth mentioning that for OH-cube, OMe-cube, (OMe-cube)2, and for (OMe-cube)3 and the other cage catenanes described below, innumerable large single crystals have been obtained from various solvents. Unfortunately, even synchrotron radiation did not provide sufficient resolution to elucidate the solid-state structures.
Investigation of the driving force for catenation
We were interested in obtaining further insight into the driving force of the unique catenation of methoxy cage OMe-cube to dimer (OMe-cube)2 and even to trimer (OMe-cube)3 and why we do not see any such catenation for the hydroxyl-substituted OH-cube at any concentration. Due to the triply interlocked catenation of dimer (OMe-cube)≡(OMe-cube) in favour of a possible singly interlocked dimer (OMe-cube)–(OMe-cube), the aforementioned π–π-stacking as driving force—found for almost all the interlocked organic cages previously described in the literature—was excluded (see above), otherwise singly interlocked catenation should have been formed preferably. In addition for OH-cube a higher tendency of dimerization would have been expected than for OMe-cube, because intramolecular hydrogen-bonding of the hydroxyl imine stiffens the π-backbone and strongly enhances intermolecular π–π-stacking40. This assumption is strengthened by the fact that a wide range of reaction conditions (different acid concentrations, different concentrations of reactants, different solvents, different and elevated temperatures, different reaction times (up to several months)) yielded no substantial catenane formation for OH-cube (see Supplementary Figs. 398, 400, 401 and 510). A kinetic formation driven by precipitation was also ruled out, because the reaction mixtures of 1, 2 and OH-cube were at all times clear solutions39. Furthermore, mixing OH-cube and 15N-labelled *OH-cube in a 1:1 ratio and treating this mixture under reaction conditions gave the two highest MALDI–TOF mass peaks at m/z 5,297.56 (corresponding to OH-cube-15N12) and m/z 5,320.54 (corresponding to (OH-cube-15N12 + Na), suggesting a complete scrambling in solution, supporting the thermodynamic formation of OH-cube and confirming that it is not a kinetic trap (Supplementary Figs. 510 and 511)41.
Since π-stacking was ruled out as a driving force, we first hypothesized that dipole–dipole interactions (so-called Keesom interactions)42 of the methoxy groups may be responsible for the catenation, as found, for example, in single crystals of methoxy-substituted π-systems (d(MeO⋯CH3O) = 3.1 Å)43. In this respect, it is worth mentioning that the bridged cage catenane reported by Greenaway et al., when originally achieving cages based on dimethoxy terephthaldehyde 3 (ref. 31). could rely on such weak interactions, although a closer look at the X-ray structure shows the same methoxy–methoxy interaction motif, albeit with a larger distance between the functional groups of d(MeO⋯CH3O) = 3.5 Å (Supplementary Fig. 513). Conformational analysis by semi-empirical calculations (Supplementary Section 19) of OMe-cube as well as nuclear Overhauser enhancement spectroscopy (NOESY) cross-peaks between imine CH and the aromatic TBTQ protons revealed a low barrier of rotation of the linker units at room temperature, which is also present in the triply interlocked dimer (OMe-cube)2, allowing the mechanically interlocked molecule to adopt conformations that have three such methoxy–methoxy interactions (Fig. 6c). According to this assumption (Supplementary Fig. 513d,e), one methoxy group per dialdehyde unit should be enough to foster catenation, and indeed reacting dialdehyde 12 (with only one methoxy group present) with triamine 1 in CD2Cl2 clearly gave catenated (H/OMe-cube)2, as determined by mass spectrometry (Supplementary Fig. 345).

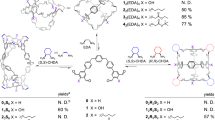
a, Reactions of various dialdehydes with TBTQ triamine 1, giving cages (left side) or catenated cages (right side). aYields are given for optimized reactions. b, Summary of interactions resulting in catenane formation or their exclusion. c, Possible conformational arrangement of the two interlocked cages with spatial arrangement of methoxy groups interacting via weak dipole–dipole forces (green dotted lines). d, Gibbs enthalpy for catenation events determined by NMR spectroscopy.
If methoxy groups are absent, no catenane formation should occur. Thus triamine 1 was reacted with non-substituted terephthalaldehyde 4 (Fig. 6a) under different conditions (various solvents, Supplementary Fig. 405) and no catenane formation was observed. Pure H-cube was isolated in 90% from THF. By adding two methyl substituents instead of two methoxy groups to the aldehyde (5) still almost no catenane formation is observed by 1H NMR (Supplementary Fig. 406) and monomeric Me-cube is formed in 84% yield. As soon as the alkyl substituents at the dialdehyde precursor (6) get longer (here ethyl), the possibility of intermolecular dispersion interactions44 (Fig. 6b) is slightly increased and now some catenane (Et-cube)2 was found by 1H NMR spectroscopy as well as mass spectrometry (Supplementary Fig. 407) besides monomeric Et-cube (which still is the main product). Comparing the different results of Me-cube versus Et-cube, based on the simple elongation of the alkyl chains by one methylene unit each, electronic effects to foster π–π-stacking can again be ruled out, because the methyl as well as the ethyl substituents have almost the same Hammett parameters (σm(Me) = −0.07; σm(Et) = −0.07; σp(Me) = −0.17; σp(Et) = −0.15)45. As for OMe-cube and (OMe-cube)2, the ratio of catenane (Et-cube)2 versus monomeric cage Et-cube was also strongly solvent dependent for the reaction of triamine 1 and aldehyde 6 and in THF the amount of catenane was higher than, for example, in CHCl3 and both compounds (monomer and catenane) were selectivity achieved by adjusting the conditions. The reaction in CHCl3 at room temperature gave monomeric cage Et-cube in 75% isolated yield, whereas running the reaction in THF gave after separation 35% of the catenated dimer (Et-cube)2 in pure form.
To further exclude pure electronic effects for a probable π-stacking, we reacted triamine 1 with diethoxy- and diisopropoxy dialdehydes 7 and 8 (Fig. 6a), where the substituents have comparable Hammett parameters to those in dimethoxy dialdehyde 3 (σm(OMe) = 0.12; σm(OEt) = 0.10; σm(OiPr) = 0.10), but are of different steric demand. Whereas for the diethoxy dialdehyde 7 some catenane formation of (OEt-cube)2 was observed, for diisopropoxy dialdehyde 8 no catenane (OiPr-cube)2 was observed by 1H NMR spectroscopy (Supplementary Figs. 408 and 409), supporting once more the hypothesis that the catenane formation is mainly driven by weak interactions derived from the substituents rather than by π-stacking, and in case of the latter steric repulsion is stronger than the weak attraction (Fig. 6b; Charton steric parameters for Me, Et and iPr are νMe = 0.52, νEt = 0.56, νiPr = 0.76)46.
By increasing these weak interactions, the equilibrium may be shifted towards the interlocked structures. By using dimethylthioether 9 in the condensation with triamine 1 in CDCl3, the triply interlocked catenated dimer (SMe-cube)2 was formed almost exclusively (Fig. 6a and Supplementary Fig. 410). Harsher conditions were needed to push the system to the trimeric cage (SMe-cube)3, which was isolated in 58% yield by using CD2Cl2 as a solvent in combination with elevated temperature (80 °C, screw-capped vessel, 4 days). By two-dimensional NMR spectroscopy, the same linear and interwoven catenation motif was found as for (OMe-cube)3 (Supplementary Fig. 495). Switching the solvent system to TCE, monomeric SMe-cube was isolated in 80% yield. Again, to rule out electronic effects based on the thioalkyl substituent donating to the π-system of the aromatic dialdehyde, di-tert-butylthioether-substituted dialdehyde 10 with two sterically demanding tert-butyl groups was investigated in the reaction (νMe = 0.52 versus νtBu = 1.24)46. As expected, only clean monomeric SC(CH3)3-cube was formed and isolated in 75% yield (Supplementary Fig. 411). Finally, we investigated the reaction of dibromo dialdehyde 11 with triamine 1, to see whether halogen bond formation47 can also induce catenation. Although the mass spectrum of the reaction mixture in CD2Cl2 showed a pronounced peak at m/z 13,591.6, which is the double that of the monomeric Br-cube (m/z 6,796.4), the correlated 1H NMR spectrum showed only small detectable peaks of interlocked species and mainly contained signals of pure monomeric Br-cube (Supplementary Fig. 412). However, in contrast to all other reactions, here a precipitate of very low solubility was formed, which may contain insoluble (Br-cube)2.
Thermodynamic studies
To correlate the weak interactions responsible for catenation, the systems where catenation occurred have been studied by concentration-dependent NMR spectroscopy (Supplementary Information, section 12), to estimate the Gibbs enthalpy of cage to catenane transformation. With ΔG298 = −26.7 kJ mol−1 the reaction of 2 SMe-cube → (SMe-cube)2 is about 6 kJ mol−1 higher than for the methoxy cages 2 OMe-cube → (OMe-cube)2 (ΔG298 = −20.8 kJ mol−1) and almost 10 kJ mol−1 higher than found for the ethoxy cages 2 OEt-cube → (OEt-cube)2 (ΔG298 = −15.7 kJ mol−1) (Fig. 6d), Unfortunately, in CDCl3 the amount of (Et-cube)2 in relation to Et-cube was too small to determine reliable numbers by this method. As mentioned repeatedly above, the chosen solvents had clear impacts on whether catenation occurred or not. Therefore, we looked at the van’t Hoff plots of temperature-dependent 1H NMR measurements of the equilibria 2 OMe-cube ↔ (OMe-cube)2 and 2 SMe-cube ↔ (SMe-cube)2 to obtain further insights into whether the processes of catenation are enthalpy- or entropy-driven reactions. In both investigated cases the catenation is an entropy-driven reaction (ΔSOMe = +114.3 J K−1 mol−1 and ΔSSMe = +185.3 J K−1 mol−1), rather than an enthalpy-driven reaction (ΔHOMe = +13.1 kJ mol−1 and ΔHSMe = +29.4 kJ mol−1), which also explains the temperature dependency of the reaction. This further suggests that solvophobic effects48 are dominating the catenation process at least in the investigated solvent systems. These solvophobic effects are dependent on the polarity of side chains49,50, as observed here, and need to be investigated further for such systems.
Conclusions
We observed the formation of dimeric and trimeric cage catenanes based on the weak interactions of the substituents of the constituent 1,4-disubstituted terephthaldehydes. Whereas π-stacking was ruled out as a driving force, Keesom and London dispersion interactions between the substituents and with the solvent were considered. Changing the methoxy groups to less polar ethyl groups decreased catenane formation substantially. In cases where there is only a methyl substituent or no substituent at the dialdehyde, the intermolecular forces are too weak to foster catenane formation. Finally, dialdehydes with thiomethyl substituents were beneficial for catenane formation and indeed a clear reaction to (SMe-cube)2 was observed, having a difference of |ΔG°| of ∼6 kJ mol−1 for catenane formation compared with (OMe-cube)2. Solvent effects play a crucial role in the cases where dimeric and trimeric catenation was observed. Both systems (with OMe and SMe substituents) showed the same trends. In TCE, monomeric cages OMe-cube and SMe-cube were formed selectively, whereas in DCM at elevated temperatures the clean formation of trimeric catenanes (OMe-cube)3 and (SMe-cube)3 was observed. Comparing the coherence energy densities of the solvents (TCE = 98.0 cal cm−3 versus DCM = 93.7 cal cm−3) in combination with the data from van’t Hoff plot analysis revealed that solvophobic effects may play a major role because reactions toward catenated cages are entropically favoured.
This motif of weak dispersion interactions in combination with solvophobic effects as a driving force for catenation of shape-persistent organic cages offers an approach for further study of the influence of subtle structural changes in combination with chosen solvents as reaction media to understand events of dynamic covalent chemistry of larger and more complex structures and to construct, for example, poly[n]catenated cages with n > 3, to create cages of higher molecular volumes.
Methods
Synthesis of (OMe-cube)2
To a solution of TBTQ 1 (20 mg, 0.043 mmol) and 2,5-dimethoxy-terephthaldehyde 3 (12.6 mg, 0.0649 mmol) in deuterated chloroform (4 ml) in a screw-capped 8 ml glass vial, a catalytic amount of TFA (0.4 µl, 0.0052 mmol) was added and the reaction mixture was stirred at r.t. for 3 days. Afterwards, the crude reaction mixture was washed with aqueous K2CO3 solution (0.25 M, 3 × 2 ml), dried over Na2SO4 and concentrated under reduced pressure. The resulting red solid was immediately dissolved in DCM and purified by r-GPC (DCM, 30 °C, 5 ml min−1) to give 14 mg (46%) of (OMe-cube)2 as a yellow solid. Melting point, 315 °C (decomposed). 1H NMR (600 MHz, CD2Cl2): δ (ppm) = 8.90 (s, 6H, HC=N), 8.87 (s, 6H, HC=N), 8.85 (s, 12H, HC=N), 8.82 (s, 6H, HC=N), 8.78 (s, 6H, HC=N), 8.65 (s, 6H, HC=N), 8.38 (s, 6H, HC=N), 8.03 (s, 6H, Ar–H), 7.88 (s, 6H, Ar–H), 7.79 (s, 6H, Ar–H), 7.71 (s, 6H, Ar–H), 7.69 (12H, Ar–H), 7.53 (d, 3J = 8.4 Hz, 6H, TBTQ Ar–H), 7.41 (d, 3J = 8.4 Hz, 6H, TBTQ Ar–H), 7.38 (d, 3J = 8.4 Hz, 6H, TBTQ Ar–H), 7.36–7.32 (m, 6H, TBTQ–Ar–H and Ar–H), 7.27 (s, 6H, TBTQ–Ar–H), 7.24 (s, 12H, Ar–H, TBTQ–Ar–H), 7.21 (s, 6H, TBTQ–Ar–H), 7.19 (s, 6H, TBTQ Ar–H), 7.17–7.14 (m, 6H, TBTQ–Ar–H), 7.08 (d, 3J = 8.4 Hz, 6H, TBTQ–Ar–H), 7.03–6.97 (m, 18H, TBTQ–Ar–H), 6.89 (s, 6H, TBTQ–Ar–H), 6.83 (d, 3J = 8.4 Hz, 6H, TBTQ–Ar–H), 6.78 (d, 3J = 8.4 Hz, 6H, TBTQ–Ar–H), 6.77 (d, 3J = 8.4 Hz, 6H, TBTQ–Ar–H), 6.72 (s, 6H, TBTQ–Ar–H), 6.59 (d, 3J = 8.4 Hz, 12H, TBTQ–Ar–H), 6.26 (d, 3J = 7.8 Hz, 6H, TBTQ–Ar–H), 4.04 (s, 18H, OCH3), 3.94 (s, 18H, OCH3), 3.90 (s, 18H, OCH3), 3.86 (s, 18H, OCH3), 3.82 (s, 18H, OCH3), 3.67 (s, 18H, OCH3), 3.11 (s, 18H, OCH3), 2.85 (s, 18H, OCH3), 2.30–1.74 (m, 96H, –CH2CH2CH3) 1.71 (s, 30H), 1.64 (s, 18H), 1.35–1.09 (m, 84H, –CH2CH2CH3), 1.0–0.90 (m, 126H, –CH2CH2CH3), 0.77–0.72 (m, 12H, –CH2CH2CH3), 0.48 (t, 3J = 7.2 Hz, 18H, –CH2CH2CH3). 13C NMR (151 MHz, CD2Cl2): δ (ppm) = 156.5, 155.7, 155.3, 155.2, 155.0, 154.6, 154.31, 154.26, 154.2, 153.9, 153.8, 153.1, 152.7, 152.5, 152.6, 152.3, 152.2, 150.1, 149.8, 149.64, 149.6, 149.5, 149.4, 149.3, 149.1, 146.7, 146.6, 146.4, 146.1, 146.0, 145.5, 145.4, 129.0, 128.9, 128.6, 128.55, 128.49, 127.8, 125.0, 124.6, 124.42, 124.36, 124.2, 124.0, 119.8, 119.7, 119.4, 119.04, 118.96, 118.8, 118.5, 118.4, 118.1, 117.5, 117.3, 116.1, 116.0, 111.2, 110.2, 109.83, 109.77, 109.63, 109.58, 73.5, 73.2, 73.0, 67.39, 67.35, 67.33, 67.23, 67.18, 66.9, 56.71, 56.68, 56.65, 56.61, 56.6, 56.4, 56.1, 55.4, 41.8, 41.2, 41.1, 41.0, 21.3, 21.1, 21.0, 20.9, 20.8, 20.3, 15.5, 15.4, 15.32, 15.29, 15.0, 14.9. Fourier transform infrared spectroscopy (neat, attenuated total reflectance): 𝜈̃ (cm−1) = 2,999 (w), 2,957 (m), 2,925 (m), 2,870 (m), 2,853 (m), 1,734 (w), 1,616 (m), 1,593 (m), 1,492 (s), 1,482 (s), 1,465 (s), 1,410 (s), 1,373 (m), 1,211 (s), 1,140 (m), 1,043 (s), 974 (w), 882 (m), 821 (m), 701 (w). Ultraviolet–visible spectroscopy (CH2Cl2): λmax (nm) = 296, 406. MALDI–TOF (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB)): m/z [M]+ calculated for C752H768N48O48, 11,245.94; found, 11,245.57. Elemental analysis: calculated for C752H768N48O48·33CH2Cl2, C 67.11, H 5.98, N 4.79; found C 66.94, H 5.91, N 4.86.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. The original experimental data to this paper has been deposited in the repository heiDATA and can be downloaded via https://heidata.uni-heidelberg.de/privateurl.xhtml?token=96ab88cd-5fc2-4b2c-b836-522dab565ba2
Change history
04 January 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41557-022-01133-6
References
Wasserman, E. The preparation of interlocking rings: a catenane. J. Am. Chem. Soc. 82, 4433–4434 (1960).
Forgan, R. S., Sauvage, J.-P. & Stoddart, J. F. Chemical topology: complex molecular knots, links, and entanglements. Chem. Rev. 111, 5434–5464 (2011).
Gil-Ramírez, G., Leigh, D. A. & Stephens, A. J. Catenanes: fifty years of molecular links. Angew. Chem. Int. Ed. 54, 6110–6150 (2015).
Erbas-Cakmak, S., Leigh, D. A., McTernan, C. T. & Nussbaumer, A. L. Artificial molecular machines. Chem. Rev. 115, 10081–10206 (2015).
Kay, E. R., Leigh, D. A. & Zerbetto, F. Synthetic molecular motors and mechanical machines. Angew. Chem. Int. Ed. 46, 72–191 (2007).
Dattler, D. et al. Design of collective motions from synthetic molecular switches, rotors, and motors. Chem. Rev. 120, 310–433 (2020).
Schill, G. & Lüttringhaus, A. The preparation of catena compounds by directed synthesis. Angew. Chem. Int. Ed. 3, 546–547 (1964).
Dietrich-Buchecker, C. O., Sauvage, J. P. & Kintzinger, J. P. Une nouvelle famille de molecules: les metallo-catenanes. Tetrahedron Lett. 24, 5095–5098 (1983).
Dietrich-Buchecker, C. O., Sauvage, J. P. & Kern, J. M. Templated synthesis of interlocked macrocyclic ligands: the catenands. J. Am. Chem. Soc. 106, 3043–3045 (1984).
Chichak, K. S. et al. Molecular Borromean rings. Science 304, 1308–1312 (2004).
Ayme, J.-F. et al. A synthetic molecular pentafoil knot. Nat. Chem. 4, 15–20 (2012).
Zhang, L. et al. Stereoselective synthesis of a composite knot with nine crossings. Nat. Chem. 10, 1083–1088 (2018).
Leigh, D. A. et al. Tying different knots in a molecular strand. Nature 584, 562–568 (2020).
Leigh, D. A. et al. A molecular endless (74) knot. Nat. Chem. 13, 117–122 (2021).
Leigh, D. A., Pritchard, R. G. & Stephens, A. J. A Star of David catenane. Nat. Chem. 6, 978–982 (2014).
Wu, Q. et al. Poly[n]catenanes: synthesis of molecular interlocked chains. Science 358, 1434–1439 (2017).
Fujita, M., Fujita, N., Ogura, K. & Yamaguchi, K. Spontaneous assembly of ten components into two interlocked, identical coordination cages. Nature 400, 52–55 (1999).
Frank, M., Johnstone, M. D. & Clever, G. H. Interpenetrated cage structures. Chem. Eur. J. 22, 14104–14125 (2016).
Zhu, R. et al. Catenation and aggregation of multi-cavity coordination cages. Angew. Chem. Int. Ed. 57, 13652–13656 (2018).
Bloch, W. M., Holstein, J. J., Dittrich, B., Hiller, W. & Clever, G. H. Hierarchical assembly of an interlocked M8L16 container. Angew. Chem. Int. Ed. 57, 5534–5538 (2018).
Ronson, T. K., Wang, Y., Baldridge, K., Siegel, J. S. & Nitschke, J. R. An S10-symmetric 5-fold interlocked [2]catenane. J. Am. Chem. Soc. 142, 10267–10272 (2020).
Wang, L., Vysotsky, M. O., Bogdan, A., Bolte, M. & Böhmer, V. Multiple catenanes derived from calix[4]arenes. Science 304, 1312–1314 (2004).
Li, Y. et al. Sulfate anion templated synthesis of a triply interlocked capsule. Chem. Commun. 46, 7134–7136 (2009).
Hasell, T. et al. Triply interlocked covalent organic cages. Nat. Chem. 2, 750–755 (2010).
Zhang, G., Presly, O., White, F., Oppel, I. M. & Mastalerz, M. A shape-persistent quadruply interlocked giant cage catenane with two distinct pores in the solid state. Angew. Chem. Int. Ed. 53, 5126–5130 (2014).
Zhang, G., Presly, O., White, F., Oppel, I. M. & Mastalerz, M. A permanent mesoporous organic cage with an exceptionally high surface area. Angew. Chem. Int. Ed. 53, 1516–1520 (2014).
Wang, Q. et al. Solution-phase dynamic assembly of permanently interlocked aryleneethynylene cages through alkyne metathesis. Angew. Chem. Int. Ed. 54, 7550–7554 (2015).
Li, H. et al. Quantitative self-assembly of a purely organic three-dimensional catenane in water. Nat. Chem. 7, 1003–1008 (2015).
Li, P. et al. De novo construction of catenanes with dissymmetric cages by space-discriminative post-assembly modification. Angew. Chem. Int. Ed. 59, 7113–7121 (2020).
Xu, S. et al. Catenated cages mediated by enthalpic reaction intermediates. CCS Chemistry 7, 1838–1850 (2021).
Greenaway, R. L. et al. High-throughput discovery of organic cages and catenanes using computational screening fused with robotic synthesis. Nat. Commun. 9, 2849 (2018).
Beaudoin, D., Rominger, F. & Mastalerz, M. Chiral self-sorting of [2+3] salicylimine cage compounds. Angew. Chem. Int. Ed. 56, 1244–1248 (2017).
Wagner, P. et al. Chiral self-sorting of giant cubic [8+12] salicylimine cage compounds. Angew. Chem. Int. Ed. 60, 8896–8904 (2021).
Xu, D. & Warmuth, R. Edge-directed dynamic covalent synthesis of a chiral nanocube. J. Am. Chem. Soc. 130, 7520–7521 (2008).
Beaudoin, D., Rominger, F. & Mastalerz, M. Synthesis and chiral resolution of C3-symmetric tribenzotriquinacenes. Eur. J. Org. Chem. 2016, 4470–4472 (2016).
Beaudoin, D., Rominger, F. & Mastalerz, M. Chiral self-sorting of [2+3] salicylimine cage compounds. Angew. Chem. 129, 1264–1268 (2017).
Bhat, A. S. et al. Transformation of a [4+6] salicylbisimine cage to chemically robust amide cages. Angew. Chem. Int. Ed. 58, 8819–8823 (2019).
Schneider, M. W., Oppel, I. M., Griffin, A. & Mastalerz, M. Post-modification of the interior of porous shape-persistent organic cage compounds. Angew. Chem. Int. Ed. 52, 3611–3615 (2013).
Schick, T. H. G., Rominger, F. & Mastalerz, M. Examination of the dynamic covalent chemistry of [2 + 3]-imine cages. J. Org. Chem. 85, 13757–13771 (2020).
Gilli, G., Bellucci, F., Ferretti, V. & Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the β-diketone fragment. J. Am. Chem. Soc. 111, 1023–1028 (1989).
Holsten, M. et al. Soluble congeners of prior insoluble shape-persistent imine cages. Chem-Eur J 27, 9383–9390 (2021).
Keesom, W. H. The second viral coefficient for righid spherical molecules, whose mutual attraction is equivalent to that of a quadruplet placed at centre. In KNAW Proceedings Vol. 18, Issue 1, 636–648 (KNAW, 1915).
Ueberricke, L. et al. Triptycene end-capped quinoxalinophenanthrophenazines (QPPs): influence of substituents and conditions on aggregation in the solid state. Chem. Eur. J. 25, 11121–11134 (2019).
Wagner, J. P. & Schreiner, P. R. London dispersion in molecular chemistry—reconsidering steric effects. Angew. Chem. Int. Ed. 54, 12274–12296 (2015).
Hansch, C., Leo, A. & Taft, R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195 (1991).
Charton, M. Nature of the ortho effect. II. Composition of the Taft steric parameters. J. Am. Chem. Soc. 91, 615–618 (1969).
Cavallo, G. et al. The halogen bond. Chem. Rev. 116, 2478–2601 (2016).
Garza, A. J. Solvation entropy made simple. J. Chem. Theory Comput. 15, 3204–3214 (2019).
Yang, L., Adam, C., Nichol, G. S. & Cockroft, S. L. How much do van der Waals dispersion forces contribute to molecular recognition in solution? Nat. Chem. 5, 1006–1010 (2013).
Yang, L., Adam, C. & Cockroft, S. L. Quantifying solvophobic effects in nonpolar cohesive interactions. J. Am. Chem. Soc. 137, 10084–10087 (2015).
Acknowledgements
The authors thank the European Research Council (ERC) in the frame of the consolidators grant CaTs n DOCs (grant number 725765). We also thank J. Holstein (Universität Dortmund, Germany), S. M. Elbert, M. Brückner and F. Rominger (Ruprecht-Karls-Universität Heidelberg), and D. Fenske (Karlsruhe Institute of Technology) for their efforts in obtaining single-crystal X-ray structures of cages and catenanes described here. Support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant number INST 40/5751 FUGG (JUSTUS 2 cluster) is acknowledged.
Author information
Authors and Affiliations
Contributions
B.P.B. found the catenation reaction. M.M. and B.P.B. conceived and planned the project. B.P.B. did all the experimental work. M.M. and B.P.B. analysed and interpreted the results. T.K. performed quantum chemical calculations. J.G. was responsible for the NMR experiments, especially in helping to elucidate the structure of catenanes. J.H.G. measured all the mass spectra. M.M. and B.P.B. prepared the manuscript, which was edited by all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks Tomáš Šolomek and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Information
Supplementary Data
Xyz files of computational data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benke, B.P., Kirschbaum, T., Graf, J. et al. Dimeric and trimeric catenation of giant chiral [8 + 12] imine cubes driven by weak supramolecular interactions. Nat. Chem. 15, 413–423 (2023). https://doi.org/10.1038/s41557-022-01094-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-022-01094-w
This article is cited by
-
Mechanically rigid metallopeptide nanostructures achieved by highly efficient folding
Nature Synthesis (2024)
-
Unlocking the geometry of bonding
Nature Synthesis (2024)
-
Computationally guided synthesis of a hierarchical [4[2+3]+6] porous organic ‘cage of cages’
Nature Synthesis (2024)
-
Self-similar chiral organic molecular cages
Nature Communications (2024)
-
Complexation-driven assembly of imine-linked helical receptors showing adaptive folding and temperature-dependent guest selection
Nature Communications (2024)
