
Abstract
The AS04-adjuvanted human papillomavirus (HPV)16/18 vaccine, an L1-based vaccine, provides strong vaccine efficacy (VE) against vaccine-targeted type infections, and partial cross-protection to phylogenetically-related types, which may be affected by variant-level heterogeneity. We compared VE against incident HPV31, 33, 35, and 45 detections between lineages and SNPs in the L1 region among 2846 HPV-vaccinated and 5465 HPV-unvaccinated women through 11-years of follow-up in the Costa Rica HPV Vaccine Trial. VE was lower against HPV31-lineage-B (VE=60.7%;95%CI = 23.4%,82.8%) compared to HPV31-lineage-A (VE=94.3%;95%CI = 83.7%,100.0%) (VE-ratio = 0.64;95%CI = 0.25,0.90). Differential VE was observed at several lineage-associated HPV31-L1-SNPs, including a nonsynonymous substitution at position 6372 on the FG-loop, an important neutralization domain. For HPV35, the only SNP-level difference was at position 5939 on the DE-loop, with significant VE against nucleotide-G (VE=65.0%;95%CI = 28.0,87.8) but not for more the common nucleotide-A (VE=7.4%;95%CI = −34.1,36.7). Because of the known heterogeneity in precancer/cancer risk across cross-protected HPV genotype variants by race and region, our results of differential variant-level AS04-adjuvanted HPV16/18 vaccine efficacy has global health implications.
Similar content being viewed by others
Introduction
The prophylactic AS04-adjuvanted human papillomavirus (HPV)16/18 vaccine (Cervarix®, GlaxoSmithKline Biologicals; Rixensart, Belgium) contains virus-like particles of HPV16/18 that self-assemble from pentamers of the L1 major capsid proteins, displaying epitopes that induce high titers of neutralizing antibodies1. These antibodies provide strong protection against vaccine-type infections in HPV-naïve people2,3. Some carcinogenic HPV genotypes have similarities in epitopes to vaccine-targeted genotypes, which allow for partially cross-reactive antibody responses4,5. Namely, HPV31, 33, and 35 are phylogenetically related to HPV16, while HPV45 is phylogenetically related to HPV186,7. Due to these similarities, Cervarix provides moderate cross-protection against HPV31, 33, 45, and possibly 358,9. Results from the Costa Rica HPV Vaccine Trial (CVT) demonstrate a significant combined vaccine efficacy (VE) of 64% against HPV31/33/45 two to eleven years following three doses of Cervarix and a significant VE of 23% against HPV35 among women without evidence of prevalent infection by these types at the time of vaccination8.
However, VE against cross-protected types may be influenced by viral genetic variation in lineages, sublineages, and/or single nucleotide polymorphisms (SNPs) within each type, all of which are referred to as “variants.” HPV lineages are distinct groups of evolutionarily related viral isolates within an HPV type, usually visualized as a branch on a phylogenetic tree, that differ from one another by 1%-10% of their L1 genome sequence7,10. Nested within lineages of a given type are evolutionarily related sublineages that differ from one another by 0.5%–1% of the whole genome7,10. Lineages and sublineages are defined by a unique set of stable, highly correlated nucleotide changes across the viral genome (termed lineage-associated SNPs). HPV variation can also occur as individual SNPs (variation not linked to a lineage or sublineage), and recent HPV genome sequencing studies have revealed that high-risk HPV types have tremendous genome diversity11,12,13,14,15. HPV genetic variation at each of these levels includes genetic changes in the relatively conserved L1 gene region, potentially leading to heterogeneity of the efficacy of Cervarix, an L1-based vaccine. For instance, some lineages of HPV31 and 45 have varying degrees of sensitivity to neutralization by vaccine-induced antibodies4,16, which may be attributed to one L1 residue changing the structural conformation of the capsid protein, increasing vaccine-induced antibody recognition4.
Epidemiological data from vaccine trials are invaluable to evaluate whether variant-level differences in VE may explain partial protection for specific types. We previously evaluated differences in the cross-protection of Cervarix by lineage during four years following vaccination17. Here, we expand our prior analysis by evaluating VE of Cervarix across lineages and L1-SNPs of HPV31, 33, 35, and 45 through 11 years post-vaccination in CVT.
Results
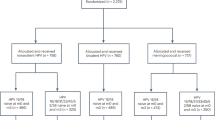
The analytical cohort included 2846 HPV-vaccinated and 5465 HPV-unvaccinated women [2909 from the HAV-vaccine arm and 2556 from the unvaccinated control group (UCG)] (Fig. 1). At the fourth-year study visit, both groups had the same median age (26 years, interquartile range = 24–28 years) (Table 1). Overall, the cumulative incidence of HPV31 detection in HPV-vaccinated women was 11.8 (95%CI = 7.4,16.1) per 1000 women compared to 59.3 (95%CI = 50.1,68.6) in HPV-unvaccinated women, corresponding to a VE of 80.2% (95%CI = 71.4%,87.4%) (Fig. 2). By lineage, vaccine protection against HPV31-lineage-B (VE = 60.7%;95%CI = 23.4%,82.8%) was lower compared to HPV31-lineage-A (VE = 94.3%;95%CI = 83.7%,100.0%) (VE ratio = 0.64;95%CI = 0.25,0.90) (Figs. 2, 3). VE against HPV31-lineage C-was 80.2% (95%CI = 68.0%,89.9%) and did not differ from lineage-A (VE ratio = 0.85; 95%CI = 0.71,1.00) or lineage-B (VE ratio = 1.32;95%CI = 0.92,3.30). VE against HPV31 detection differed for several L1-SNPs, all of which were lineage-associated (i.e., part of the known linked haplotype that defines one or more of the lineages), including positions 5921, 6238, 6367, 6372, 6772, 6796, and 6862 (Fig. 3). At all of these positions, VE was higher for nucleotides that were associated with lineages-A or A/C compared to lineage-B, or lineage-A alone compared to lineages-B/C.

This figure describes the flow of study participants included in the analysis from both the randomized trial phase of the Costa Rica HPV Vaccine Trial and the observational long-term follow-up phase through 11 years of follow up. The final analytical sample for the HPV vaccinated group and HPV-unvaccinated group for the full 11-year period is shown in the bottom most boxes.

This figure shows the HPV vaccine efficacy, indicated by the “X” symbol, against incident HPV31 detections overall and by lineage and SNP, and the corresponding 95% CIs. The bolded estimates indicate statistical significance. For each SNP, both nucleotides are shown. The analytical cohort includes all women who received all three doses of either Cervarix or Havrix, and all women in the unvaccinated control group who had at least one HPV DNA test result after two years post-enrollment and who did not have an outcome of interest during the initial two-year period. CI confidence interval, HPV human papillomavirus, SNP single nucleotide polymorphism.

This figure shows the HPV vaccine efficacy ratios, indicated by the circle symbols, against incident HPV31 detections by lineage and SNP, and the corresponding 95% CIs. The bolded estimates indicate statistical significance. For each SNP, both nucleotides are shown. The closed circles represent the vaccine efficacy ratio comparing vaccine efficacy against a specific variant compared to the vaccine efficacy against the reference group (open circles). The analytical cohort includes all women who received all three doses of either Cervarix or Havrix, and all women in the unvaccinated control group, who had at least one HPV DNA test result after two years post-enrollment and who did not have an outcome of interest during the initial two-year period. CI confidence interval, HPV human papillomavirus; SNP single nucleotide polymorphism.
HPV31 L1-SNP position 6372 was nonsynonymous and resulted in an amino acid change from Threonine (Thr) to asparagine (Asn) at position 274 (T > N) (Table 2). Our superimposed homology models illustrate that this position is located on the FG neutralization loop (Fig. 4). Both Thr and Asn residues are polar but not charged and can form hydrogen bonds, but their side chains differ, as they belong to different amino acid functional groups (hydroxyl versus amide).

This figure shows the superimposed homology models of HPV31 L1, which has an amino acid change at position 274 from Thr (threonine) to Asn (asparagine), and presents the vaccine efficacy estimates for each amino acid with the vaccine efficacy ratio between the two. The tan color on the figure represents threonine, while the blue color represents asparagine. CI confidence interval, HPV human papillomavirus.
The overall cumulative incidence of incident HPV33 and 35 detection was similar between women in the HPV-vaccine arm and HPV-unvaccinated group, resulting in a lack of significant VE (Fig. 5). The lineage/sublineage-stratified analyses for HPV33 and 35 were underpowered due to small case counts of HPV33-lineage-B infections and HPV35-sublineage-A2 infections, irrespective of vaccination group. While no differential VE was observed for HPV35 by sublineage, differences in VE were detected at L1-SNP position 5939 (VE-ratio = 0.11;95%CI = -0.60,0.73) (Fig. 6), a synonymous substitution at amino acid position 113 located on the DE loop (Table 2). The G nucleotide of SNP 5939 had a significant VE of 65.0% (95%CI = 28.0%,87.8%), while VE was not significant for the more common A nucleotide (VE = 7.4%;95%CI = −34.1%,36.7%) (Fig. 5). This is an independent SNP that is not associated with a sublineage. For HPV45, high VE was observed against incident detection overall (VE = 84.7%;95%CI = 76.0,91.6) (Fig. 5) and remained similarly high for lineages-A and B (Fig. 6).

This figure shows the HPV vaccine efficacy, indicated by the “X” symbol, against incident HPV33, 35, and 45 detections overall and by variant, and the corresponding 95% CIs. The bolded estimates indicate statistical significance. The analytical cohort includes all women who received all three doses of either Cervarix or Havrix, and all women in the unvaccinated control group, who had at least one HPV DNA test result after two years post-enrollment and who did not have an outcome of interest during the initial two-year period. CI confidence interval, HPV human papillomavirus; SNP single nucleotide polymorphism.

This figure shows the HPV vaccine efficacy ratios, indicated by the circle symbols, against incident HPV33, 35, and 45 detections by variant, and the corresponding 95% CIs. The bolded estimate indicates statistical significance. The closed circles represent the vaccine efficacy ratio comparing vaccine efficacy against a specific variant compared to the vaccine efficacy against the reference group (open circles). The analytical cohort includes all women who received all three doses of either Cervarix or Havrix, and all women in the unvaccinated control group, who had at least one HPV DNA test result after two years post-enrollment and who did not have an outcome of interest during the initial two-year period. CI confidence interval, HPV Human papillomavirus; SNP single nucleotide polymorphism.
Discussion
In this analysis of long-term variant-level cross-protection of the AS04-adjuvanted bivalent HPV vaccine, we discovered that overall cross-protection against incident HPV31 detection is weighted by the very high VE against lineage-A (94%) and partial protection against lineage-B (60%) and lineage-C (80%). We identified a potentially important HPV31 SNP at position 6372, located within the FG loop of L1, which is an essential neutralization domain18,19,20. We also observed differential VE against HPV35 at SNP position 5939, which is located within another neutralization region although, this substitution was synonymous and its potential function is unclear. Lastly, we confirmed VE against HPV45 was high ( > 80%) and did not significantly differ across lineages.
The significant non-synonymous substitution we identified within the HPV31 L1 region likely resulted in changes in epitopes recognized by the vaccine-induced antibodies, leading to variant-level differences in protection; however, we did not conduct analyses of in vitro neutralization of variant-specific pseudoviruses to confirm that the amino acid change was the exact mechanism for differential VE. We observed that the Thr residue at position 274, which is associated with HPV31-lineage-A, had significantly higher VE compared to the Asn residue, which was present only in HPV31-lineages-B/C. A study noted that although the change at position 274 from Thr to Asn is relatively subtle, it still resulted in small local structural changes that could contribute to differences in distal epitope recognition because of its location on the tip of the L1 FG loop4. Our observation of significantly different VE between variants at this position suggests that this particular nonsynonymous substitution may partially explain heterogeneity in cross-protection against HPV31 infections by lineage.
Our discovery of variant-level differences in VE is clinically important because of heterogeneity in risk of precancer/cancer reported for cross-protected HPV genotype lineages/sublineages11,15,21,22. Both HPV31-lineages-A and B are associated with increased precancer/cancer risk compared to lineage-C15, thus, our observation of high VE against HPV31-lineage-A is promising but, lower VE against HPV31-lineage-B is concerning.
For HPV35, prior studies have shown that cervical precancer/cancer risk may differ via interactions of sublineage and race, with A1 associated with higher risk of CIN2+ among White women, and A2 being more prevalent and associated with CIN3+ among African-American women11,23. Our HPV35 sublineage-stratified analysis was limited by the low prevalence of one of the two sublineages in Costa Rica. However, we note at the SNP-level, VE differed at HPV35 L1-SNP position 5939, with significantly moderate VE against nucleotide-G but, a lack of significant VE against the more common nucleotide-A. While this observation would have important implications due to the possibility of HPV35-caused cervical cancer in world regions with high cervical cancer burden, this position was synonymous, meaning the biological mechanism for how it impacted VE is unclear. In a previous large characterization of HPV35 variation, we observed that nucleotide-A at HPV35 position 5939 (for which we observed no significant VE) was present in all women with an A2 sublineage infection and variably present in women with an A1 sublineage infection11,23, emphasizing the high prevalence of this nucleotide among people with HPV35 infections. Therefore, if differential variant-level VE against HPV35 is true, it poses important implications for global HPV vaccine implementation, particularly for Sub-Saharan African countries, where HPV35 may cause up to 10% of cervical cancers24, and where much of the cervical cancer burden resides25.
Given the long-term follow-up of our study, our results inherently demonstrate that cross-protection afforded by the AS04-adjuvanted bivalent HPV vaccine persists for at least 11 years, particularly for HPV31 and 45 infections. This conclusion supports our recent evaluation of the durability of cross-protection of Cervarix, which showed no evidence of waning cross-protection over 11 years8. These observations differ from prior studies that have suggested decreasing VE against HPV31 and 45 infections over time26.
Our study was limited by the low prevalence of HPV33-lineage-B and HPV35-sublineage-A2 in Costa Rica, resulting in a lack of power to detect differences in VE between HPV33 and 35 lineages/sublineages. We only included women with three doses because the one-dose group was limited in sample size and variant-level outcomes were not available for this group; however, one-dose cross-protection against some HPV genotypes has been observed for Cervarix, with strong protection durability over time, which provides positive global health implications8. Another limitation is that the SNP-level findings were predetermined in the discovery phase due to restricting to L1-SNPs with different allele frequencies by vaccination status. In this work, we took an exploratory approach and did not adjust for multiple comparisons; this investigation should be replicated in independent datasets, especially in world regions with higher prevalence of HPV33 and 35. An important consideration is that our outcomes of interests were incident, HPV31, 33, 35, and 45 detections, rather than persistence. We calculated cumulative incidence over 11-years of follow-up, which is a different metric than what is presented in global reports of HPV genotype distributions which report prevalence estimates. The majority (84%) of the Costa Rican population is White or Mestizo; thus, our results may not be generalizable to populations with more racial and ethnic diversity.
While our findings are robust and valid, VE estimates may vary depending on the prevalence of HPV genotypes and variants in the population, as the distribution of HPV genotypes and their variants differ by region, race, and ethnicity11,15. Specifically, the global prevalence of HPV31, 33, 35, and 45 infections ranges from 0.5%-1.2% in women with normal cytology27 and 1.0%–1.8% in men28, depending on the HPV genotype. In Costa Rica, the prevalence of HPV31, 33, 35, and 45 infections ranges from 0.5%-1.1% in women with normal cytology, which is comparable to the global prevalence27. Among women with normal cytology, the highest HPV prevalence is in Oceania (22%) and Africa (21%)29, but the distribution of HPV lineages/sublineages varies by world region15,29. Additionally, as noted above, HPV genotype distributions and their variants vary by race and ethnicity, such as HPV35 being more common among women with African ancestry11,15.
This is the most robust analysis of HPV VE by cross-protected genotype variants with the longest follow-up of HPV-vaccinated women. We used a longer exclusion period for event-counting (2 years) to reduce the potential for misclassifying prevalent infections present at enrollment. We comprehensively utilized viral whole-genome sequencing, instead of HPV targeted sequencing of the URR/E6 regions like in our prior analysis17, enabling precise classification of lineages/sublineages and allowing us to evaluate associations with individual nucleotide variants in the L1 sequence. We also focused solely on L1-SNPs to reduce the possibility of false positive chance findings, as the L1 region is highly relevant to analyses of the AS04-adjuvanted bivalent HPV vaccine, an L1-based vaccine. Although variations in other regions of the HPV genome, such as L2, can also affect endpoints, these variations would presumably apply to infections in both vaccinated and unvaccinated groups (i.e., non-differential). Because the HPV vaccine is composed on the L1 protein alone, variations in the L1 gene region might directly affect vaccine protection by impacting the sterilizing immunity induced by vaccination.
Our observations would not be applicable for multivalent HPV vaccines that directly target these cross-protected HPV genotypes and for higher valency HPV vaccines that use the AS04-adjuvant, which is the proposed mechanism that provides cross-protection of phylogenetically related types. Currently, a candidate adjuvanted nonavalent HPV vaccine targeting HPV6/11/16/18/31/33/45/52/58 is being evaluated30, which might offer even greater cross-protection than the AS04-adjuvanted bivalent HPV vaccine given the inclusion of more HPV types in the nonavalent vaccine that may offer increased opportunity for similarities with more phylogenetically related HPV types. However, many countries have used the bivalent HPV vaccine for many years and continue to use this vaccine; thus, our observations are still relevant for many birth cohorts with bivalent HPV vaccination. Importantly, the phenomenon regarding differential protection afforded by the AS04-adjuvanted bivalent HPV vaccine against variants of HPV31 and 35 is interesting and warrants further investigation, particularly in other vaccine clinical trials for external validation.
Methods
CVT study design
CVT is a double-blinded, randomized phase III clinical trial aimed to evaluate the efficacy of Cervarix against HPV infections and HPV-associated neoplasia31. During 2004–2005, before Cervarix licensure, 7466 Costa Rican women aged 18–25 years who resided in Guanacaste and Puntarenas provinces were enrolled and randomized (1:1) to receive three doses of either Cervarix or the control hepatitis A virus (HAV) Havrix® vaccine, administered intramuscularly in 0.5 mL doses at 0, 1, and 6 months. Randomization was conducted using a blocked randomization method with permuted block sizes of 14, 16, and 18. After four years, participants in the HPV-vaccine arm were invited to enroll in the observational long-term follow-up phase, extending follow-up through 11 years32. Women in the HAV-vaccine arm were offered the HPV vaccine before exiting the study and a new screening-only cohort was recruited and enrolled, including HPV-unvaccinated women from similar birth cohorts and geographical regions as women in the original control arm. The new control group is referred to as the “unvaccinated control group” (UCG). These studies are registered with ClinicalTrials.gov, NCT00128661 and NCT00867464. All participants provided written, informed consent. All research activity for CVT was approved by Institutional Review Boards of Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud in Costa Rica and the US National Cancer Institute (Bethesda, MD, USA).
Sample collection
During the randomized trial phase, serum samples were collected at enrollment and at annual follow-up visits. For sexually experienced women, cervical samples were collected using a Cervex-Brush (Rovers Medical Devices BV, Oss, Netherlands) and rinsed in PreservCyt solution (Hologic, Marlborough, MA, USA) for cytology and HPV DNA testing. During the observational long-term follow-up phase (LTFU), participants in both the HPV-vaccine arm and unvaccinated control group (UCG) were seen biennially, and cervical samples were collected at each of these routine clinic visits. In both the trial phase and the LTFU phase, women with low-grade cervical abnormalities were seen every six months, while women with evidence of high-grade cervical abnormalities were referred to colposcopy for evaluation and treatment, as needed.
Outcomes
The primary outcomes were incident detection of HPV31, 33, 35, and/or 45 in cervical exfoliated cells. Each HPV-type was treated as an independent analysis; women who were infected with multiple cross-protected HPV types could contribute to each analysis. Lineage and SNPs were only determined at a single point (the first time at which infection with the corresponding genotype was detected with a valid lineage assignment). During quality control, 37% of infections were excluded due to sequencing failure, insufficient coverage or poor-quality reads across the HPV genome, or a within-type lineage coinfection that could not be resolved or had an ambiguous position in the phylogenetic tree. Before the main analyses, we conducted analyses to determine whether selection bias was introduced, including comparisons of: the proportion of samples with sequencing results by HPV genotype across vaccination status (Supplementary Table 1); woman-level characteristics by sequencing result status (Supplementary Table 2); and the distribution of HPV positivity by vaccination status across sequencing result status (Supplementary Table 3). These metrics confirmed that our analytical samples were not substantially different from excluded samples, thus, our results are assumed to be generalizable to the full study population.
Analytical cohort
We included women with at least one HPV DNA test result after two years post-enrollment who did not have an HPV type-specific outcome of interest during the initial two-year period to minimize misclassification of infections present at the time of recruitment that were not detected by our HPV genotyping assay. The two-year period was used because most (80%-90%) HPV infections spontaneously clear within two years33. The analytical cohort for the randomized trial phase (years 1–4) included women who received three doses of Cervarix (HPV-vaccine arm), and three doses of Havrix (HAV-vaccine arm); women who were free of type-specific infections in the two years following recruitment were genotyped at years 3 and 4. The analytical cohort for the observational LTFU phase (years 4-11) included women who received three doses of Cervarix during the randomized trial phase (HPV-vaccine arm) and women UCG; women who were free of type-specific infections during two years following their enrollment into the long-term follow-up phase (i.e., event-free during years 4-7) were genotyped at years 9 and 11.
HPV DNA detection and genotyping
During the trial phase, DNA was extracted from cervical samples and tested for HPV DNA detection and genotyped using the SPF10 polymerase chain reaction primer system and DNA enzyme immunoassay detection of amplimers (DEIA) system (DDL Diagnostic Laboratory, Delft, Netherlands). Specifically, primers were used to amplify a fragment from the L1 region of HPV genotypes, after which the DEIA system detected the amplified products. All DEIA-positive SPF10 amplimers were used to identify HPV genotype by reverse hybridization with the LiPA25 HPV line probe assay (Labo Bio-medical Products, Rijswijk, Netherlands). During the long-term follow-up, HPV DNA detection was replaced with TypeSeq (National Cancer Institute Cancer Genomics Research Laboratory, Frederick, MD, USA) and genotyping was done using Ion S5 next-generation sequencing, followed by a custom Torrent Suite plugin analysis (Thermo Fisher Scientific, Waltham, MA, USA). A prior comparison of LiPA25 and TypeSeq found that the percent agreement between both genotyping methods was high for the four HPV types (HPV 31/33/35/45) in our study (total agreement range = 99.5%-99.8%; positive agreement range = 88.2%–93.3%)34. More detailed HPV DNA detection and genotyping methods are presented elsewhere31,32.
HPV viral whole-genome sequencing
The first positive cervical sample of HPV31, 33, 35, and 45 for each woman was HPV whole-genome sequenced using custom Thermo Fisher Ion Torrent AmpliSeq HPV type-specific panels to amplify the entire genome, as previously described35. Briefly, custom overlapping degenerate primers were created to cover the entire viral genome of HPV31, 33, 35, or 45. After amplification, an Ion Torrent adapter-ligated library was generated following the manufacturer’s Ion AmpliSeq Library Preparation kit 2.0-96LV protocol with slight modifications (Life Technologies, Part #4480441). Raw sequencing reads were quality and adaptor trimmed and aligned to the HPV type-specific reference sequence from PAVE36 using the Torrent Mapping Alignment Program v5.0.1337. Individual nucleotide variants of each HPV type were called using the Torrent Variant Caller v.5.0.3 and annotated using snpEff v.3.6 c38.
Classification of HPV variants
A consensus whole-genome sequence was created for each sample and HPV type and combined with type-specific lineage reference sequences from GenBank39. Phylogenetic trees were constructed using RAxML MPI v7.2.8.2740 and MEGAX41 with 1000 bootstraps. Lineages were assigned based on the tree topology and sample proximity to the reference lineage genomes and confirmed with individual SNP patterns. For each HPV type, lineages were categorized as: HPV31-lineage-A, B, or C; HPV33-lineage-A or B; HPV35-sublineage-A1 or A2; and HPV45-lineage-A or B. Notably, HPV35 consists predominately of one main lineage-A that is divided into two sublineages, A1 and A27, but there is also a B lineage found in Asia11, but is not observed in our study population. For each HPV type, individual SNPs or variable positions within the L1 region were evaluated if they were observed in at least two samples. For each HPV type, the L1 region boundaries are presented in Supplementary Table 4. For L1-SNPs, we compared allele frequencies at each nucleotide position between HPV-vaccinated women and HPV-unvaccinated women using the most common nucleotide at each position as the referent group (Supplementary Table 5).
Statistical analysis
Balanced baseline characteristics of women in the HPV-vaccine arm and HAV-vaccine arm have been demonstrated31. When the UCG was enrolled (the fourth year study visit of CVT), characteristics of the original control group were compared with UCG32. In the present study, among women who met our inclusion criteria, we described characteristics at the year four study visit (month 48 visit for the HPV-vaccine arm and HAV-vaccine arm and the baseline visit for UCG) by all vaccination status groups, including the HPV-vaccine arm, combined HPV-unvaccinated group, HAV-vaccine arm alone, and UCG alone.
For each HPV type irrespective of variant and stratified by variant, we reported the cumulative incidence over 11 years of follow-up, VE [100 x (1-relative risk)], and asymptotic 95% confidence intervals (95%CIs). We calculated the proportions of women in the randomized trial phase who were not infected through year 2 but tested positive for a specific genotype later, namely,
and
For the LTFU phase, we calculated
and
We calculated relative risks for the trial phase, pTrialHPV-vaccine arm / pTrialHAV-vaccine arm, and for the LTFU phase, pLTFUHPV-vaccine arm / pLTFUUCG. To get a combined probability of infection for the control groups over the full 11-year follow-up period, we calculated pcontrol = pTrialHAV-vaccine arm + (1-pTrialHAV-vaccine arm) x pLTFUUCG. Likewise, pHPV-vaccine arm = pTrialHAV-vaccine arm + (1-pTrialHAV-vaccine arm) x pLTFUHAV-vaccine arm. The combined relative risk was calculated by pHPV-vaccine arm / pcontrol. We resampled women with replacement to obtain bootstrap distributions of vaccine efficacy and vaccine efficacy ratio estimates42.
We identified ten individual L1-SNPs that were significantly associated with vaccination status (nine for HPV31 and one for HPV35) (Supplementary Table 5), for which VE was calculated stratified by nucleotide (e.g., nucleotide-T and nucleotide-C at SNP position 6862). To evaluate whether VE differed across variants, we calculated VE ratios between groups (e.g., HPV31-lineage-B compared to HPV31-lineage-A, or nucleotide-T compared to nucleotide-C at SNP position 6862). Statistical significance was determined if the VE 95%CI did not include zero, and VE ratio 95%CI did not include 1.0. We translated and annotated each DNA change (SNP) observed in L1 and mapped them to L1 loops to determine whether the SNP was located within a neutralization domain. Three-dimensional homology models for the only non-synonymous L1-SNP position 6372 (amino acid position 274) were created using the SWISS-MODEL (https://swissmodel.expasy.org/) to visualize potential structural changes related to this amino acid change.
Recognizing that the analysis includes different control groups over two study periods, we conducted a sensitivity analysis to compare VE between study phases to justify combining periods for a cumulative VE across the full 11 years. None of the VEs were significantly different between time points (all variants p-heterogeneity > 0.05, Supplementary Table 6) and follow-up times were similar during each study phase (Supplementary Table 7).
Ethics and inclusion
Local researchers from Costa Rica were involved throughout the entire research process, including study design, study implementation, data ownership, data analysis, and manuscript preparation. The research is locally relevant to women in Costa Rica. Authorship roles and responsibilities were agreed upon prior to the conduct of this analysis. The project and resulting manuscript were reviewed by the Costa Rica HPV Vaccine Trial group.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Participant data can be shared with outside collaborators for research to understand more about the performance of the HPV vaccine, immune response to the vaccine, and broader study factors associated with the natural history of HPV infection and risk factors for infection and disease. Outside collaborators can apply to access our protocols and data from the blinded phase of the Costa Rica Vaccine Trial (NCT00128661). All of the L1 nucleotide sequences used in our study have been deposited in GenBank (accession numbers PP791977 - PP792123, PP792124 - PP792208, PP792209 - PP792340, and PP792341 - PP792520). A trial summary, current publications, and contact information are available online at: https://dceg.cancer.gov/research/who-we-study/cohorts/costa-rica-vaccine-trial.
References
Schiller, J. & Lowy, D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine 36, 4768–4773 (2018).
Hildesheim, A. et al. Efficacy of the HPV-16/18 Vaccine: Final according to protocol results from the blinded phase of the randomized Costa Rica HPV-16/18 Vaccine Trial. Vaccine 32, 5087–5097 (2014).
Paavonen, J. et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet Lond. Engl. 374, 301–314 (2009).
Bissett, S. L. et al. Naturally Occurring Capsid Protein Variants of Human Papillomavirus Genotype 31 Represent a Single L1 Serotype. J. Virol. https://doi.org/10.1128/JVI.00842-15 (2015).
Carter, J. J. et al. Identification of human papillomavirus type 16 L1 surface loops required for neutralization by human sera. J. Virol. 80, 4664–4672 (2006).
Chen, Z. et al. Evolution and Taxonomic Classification of Human Papillomavirus 16 (HPV16)-Related Variant Genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLOS ONE 6, e20183 (2011).
Burk, R. D., Harari, A. & Chen, Z. Human papillomavirus genome variants. Virology 445, 232–243 (2013).
Tsang, S. H. et al. Durability of Cross-Protection by Different Schedules of the Bivalent HPV Vaccine: The CVT Trial. JNCI J. Natl Cancer Inst. 112, 1030–1037 (2020).
Wheeler, C. M. et al. Cross-protective efficacy of HPV-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by non-vaccine oncogenic HPV types: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 13, 100–110 (2012).
Nelson, C. W. & Mirabello, L. Human papillomavirus genomics: Understanding carcinogenicity. Tumour Virus Res. 15, 200258 (2023).
Pinheiro, M. et al. Association of HPV35 with cervical carcinogenesis among women of African ancestry: Evidence of viral-host interaction with implications for disease intervention. Int. J. Cancer 147, 2677–2686 (2020).
Mirabello, L. et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 170, 1164–1174.e6 (2017).
van der Weele, P., Meijer, C. J. L. M. & King, A. J. Whole-Genome Sequencing and Variant Analysis of Human Papillomavirus 16 Infections. J. Virol. 91, e00844–17 (2017).
van der Weele, P., Meijer, C. J. L. M. & King, A. J. High Whole-Genome Sequence Diversity of Human Papillomavirus Type 18 Isolates. Viruses 10, 68 (2018).
Pinheiro, M. et al. Phylogenomic Analysis of Human Papillomavirus Type 31 and Cervical Carcinogenesis: A Study of 2093 Viral Genomes. Viruses 13, 1948 (2021).
Godi, A. et al. Naturally Occurring Major and Minor Capsid Protein Variants of Human Papillomavirus 45 (HPV45): Differential Recognition by Cross-Neutralizing Antibodies Generated by HPV Vaccines. J. Virol. https://doi.org/10.1128/JVI.02859-15 (2015).
Harari, A. et al. Cross-protection of the Bivalent Human Papillomavirus (HPV) Vaccine Against Variants of Genetically Related High-Risk HPV Infections. J. Infect. Dis. 213, 939–947 (2016).
Dehghani, B., Hasanshahi, Z., Hashempour, T. & Motamedifar, M. The possible regions to design Human Papilloma Viruses vaccine in Iranian L1 protein. Biol. (Bratisl.) 75, 749–759 (2020).
Bissett, S. L., Godi, A. & Beddows, S. The DE and FG loops of the HPV major capsid protein contribute to the epitopes of vaccine-induced cross-neutralising antibodies. Sci. Rep. 6, 39730 (2016).
Carter, J. J., Wipf, G. C., Benki, S. F., Christensen, N. D. & Galloway, D. A. Identification of a human papillomavirus type 16-specific epitope on the C-terminal arm of the major capsid protein L1. J. Virol. 77, 11625–11632 (2003).
Schiffman, M. et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 70, 3159–3169 (2010).
Xi, L. F. et al. Association of human papillomavirus type 31 variants with risk of cervical intraepithelial neoplasia grades 2-3. Int. J. Cancer 131, 2300–2307 (2012).
Williams, E. A. et al. Prevalence of High-Risk Nonvaccine Human Papillomavirus Types in Advanced Squamous Cell Carcinoma Among Individuals of African vs Non-African Ancestry. JAMA Netw. Open 4, e216481 (2021).
Denny, L. et al. Human papillomavirus prevalence and type distribution in invasive cervical cancer in sub-Saharan Africa. Int. J. Cancer 134, 1389–1398 (2014).
Singh, D. et al. Global estimates of incidence and mortality of cervical cancer in 2020: a baseline analysis of the WHO Global Cervical Cancer Elimination Initiative. Lancet Glob. Health 11, e197–e206 (2023).
Malagón, T. et al. Cross-protective efficacy of two human papillomavirus vaccines: a systematic review and meta-analysis. Lancet Infect. Dis. 12, 781–789 (2012).
ICO/IARC Information Centre on HPV and Cancer. Costa Rica Human Papillomavirus and Related Cancers, Fact Sheet 2023. https://hpvcentre.net/statistics/reports/CRI_FS.pdf (2023).
Bruni, L. et al. Global and regional estimates of genital human papillomavirus prevalence among men: a systematic review and meta-analysis. Lancet Glob. Health 11, e1345–e1362 (2023).
Kombe Kombe, A. J. Epidemiology and burden of human papillomavirus and related diseases, molecular pathogenesis, and vaccine evaluation. Front. Public Health 8, 552028 (2020).
GlaxoSmithKline. A Phase 1/2 Randomized, Observer-Blinded, Multi-Country Study to Evaluate Safety and Immunogenicity of Investigational Adjuvanted Human Papillomavirus Vaccine in Females (16 to 26 Years of Age). https://clinicaltrials.gov/study/NCT05496231 (2023).
Herrero, R. et al. Rationale and design of a community-based double-blind randomized clinical trial of an HPV 16 and 18 vaccine in Guanacaste, Costa Rica. Vaccine 26, 4795–4808 (2008).
Gonzalez, P. et al. Rationale and design of a Long Term Follow-up study of women who did and did not receive HPV 16/18 vaccination in Guanacaste, Costa Rica. Vaccine 33, 2141–2151 (2015).
Adebamowo, S. N. et al. Different human papillomavirus types share early natural history transitions in immunocompetent women. Int. J. Cancer 151, 920–929 (2022).
Wagner, S. et al. Evaluation of TypeSeq, a Novel High-Throughput, Low-Cost, Next-Generation Sequencing-Based Assay for Detection of 51 Human Papillomavirus Genotypes. J. Infect. Dis. 220, 1609–1619 (2019).
Cullen, M. et al. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res. 1, 3–11 (2015).
Van Doorslaer, K. et al. The Papillomavirus Episteme: a central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 41, D571–D578 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6, 80–92 (2012).
National Institute of Allergy and Infectious Diseases. PaVE: The Papillomavirus Episteme. https://pave.niaid.nih.gov/.
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Efron, B. The Jackknife, the Bootstrap and Other Resampling Plans. (Society for Industrial and Applied Mathematics, 1982). https://doi.org/10.1137/1.9781611970319.
Acknowledgements
This work was supported by the Intramural Research Program of the National Cancer Institute at the US National Institutes of Health. The Costa Rica HPV Vaccine Trial is a long-standing collaboration between investigators in Costa Rica and the US National Cancer Institute (NCI). The trial is sponsored and funded by the NCI (contract N01-CP-11005) with funding support from the National Institutes of Health Office of Research on Women’s Health. GlaxoSmithKline Biologicals provided vaccine and support for aspects of the trial associated with regulatory submission needs of the company under a Clinical Trials Agreement (FDA BB-IND 7920) during the four-year, randomised blinded phase of the study, but had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. The NCI and Costa Rica investigators are responsible for the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation of the manuscript. All authors have access to the raw data. We extend a special thanks to the women of Guanacaste and Puntarenas, Costa Rica, who gave of themselves in participating in this effort. In Costa Rica, we acknowledge the tremendous effort and dedication of the staff involved in this project; we would like to specifically acknowledge the meaningful contributions by Carlos Avila, Loretto Carvajal, Cristian Montero, and Diego Guillen. In the United States, we extend our appreciation to the team from Information Management Services (IMS) responsible for the development and maintenance of the data system used in the trial and who serve as the data management center for this effort, especially Jean Cyr, Julie Buckland, and John Schussler. We thank Dr. Diane Solomon (CVT: medical monitor & QC pathologist) for her invaluable contributions during the randomized blinded phase of the trial and the design of the LTFU and Nora Macklin (CVT) and Kate Torres (LTFU) for the expertise in coordinating the study. We thank the members of the Data and Safety Monitoring Board charged with protecting the safety and interest of participants during the randomized, blinded phase of our study (Steve Self, Chair, Adriana Benavides, Luis Diego Calzada, Ruth Karron, Ritu Nayar, and Nancy Roach) and members of the external Scientific HPV Working Group who have contributed to the success of our efforts over the years (Henriette Raventós, Chair, Diane Davey, Gypsyamber D’Souza, Anne Gershon, Sylvia Lara, Wasima Rida, Maria del Rocio Saenz, Richard Roden, and Margaret Stanley).
Funding
Open access funding provided by the National Institutes of Health.
Author information
Authors and Affiliations
Consortia
Contributions
J.Z.S.: Conceptualization, Formal analysis, Visualization, Writing – original draft; C.P.: Resources, Investigation, Project administration, Writing – review & editing; M.P.: Data curation, formal analysis, Writing – review & editing; R.H.: Resources, Project administration, Writing – review & editing; A.H.: Conceptualization, Methodology, Validation, Writing – review & editing; D.L.: Methodology, Writing – review & editing; M.H.G.: Methodology, Writing – review & editing; B.R.: Investigation, Project administration, Writing – review & editing; J.T.S.: Validation, Writing – review & editing; M.Z.: Investigation, Project administration, Writing – review & editing; S.M.: Data curation, Investigation; L.B.: Data curation, Investigation; K.J.: Data curation, Investigation; J.S.: Formal analysis, Methodology, Software, Writing – review & editing; R.O.: Investigation, Project administration, Writing – review & editing; J.F.: Visualization, Software, Writing – review & editing; Z.L.: Software, Writing – review & editing; D.R.L.: Validation, Writing – review & editing; S.H.T.: Conceptualization, Writing – review & editing; A.C.R.: Investigation; MS: Writing – review & editing; C.B.H.: Visualization, Writing – review & editing, L.J.C.: Writing – review & editing; J.B.: Visualization, Writing – review & editing; A.R.K.: Conceptualization, Formal analysis, Resources, Supervision, Validation, Writing – review & editing; L.M.: Conceptualization, Data curation, Formal analysis, Resources, Validation, Writing – review & editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Maísa Pinheiro is currently an employee of GlaxoSmithKline and Sabrina H. Tsang is currently an employee of Merck Sharp & Dohme Corp, a subsidiary of Merck & Co, Inc (Kenilworth, New Jersey), but they both completed all work associated with this project while employed at the National Cancer Institute. John T. Schiller and Douglas R. Lowy report that they are named inventors on US Government owned HPV vaccine patents with expired licenses to GlaxoSmithKline and Merck. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shing, J.Z., Porras, C., Pinheiro, M. et al. Differential long-term bivalent HPV vaccine cross-protection by variants in the Costa Rica HPV vaccine trial. npj Vaccines 9, 101 (2024). https://doi.org/10.1038/s41541-024-00896-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-024-00896-y
