
Abstract
Successful drug discovery is ultimately contingent on the availability of workable, relevant, predictive model systems. Conversely, for cardiac muscle, the lack of human preclinical models to inform target validation and compound development has likely contributed to the perennial problem of clinical trial failures, despite encouraging non-human results. By contrast, human cardiomyocytes produced from pluripotent stem cell models have recently been applied to safety pharmacology, phenotypic screening, target validation and high-throughput assays, facilitating cardiac drug discovery. Here, we review the impact of human pluripotent stem cell models in cardiac drug discovery, discussing the range of applications, readouts, and disease models employed, along with the challenges and prospects to advance this fruitful mode of research further.
Similar content being viewed by others


The unmet need: resuscitating cardiac drug discovery
Notwithstanding decades of aggressive risk factor reduction, as well as transforming health care delivery systems to restore coronary flow urgently in myocardial infarction, the prevalence of heart disease remains predominant—wholly unchanged in rank as the single leading cause of death and disability for both men and women worldwide1,2. Ischemic heart disease remains, within this, the preponderant form1,2. From 2007 through 2017, the age-adjusted death rates from ischemic heart disease fell 9.7%, which is gratifying progress, yet to focus on this one metric would be quite misleading. Indeed, during this same period, the actual number of deaths from ischemic heart disease increased 22.3%, and years of life lost increased to the same degree2. Similar concerns arise from the adverse trends in “disability-adjusted life-years,” a measure of healthy life expectancy1. In short, progress in allaying ischemic heart disease is stymied, at the epidemiological level, and the burden—to patients, their families, care-givers, and health care systems—remains stupefying. The pandemic continues, unabated.
In this context, far from subsiding, the need remains paramount for novel, transformative therapies to mitigate heart muscle cell death and dysfunction. However, as measured by the number of new cardiac drugs that enter clinical practice, making innovation workable has shown dismaying lack of success3,4. From 2011 to 2019, the US Food and Drug Administration approved nearly 90 novel drugs for cancer but, astoundingly, a mere four that target cardiac muscle directly (Fig. 1): sacubitril/valsartan for heart failure5, ivabradine for heart failure6,7, deferiprone for transfusional iron overload in thalassemia8, and tafamidis meglumine for cardiomyopathy in transthyretin amyloidosis9. (Additional drugs approved for broader cardiac indications were those for which cardiac muscle is neither the mechanistic focus of disease nor the site of the corresponding benefit, such as anti-platelet agents, Factor Xa inhibitors, and anti-hyperlipidemics.)

Shown, by year, are the cumulative US Food and Drug Administration NME approvals for cancer chemotherapy (consistently, 20–25% of the total approvals each year), compared with the paucity of new drugs targeting cardiac muscle110. See text for details.
Many drugs fail to reach approval for reasons of efficacy, and in the realm of cardioprotection—seeking to rescue jeopardized human heart muscle beyond the benefits of reperfusion alone—such failures have been rife. Time and again, proposed counter-measures, some with highly credible support from whole-animal studies of myocardial infarction, failed to show the anticipated efficacy10,11,12,13,14,15. But, there is a disturbing paucity even of new cardiac drug candidates put forward into early phase development, i.e., the number of New Molecular Entity (NME) applications seeking to initiate human trials3. In short, both ends of the cardiac drug pipeline are dry.
What are the barriers to trying more often, more effectively? Which, if any, might be remediable? Among the myriad obstacles most cited are: the drug development costs to bring a new agent to market; regulatory uncertainties; discrepancies in philanthropy and research grant support; the lack of appealing biological targets; commercial viability; the length, size, and complexity of trials required; risks of reliance on surrogate endpoints; and, of course, poor return on investment, given the frequency and cost of clinical failures3,16. The net result is, consequently, few novel “first-in-class” therapies. Expert think tank recommendations have focused on the trials eco-system (reducing operating costs, streamlining trial design, embracing adaptive design, funding early-phase development more assiduously) but, appropriately, also call for strengthening “novel scientific methods to further define the pathophysiology”3.
One key driver of failure in human cardiac trials is, likely, the recurring lack of systematic human preclinical data for target validation and compound development, i.e., a gap in the information available to de-risk the proposition before ever entering human trials. Model organisms, ranging from the traditional to bespoke genetic lines, are instructive, to be sure, but have, in the aggregate, failed thus far to show sufficient predictive power for efficient translation of targets and drugs to benefit human health17. The abysmal track record for cardiac drug development speaks for itself. Drawing again on oncology as an instructive comparison, cancer drugs enter human trials having first, at a very early stage, been vetted in dozens to hundreds of well-characterized human cancer cell lines, available as turnkey resources in laboratories worldwide17,18,19. Highlighted by the National Cancer Institute’s pioneering 60 human tumor cell line screen20 and by later, larger initiatives including the Cancer Cell Line Encyclopedia21,22, the role played by human cancer cells in cancer drug discovery is central, essential, and incontrovertible. In contrast, human heart muscle has never been available for equivalent proof-of-concept studies, other than sporadically—through biopsies and explanted hearts—never the routinized, scalable, renewable resource required for library screening, systematic compound development, and pull-through to translation. Apart from this want of starting material, even short-term expansion of adult human cardiomyocytes in primary culture is thwarted by the terminally differentiated myocytes’ characteristic state of growth arrest.
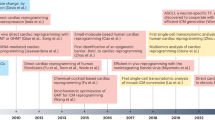
By contrast, human cardiac muscle cells made in limitless quantities from human pluripotent stem cells (hPSC-CMs) now provide unprecedented access to “heart disease in a dish,” with encouraging potential to accelerate the present tepid pace of cardiac drug discovery23,24,25,26,27,28,29,30,31,32 (Fig. 2; Table 1). Cultured from the inner cell mass of the blastocyst, human embryonic stem cells (hESCs) are the native “gold standard” for human pluripotency33, have all the requisite properties including efficient cardiomyocyte creation34, and have even progressed into primate trials of cardiomyocyte grafting35. Human induced pluripotent stem cells (hiPSCs) are made instead from adult somatic cells reprogrammed with ESC transcription factors36, augmented or replaced by chemical reprogramming. Compared to hESCs, hiPSCs have similarly well-proven cardiogenic potential34 but pose fewer ethical or religious concerns, and make possible the interrogation of patient-specific genetic variants24,37, in addition to modeling the pandemic forms of acquired heart disease that have greatest public health significance. Here, except if otherwise noted, we refer to hPSCs, for ESCs and iPSCs collectively.

Diverse cardiac disorders have been successfully modeled in hPSC-CMs, beyond merely the patient-specific mutations for which this technology was first used, with broad applicability now demonstrated for the widespread, acquired forms of human heart disease. Concurrently, the readouts relevant to cardiac drug development have expanded beyond the arrhythmias first studied, to encompass the full spectrum of molecular and functional cardiomyocyte phenotypes including mechanical performance, energetics, myocyte formation, and myocyte loss. The impact on drug development has been manifested initially through more predictive safety pharmacology (including the improved profiling of non-cardiac drugs) and through human preclinical studies of approved agents, toward novel applications. In the development of novel agents, hPSC-CMs can augment not only target-based approaches, as platforms for validation by gene silencing and the investigation of new chemical series, but also as a human substrate for mechanistically agnostic, phenotype-driven screens. Diverse approaches promote cardiomyocyte maturation and fidelity to the adult human heart itself, which remains an acknowledged limitation of these models.
hPSC-derived cardiomyocytes for predictive toxicology: canaries in a coal mine
Historically, the unmet scientific need to consider hPSC-CMs in the context of drug development begins with safety pharmacology: namely, the failure of conventional animal models to predict drug toxicities38. In the USA, roughly one in seven approved compounds is later withdrawn from clinical use, 28% of these for adverse cardiac events, including potential lethal arrhythmias, muscle cell death, and heart failure39. Using microelectrode arrays (MEA), pioneering studies by Hoffman La-Roche40, GlaxoSmithKline41 and Johnson and Johnson42 profiled 10 to 30 compounds, for their pro-arrhythmic effects in hPSC-CMs. All three surveys concluded that the relevant pharmacology was captured in these human cardiomyocyte assays, with obvious inherent advantages over non-cardiac cells that are engineered to express a single human cardiac ion channel such as hERG (substrate for the ventricular arrhythmia Torsade de Pointes [TdP]). The hPSC-CMs enabled very high accuracy over the relevant range of concentrations, compared to standard lower throughput, higher cost ex vivo methods (ventricular wedge, Purkinje fiber, Langendorff). Indeed, hPSC-CMs were superior to the routine preclinical models for detecting some key parameters of risk, such as effects on repolarization akin to human QTc prolongation41.
Building on these encouraging findings, more systematic use of hPSC-CMs has been promoted, together with in silico modeling and other prediction tools, by the Comprehensive In Vitro Proarrhthymia Assessment (CiPA) initiative25,32,43. Explicitly, hPSC-CMs are viewed as “more complete ‘biological integrators’ that detect not only complex effects of drugs on multiple cardiac currents, but also modulatory effects on currents elicited through signaling pathways, channel-associated subunits, altered intracellular calcium handling, additional transporters and exchangers, and potential changes in channel densities in myocytes”25. The potential utility of hPSC-CMs to predict drug-induced proarrhythmic effects was demonstrated most conclusively in a blinded, international, 10-site study of 28 drugs, using two commercially available lines and diverse electrophysiological platforms including MEAs and voltage-sensing potentiometric dyes32. The test compounds were first categorized by degree of known clinical risk for TdP, then were analyzed in blinded fashion for the prevalence of drug-induced repolarization abnormalities and arrhythmia-like events. These data, from 15 cell type × platform combinations, were then used to a construct a predictive model of TdP risk. Significant predictors in the hPSC-CMs were: arrhythmia-like event at any tested concentration, maximum prolongation at any tested concentration, and estimated prolongation at the clinical concentration of drug. These parameters in turn were fed into a composite model of TdP risk, with dichotomous outcomes (low, versus high or intermediate). The receiver operating characteristic (ROC) area under the curve (AUC) was 0.87, regardless of the cell line used or any local differences in culture method. In short, blinded studies have made it clear that measurements in hPSC-CMs are a highly reliable preclinical classifier of clinical TdP risk. Given such evidence, hiPSC-CMs have gained acceptance by industry and regulatory authorities as predictive of drug safety in humans, at least with respect to arrhythmic risk 25,32,44,45.
Two further initiatives with hPSC-CMs for safety pharmacology are the Japan iPS Cardiac Safety Assessment (JiCSA)46,47 and the CRACK IT InPulse Challenge48,49,50. Key lessons from JiCSA, which likewise is focused on arrhythmic risk, have included fidelity of the relationship between MEA-measured field potential duration and interspike interval in hPSC-CMs to the QT-RR relation deduced from clinical electrocardiograms in the Framingham Heart Study46. Human-relevant characteristics included repolarization delay at slow beating rates, reverse use-dependency, categorical analyses as potential indices of risk, and a threshold field potential duration that predicts early afterdepolarizations (EADs) and triggered activity, useful as a potential marker of risk for the onset of human Torsade de pointes46. A subsequent large-scale validation study of 60 compounds’ torsadogenic risk markedly expanded the conclusions available from CiPA32, though not yet in blinded fashion. Findings were highly concordant in iCell and Cor.4U hPSC-CMs47, despite the lines’ differences in ion channel and membrane transporter expression 51.
The InPulse academic-industry consortium, by contrast, has emphasized developing a robust in vitro platform to monitor cardiac contractility, with cells that are phenotypically mature48,49,50. (See the Challenges and Prospects section for a detailed discussion of this concern.) This project’s discoveries include a synthetic polymer that promotes the maturity of hPSC-CMs (a co-polymer of isobornyl methacrylate and tert-butylamino-ethyl methacrylate), identified using combinatorial subtrate material microarrays48, novel open-source tools for quantifying cardiomyocyte contraction49, and proof that driving contractile work in hPSC-CMs induces mitochondrial biogenesis and recapitulates the normal post-natal shift to fatty acid oxidation 50.
Other cardinal features of potential cardiotoxicity—notably, myocyte loss and ensuing heart failure—are likewise amenable to profiling in hPSC-CMs, with the cardiotoxicity of anti-cancer drugs being an especially robust area of investigation26,52,53,54. Readily applicable assays include biochemical readouts (defective ATP production, cardiac troponin release), microscopy (loss of surface membrane integrity, apoptosis), and video imaging to assess contractility (edge detection or deformation maps). As was done for arrhythmic risk, these quantitative parameters of toxicity can be integrated as a composite cardiac safety index26,54. Interestingly, interpatient variations in heart tissue transcriptomics were largely recapitulated in patient-specific hPSC-CMs, including the NRF2-PPARGC1A pathway controlling oxidative stress, and correlated well with the patient-derived cells’ functional difference in cardiotoxic responses31. In a related study, patient-specific hPSC-CMs reproduced the patients’ respective vulnerability to trastuzumab55. More recently, a network-level comparison of cardiotoxicity in hPSC-CMs used RNA sequencing to distinguish differences in the pathways engaged by anthracyclines versus tyrosine kinase inhibitors56. Here, the principal finding was the categorically different transcriptomic signatures evoked by these anti-cancer agents in human cardiomyocytes. Specifically, whereas doxorubicin (DOX) induces pathways that initiate DNA damage, tyrosine kinase inhibitors disrupt mitochondrial energetics even at non-lethal concentrations, downregulating oxidative phosphorylation and upregulating glycolysis56, metabolic reprogramming that is a common feature of hypertrophied and failing hearts57. In agreement with DNA damage as the main target for DOX, genome editing to delete topoisomerase-II beta (TOP2B) markedly reduced the vulnerability of hPSC-CMs to DOX-induced DNA double strand breaks and cell death 53.
HPSC-CMS in cardiac drug development: early exemplars
As a potential step change beyond just safety assessment and risk prediction, might hPSC-CMs also be useful as human preclinical proof of efficacy, to guide and inform experimental therapeutics? In one early study of this kind, investigators sought to mitigate the known cardiotoxicity of DOX, a mainstay of cancer chemotherapy, as mentioned, targeting DOX selectively to breast cancer using liposomes conjugated with antibody against human epidermal growth factor receptor 2 (HER2)58. By this means, cardiotoxicity was virtually abolished in hPSC-CMs58, and a Phase 1 clinical dose-escalation study confirmed the improvements expected on the basis of the pioneering human preclinical results59. No cardiac adverse events occurred in patients receiving this form of DOX alone, and the toxicity of combination therapy along with trastuzumab also was reduced59. Thus, human proof-of-principle was substantiated in hPSC-CMs (in this instance, cardiac safety of the targeted DOX), in advance of progression into human trials. Analogously, the cardiotoxicity of trastuzumab was found to be associated with defective energy metabolism in hPSC-CMs and was ameliorated by treatment with activators of AMP-activated protein kinase55, suggesting the potential for cardioprotection in this context by a current approved drug.
A further success in translational relevance is the progress made using hPSC-CMs for drug repurposing, in particular thus far where based on the cells’ fidelity to clinical phenotypes in certain hereditary heart disorders. This progress is perhaps most notable for personalized treatment of channelopathies such as long QT syndrome due to mutations in KCNH2 that disrupt intracellular trafficking of the hERG potassium channel Kv11.1 (LQT2). Lumacaftor (the protein chaperone VX-809) was evaluated in patient-specific hPSC-CMs, and shown to shorten the cell culture equivalent of QTc, as measured with multi-electrode arrays. This benefit was achieved solely in patients with Class 2 mutations (which affect channel trafficking) but not in patients with Class 1 mutations (which affect channel synthesis)60. A clinically approved drug, Orkambi, combines Lumacaftor and Ivacaftor (a CFTR potentiator), rescues the analogous defect in patients with a homozygous CFTR-F508del mutation, and was taken forward into two patients with Class 2 mutations of LQT2, shortening QTc in both, as had been hypothesized61. Concomitantly, however, this landmark first-in-human study also acknowledged several “differences between the cellular model and clinical reality,” which provide instructive caveats, including the magnitude of rescue achieved (much greater in hPSC-CMs than in the clinic) and expression of hERG (higher in hPSC-CMs than in native adult CMs) 61.
What about cardiac drug development, more broadly than just reformulation or repurposing? Miniaturization of phenotypic assays to a 384-well format makes it possible to implement high-throughput chemical or genetic screens (HTS) for target validation and drug discovery, more rooted in human cardiac biology than has been possible heretofore. In an early example of tool-building toward high-throughput studies, hPSC-CMs were implemented to model myocarditis due to coxsackievirus B3, using CVB3-luciferase as an easy bioluminescent readout of virus proliferation62. Antiviral drugs including interferon-β1 and ribavirin were shown to suppress virus production. In addition, mechanistic insights were captured in this human cardiomyocyte milieu: by microarray profiling, interferon-β1 was shown to activate a network of downstream anti-viral genes including EIF2AK2, encoding protein kinase R, an inhibitor of viral mRNA translation 62.
For cardiac muscle hypertrophy, one readily assayable readout is the induction of brain natriuretic peptide, a highly dynamic protein with especially strong diagnostic and prognostic significance in the clinical setting63. Its induction is triggered in hPSC-CMs by endothelin-1, much as in non-human models of pathological hypertrophy, and its detection by ELISA or high-content imaging can be multiplexed with other parameters such as increased cardiomyocyte size64. In an early pilot phenotypic screen, hPSC-CMs were provoked at a saturating concentration of endothelin-1, and were treated in quadruplicate at 10 concentrations with candidate inhibitors including the calcium channel blocker verapamil, a PI3K-mTOR inhibitor, BEZ-235, and a broad-spectrum histone deacetylase inhibitor64. A design feature worth noting was the use of serum-free fatty acid-supplemented media, to accelerate cardiomyocyte maturation64. More fundamentally, these experiments demonstrated that inhibitor profiling in this human platform was robust and workable at scale.
map4k4 AS A druggable target in human cardiac muscle cell death: target validation and compound development in HPSC-CMS
Many logical targets have failed in clinical trials for heart disease, perhaps especially those aiming to enhance cardiac muscle cell survival after myocardial infarction10,11,12,13,14,15. While specific shortcomings in trial design or implementation are sometimes culpable, what these failed trials have in common uniformly is that none was based on proof of efficacy in human preclinical studies, before proceeding into the clinic. The use of hPSC-CMs toward drug discovery for cardioprotection was championed in our recent study creating novel inhibitors of the stress-activated kinase MAP4K4 (mitogen-activated protein kinase kinase kinase kinase-4)65, an upstream member of the MAPK superfamily with connections to Jun N-terminal kinase66,67 and NFkB68, as well as to several non-canonical effectors as substrates69,70. The scientific case for MAP4K4 as a druggable target in heart muscle cell death began with human tissue characterization, finding that myocardial MAP4K4 was activated in end-stage heart failure regardless of cause (dilated, hypertrophic, ischemic, and anthracycline cardiomyopathy). MAP4K4 was likewise activated in a range of disease models in adult mouse myocardium and cultured rat cardiomyocytes, including ischemia/reperfusion injury and H2O2 as a surrogate oxidative stress. The causal role of MAP4K4 suggested by these observations was then corroborated using transgenic over-expression in mice, plus gain-of-function mutations, dominant-negative mutations, and gene silencing in rodent cardiomyocytes. Yet, even collectively, these methods—typical of the toolkit for academic target validation—leave altogether unanswered the question of whether MAP4K4 is a mechanistically sound therapeutic target in human heart muscle cell death.
Consequently, using hPSC-CMs as a human platform for target validation and proof-of-concept studies, the requirement for MAP4K4 in human cardiac cell death was affirmed by gene silencing, giving uniquely direct impetus to a small-molecule discovery program65. Throughout, several high-throughput platforms were utilized, including automated microscopy (DRAQ7 uptake, TUNEL staining for apoptosis), ATP generation, and cardiac troponin release. Protection was conferred in three wholly independent human cardiomyocyte lines, suggesting not only the reliability of hPSC-CMs as a model but also the unvarying dependence on MAP4K4 in the tested forms of cardiac cell death. Beyond these key readouts of viability, protective effects of inhibiting MAP4K4 were also proven under sublethal stress, using the Seahorse extracellular flux (XF) method to study mitochondrial function and FLIPR assays to measure calcium cycling. Cardiomyocyte viability and function (auxotonic force) were even preserved in human 3D engineered heart tissue65, a model with further maturity of structure and physiological properties71. Importantly, the pathway and compounds developed in hPSC-CMs were substantiated further by proof-of-concept studies in mice, with the MAP4K4 inhibitor reducing infarct size by more than 55% in blinded studies, even given an hour after injury65. These MAP4K4 studies demonstrate the pivotal role played by hPSC-CMs in validating a suspected target by gene silencing, then building a small-molecule program upon efficacy proven in this human platform.
Will treatments devised in human models be more likely to succeed than prior ones, upon eventual testing in the clinic? This overall hypothesis—the crux of using hPSC-CMs as a model in drug discovery—will require a decade or more to resolve empirically, as exemplars of this class progress into human trials. However, it is tantalizing to apply the “retrospectoscope”: examining the outcome, in human cardiomyocytes, of manipulating pathways whose success or failure is already known in reducing infarct size. Indeed, from this perspective, the potential predictive value of studies in hPSC-CMs is suggested by finding that human cardiac muscle cell death can be suppressed experimentally by β-adrenergic blockade (as found in METOCARD-CNIC72,73) but not by inhibiting p38 MAPK (as was ultimately true in SOLSTICE14).
Preventing cardiac muscle cell death
By contrast to more reductionist but biased target-based approaches, phenotype-driven screens, discussed here and in the following sections, require no a priori assumption about the drug’s target, can pursue wholly novel or unanticipated mechanisms of action, engage the cell type-specific signaling and transcriptional context, and encompass diverse endogenous readouts of the disease74—including cardiomyocyte survival, but also myocyte function and creation.
In a mechanistically agnostic version of the MAP4K4 cell death studies detailed above, an unbiased chemical biology screen of nearly 50,000 small molecules was performed to identify and validate compounds that protect hPSC-CMs from H2O275. Cardiomyocyte protection was demonstrated using ATP content as the primary end-point and cellular impedance as the secondary readout, a measure of monolayer integrity and contractile function. From this screen, in 1536-well format, using a 35% improvement in viability as the criterion, 220 hits were identified of which half were confirmed upon retesting. A novel compound, designated cardioprotectant-312, was found to protect hPSC-CMs from the oxidative stress of H2O2, triggering upregulation of the essential endogenous anti-oxidant, heme oxygenase-175. Of note, no protection was seen in rat H9c2 cells, embryonic heart-derived myoblasts that are commonly used in toxicology research; conversely, none of the authors’ earlier lead compounds identified in H9c2 cells76 was protective in human cardiomyocytes. Together, these vivid reciprocal disparities highlight the importance of implementing human preclinical cardiac models, in lieu of basing human trials solely on results obtained in rodent cardiomyocytes alone.
Attenuating diabetic cardiomyopathy
The power of hPSC-CMs for drug discovery to alleviate heart failure was demonstrated in two studies using patient-specific iPSCs to model diabetic cardiomyopathy57,77. To mimic the diabetic-like environment, normal hPSCs were cultured in a maturation medium to promote the substrate utilization of adult ventricular myocytes (fatty acid β-oxidation), then were subjected to glucose excess, plus endothelin-1 and cortisol77, mimicking the systemic environment. The combined effect was marked induction of brain natriuretic peptide (BNP), other molecular markers of cardiac hypertrophy, and myocyte enlargement itself. Functional abnormalities included less frequent Ca2+ transients, decreased beat amplitude, and increased beat irregularity. Lipid accumulation and peroxidation were other associated findings. The cardiomyopathic phenotype was captured, too, in cardiomyocytes derived from diabetic patient-specific iPSCs, even in the absence of the diabetic milieu 77.
This model was then used as a phenotypic drug screening platform, determining the success of a therapeutic compound as monitored by BNP production, nuclear area, and sarcomere organization (α-actinin staining). From a library of 480 compounds, 47 were identified that improved all three of these disease parameters77. These compounds encompassed diverse modes of action, including regulators of Ca2+ homeostasis (fluspirilene, thapsigargin, and the calmodulin inhibitor W7), Na+ and K+ channel blockers, phosphodiesterase inhibitors, and multiple protein kinase inhibitors (H89, K252a, SB202190). From this screen, in turn, compounds were found that rescued the reduction of Ca2+ transients in cardiomyocytes subjected to the diabetic milieu and improved the phenotype of cardiomyocytes from diabetic patients. Analogous improvement of diabetic phenotypes in hPSC-CMs were elicited by empagliflozin, an inhibitor of sodium-glucose co-transporters that are upregulated in diabetes, possibly explaining the unexpectedly improved cardiovascular mortality in trials of this compound for glycemic control78. Together, these findings support the utility of hPSC-CMs as models not merely for simple monogenic disorders, on the one hand, and for wild-type cardiomyocytes’ responses to lethal stress, on the other, but even for diabetic cardiomyopathy, a highly complex polygenic disease.
Driving cardiac muscle cell proliferation
Ultimately, myocardial infarction can be viewed as a “myocyte-deficiency disease” whose phenotype is determined not just by the extent of myocyte death but also by the lack of functionally significant restorative growth. Indeed, the plausible clinical benefits of hPSC-CMs very clearly include therapeutic grafting, as cell therapy35,79,80, Recently, though, advances in understanding the fundamental biology of cardiac growth arrest have pointed to greater plasticity that was formerly evocable, with significant potential for restarting the cardiac cell cycle therapeutically, at least in model organisms81,82. Might induced proliferation as a route to heart repair also be amenable to exploration or triage in hPSC-CMs? As one starting point, functional screening of more than 10,000 hPSC-CM organoids was undertaken to optimize diverse aspects of the culture milieu, resulting in enhanced maturation and recapitulation of the adult heart’s notorious resistance to cell cycling, driven by a shift to fatty acid oxidation83. Conversely, this successful model of implementing cardiac cell cycle arrest in hPSC-CMs was then subjected to a functional screen of 105 small molecules, resulting in the identification of novel cell cycle activators, working through the mevalonate pathway84. Thus, notwithstanding the potential immaturity of pluripotent cell-derived myocytes with respect to cell cycle control, a post-mitotic phenotype could be imposed experimentally, and means to override it discovered.
Challenges and prospects
The research advances reviewed here highlight the many benefits of hPSC-CMs as a transformative human platform for cardiac drug discovery74—accessible, scalable, faithful by a large number of clinically relevant parameters, amenable to genetic engineering, amenable to tissue engineering, able to capture patient variations, predictive of clinical success at least retrospectively, and predictive prospectively at least of success in whole-animal studies (Fig. 2; Table 1). One must, of course, temper optimism with caution. For the moment, it remains a supposition that new drugs developed by this route will achieve clinical success more reliably than drugs lacking human preclinical proof of effect. But, this hypothesis is testable—even if needing years of accumulated experience to do—while drawing ample credence from the established precedents and practices in cancer chemotherapy.
How might the predictive power of hPSC-CMs be augmented or ensured? Despite the predictive power shown even with routine 2D models, it is clear that existing lines—or, more accurately, their current embodiment in tissue culture—do not suffice to model all possible phenotypes of concern. The many acknowledged shortcomings, which have been mitigated to date only partially, include morphology (lack of sarcomere organization, T-tubules, and the normal mitochondrial density), molecular profile (weak expression of maturation-associated genes and splicing isoforms), metabolism (glycolysis, not fatty acid oxidation), contractility (lower maximum contractile force), and electrophysiology (lesser action potential upstroke velocity and amplitude)48,49,50,51,85,86,87,88,89,90,91,92,93,94,95. For instance, by comparison to adult human hearts, commercially available hPSC-CMs were uniformly deficient in the expression of KCNJ2, with higher than normal expression of HCN4 and large line-to-line variations in the other ion channels and membrane transporters assayed (CACNA1C, KCNH2, KCNQ1, SLC8A1, ATP1A1, ATP2A2)51. As a consequence, some pharmacological and pathobiological responses can be deficient or anomalous. In some reports, hPSC-CMs do not mirror the TdP risk of drugs with late sodium current effects, like ranolazine32, and hiPSC-CMs were less sensitive to hypoxia/reoxygenation than to other death signals65,96,97. Such disparities must be taken into account, whether inherent short-comings or idiosyncratic.
Efforts to enhance (further) the stem cell-derived myocytes’ predictive value center on manipulating chamber and cell sub-type specificity on the one hand, and on improving structural and functional maturity on the other. Purely pharmacological efforts at enhancing maturation in routine 2D culture include fatty acids85,86, thyroid hormone87, and inhibition of mTOR88. In another approach, transduction of the defectively expressed gene KCNJ2 has been applied to rescue the channel levels and promote aspects of fidelity directly98. More generally, however, the procedures of most proven value to enhance the maturity of hPSC-CMs include the use of 3D human engineered heart tissue (EHT), mechanical or electrical conditioning, and heart-on-chip technologies, advances discussed at length elsewhere89,90,91,92,93,94,95. The tissue engineering solutions to create more heart-like phenotypes in hPSC-CMs range in complexity from micropatterned 2D substrates to scaffolds, organoids, microfluidics, 3D bioprinting, and even the construction of hollow spheres. Apart from just geometry, key elements of these tissue engineering strategies notably include cyclic electrical or mechanical stimulation. Incorporation of other cell types can promote maturity or function, as well as the microvascularization required for oxygen delivery at larger scale than mere diffusion can confer. Indeed, 3D spheroids composed of hPSC-CMs plus hPDSC-derived endothelial cells showed progressive changes in gene expression typical of post-natal development99. Analogously, 3D culture of hPSC-CMs as engineered heart tissue in concert with cardiac fibroblasts, combined with long-term electrical stimulation, enables the development of physiological responses that are absent from the cardiomyocytes cultured routinely93,100. The reported adult-like properties included a positive force-frequency relationship, postrest potentiation of force, and inotropic beta-adrenergic responses to isoproterenol and dobutamine, as well as other compounds and pathways tested 93,100.
Although these collective efforts should be viewed with enthusiasm, progress toward standardization is confounded by the diversity of available hPSC lines, stem cell and differentiation media, physical substrata, timing, purification methods, presence or absence of serum, cell density (syncytial sheets, versus sporadic single cells), the heterogenous mixtures of cell types (atrial, ventricular, pacemaker cells, non-cardiomyocytes), and even lot-to-lot variation in ostensibly standardized cells. However, though procedures exist for the selective production of ventricular myocytes versus atrial myocytes or conduction system cells93,101,102,103,104, even these simple advances are not yet exploited uniformly. Ultimately, it is plausible that the drivers of standardization will include the emergence of consensus best practice solutions, but also adherence within the scientific community to procedures validated by multi-site initiatives such as CiPA, JiCSA, and InPulse, functioning as exemplars.
A complementary approach—obvious once the issue is raised—is also to improve the breadth of human cardiomyocytes surveyed, including but not limited to demographic features like gender and ethnic background. To illustrate, genetic determinants of susceptibility include a polymorphism in ALDH2 that predominates in East Asians and renders the carriers’ myocytes much more vulnerable to ischemic heart damage105. Analogously, screening a single commercial line failed to capture the known cardiotoxicity of rosiglitazone, an anti-diabetic drug that enhances PPARγ activity but can exacerbate heart failure106. Indeed, marked inter-patient variation was later found in the response of hPSC-CMs to rosiglitazone, including adverse effects on reactive oxygen and nitrogen species, associated with divergent transcriptomic signatures that relate to NRF2-mediated oxidative stress31. A third demographic axis, aging, may be resistant to capture in hPSC-derived models, given the rejuvenation signature imparted by reprogramming to a primitive, pluripotent state; this aspect might be addressable using directly induced cardiomyocytes, instead107,108, for which a precedent is the success using forward programming to model age-related neurodegeneration109. Much like trials in the real world, clinical trials in a dish may need to take patient recruitment into account—along with their choices of compound, regimen, and readout—in developing robust new counter-measures to combat human heart disease.
References
GBD 2017 Causes of Death Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1859–1922 (2018).
GBD 2017 DALYs and HALE Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788 (2018).
Fordyce, C. B. et al. Cardiovascular drug development: is it dead or just hibernating? J. Am. Coll. Cardiol. 65, 1567–1582 (2015).
Gromo, G., Mann, J. & Fitzgerald, J. D. Cardiovascular drug discovery: a perspective from a research-based pharmaceutical company. Cold Spring Harb. Perspect. Med 4, a014092 (2014).
Packer, M. et al. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 131, 54–61 (2015).
Fox, K., Ford, I., Steg, P. G., Tendera, M. & Ferrari, R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet 372, 807–816 (2008).
Swedberg, K. et al. Effects on outcomes of heart rate reduction by ivabradine in patients with congestive heart failure: is there an influence of beta-blocker dose?: findings from the SHIFT (Systolic Heart failure treatment with the I(f) inhibitor ivabradine Trial) study. J. Am. Coll. Cardiol. 59, 1938–1945 (2012).
Pennell, D. J. et al. Cardiovascular function and treatment in beta-thalassemia major: a consensus statement from the American Heart Association. Circulation 128, 281–308 (2013).
Maurer, M. S. et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ. Heart Fail. 8, 519–526 (2015).
Heusch, G. Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381, 166–175 (2013).
Hausenloy, D. J. & Yellon, D. M. Targeting myocardial reperfusion Injury–the search continues. N. Engl. J. Med. 373, 1073–1075 (2015).
Piot, C. et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 (2008).
Lincoff, A. M. et al. Inhibition of delta-protein kinase C by delcasertib as an adjunct to primary percutaneous coronary intervention for acute anterior ST-segment elevation myocardial infarction: results of the PROTECTION AMI Randomized Controlled Trial. Eur. Heart J. 35, 2516–2523 (2014).
Newby, L. K. et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet 384, 1187–1195 (2014).
Ottani, F. et al. Cyclosporine A in reperfused myocardial infarction: the multicenter, controlled, open-label CYCLE trial. J. Am. Coll. Cardiol. 67, 365–374 (2016).
Pammolli, F., Magazzini, L. & Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 10, 428–438 (2011).
Sharma, S. V., Haber, D. A. & Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 10, 241–253 (2010).
Gillet, J. P., Varma, S. & Gottesman, M. M. The clinical relevance of cancer cell lines. J. Natl Cancer Inst. 105, 452–458 (2013).
Gazdar, A. F., Hirsch, F. R. & Minna, J. D. From mice to men and back: an assessment of preclinical model systems for the study of lung cancers. J. Thorac. Oncol. 11, 287–299 (2016).
Shoemaker, R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823 (2006).
Rees, M. G. et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116 (2016).
Ghandi, M. et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508 (2019).
Matsa, E., Burridge, P. W. & Wu, J. C. Human stem cells for modeling heart disease and for drug discovery. Sci. Transl. Med. 6, 239ps236 (2014).
Bellin, M., Marchetto, M. C., Gage, F. H. & Mummery, C. L. Induced pluripotent stem cells: the new patient? Nat. Rev. Mol. Cell Biol. 13, 713–726 (2012).
Gintant, G., Fermini, B., Stockbridge, N. & Strauss, D. The evolving roles of human iPSC-derived cardiomyocytes in drug safety and discovery. Cell Stem Cell 21, 14–17 (2017).
Sharma, A. et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 9, eaaf2584 (2017).
Burridge, P. W. et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556 (2016).
Liang, P. et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 127, 1677–1691 (2013).
Cameron, B. J. et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 5, 197ra103 (2013).
Lee, Y. K. et al. Modeling treatment response for Lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J. Am. Heart Assoc. 6, e005677 (2017).
Matsa, E. et al. Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 19, 311–325 (2016).
Blinova, K. et al. International multisite study of human-induced pluripotent stem cell-derived cardiomyocytes for drug proarrhythmic potential assessment. Cell Rep. 24, 3582–3592 (2018).
Mascetti, V. L. & Pedersen, R. A. Human-mouse chimerism validates human stem cell pluripotency. Cell Stem Cell 18, 67–72 (2016).
Kattman, S. J. et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228–240 (2011).
Liu, Y. W. et al. Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 36, 597–605 (2018).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Chen, I. Y., Matsa, E. & Wu, J. C. Induced pluripotent stem cells: at the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 13, 333–349 (2016).
Denning, C. et al. Cardiomyocytes from human pluripotent stem cells: from laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 1863, 1728–1748 (2016).
Gwathmey, J. K., Tsaioun, K. & Hajjar, R. J. Cardionomics: a new integrative approach for screening cardiotoxicity of drug candidates. Expert Opin. Drug Metab. Toxicol. 5, 647–660 (2009).
Guo, L. et al. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 123, 281–289 (2011).
Harris, K. et al. Comparison of electrophysiological data from human-induced pluripotent stem cell-derived cardiomyocytes to functional preclinical safety assays. Toxicol. Sci. 134, 412–426 (2013).
Braam, S. R. et al. Repolarization reserve determines drug responses in human pluripotent stem cell derived cardiomyocytes. Stem Cell Res. 10, 48–56 (2013).
Gintant, G., Sager, P. T. & Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 15, 457–471 (2016).
Sala, L., Bellin, M. & Mummery, C. L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: has the time come? Br. J. Pharm. 174, 3749–3765 (2017).
Magdy, T., Schuldt, A. J. T., Wu, J. C., Bernstein, D. & Burridge, P. W. Human induced pluripotent stem cell (hiPSC)-derived cells to assess drug cardiotoxicity: opportunities and problems. Annu. Rev. Pharm. Toxicol. 58, 83–103 (2018).
Yamazaki, D. et al. Proarrhythmia risk prediction using human induced pluripotent stem cell-derived cardiomyocytes. J. Pharm. Sci. 136, 249–256 (2018).
Kanda, Y., Yamazaki, D., Osada, T., Yoshinaga, T. & Sawada, K. Development of torsadogenic risk assessment using human induced pluripotent stem cell-derived cardiomyocytes: Japan iPS Cardiac Safety Assessment (JiCSA) update. J. Pharm. Sci. 138, 233–239 (2018).
Patel, A. K. et al. A defined synthetic substrate for serum-free culture of human stem cell derived cardiomyocytes with improved functional maturity identified using combinatorial materials microarrays. Biomaterials 61, 257–265 (2015).
Sala, L. et al. MUSCLEMOTION: a versatile open software tool to quantify cardiomyocyte and cardiac muscle contraction in vitro and in vivo. Circ. Res. 122, e5–e16 (2018).
Ulmer, B. M. et al. Contractile work contributes to maturation of energy metabolism in hiPSC-derived cardiomyocytes. Stem Cell Rep. 10, 834–847 (2018).
Kodama, M. et al. Systematic expression analysis of genes related to generation of action potentials in human iPS cell-derived cardiomyocytes. J. Pharm. Sci. 140, 325–330 (2019).
Zhang, S. et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 18, 1639–1642 (2012).
Maillet, A. et al. Modeling doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci. Rep. 6, 25333 (2016).
Sharma, A. et al. Use of human induced pluripotent stem cell-derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 13, 3018–3041 (2018).
Kitani, T. et al. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation 139, 2451–2465 (2019).
Wang, H. et al. Adaptation of human iPSC-derived cardiomyocytes to tyrosine kinase inhibitors reduces acute cardiotoxicity via metabolic reprogramming. Cell Syst. 8, 412–426 (2019).
Karwi, Q. G., Uddin, G. M., Ho, K. L. & Lopaschuk, G. D. Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 5, 68 (2018).
Reynolds, J. G. et al. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol. Appl. Pharm. 262, 1–10 (2012).
Munster, P. et al. Safety and pharmacokinetics of MM-302, a HER2-targeted antibody-liposomal doxorubicin conjugate, in patients with advanced HER2-positive breast cancer: a phase 1 dose-escalation study. Br. J. Cancer 119, 1086–1093 (2018).
Mehta, A. et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Heart J. 39, 1446–1455 (2018).
Schwartz, P. J. et al. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome Type 2. Eur. Heart J. 40, 1832–1836 (2019).
Sharma, A. et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ. Res. 115, 556–566 (2014).
Mueller, C. et al. Heart failure association of the European Society of Cardiology practical guidance on the use of natriuretic peptide concentrations. Eur. J. Heart Fail. 21, 715–731 (2019).
Carlson, C. et al. Phenotypic screening with human iPS cell-derived cardiomyocytes: HTS-compatible assays for interrogating cardiac hypertrophy. J. Biomol. Screen. 18, 1203–1211 (2013).
Fiedler, L. R. et al. MAP4K4 inhibition promotes survival of human stem cell-derived cardiomyocytes and reduces infarct size in vivo. Cell Stem Cell 24, 579–591 (2019).
Wu, C., Watts, M. E. & Rubin, L. L. MAP4K4 activation mediates motor neuron degeneration in amyotrophic lateral sclerosis. Cell Rep. 26, 1143–1156 (2019).
Larhammar, M., Huntwork-Rodriguez, S., Rudhard, Y., Sengupta-Ghosh, A. & Lewcock, J. W. The Ste20 family kinases MAP4K4, MINK1 and TNIK, converge to regulate stress induced JNK signaling in neurons. J. Neurosci. 37, 11074–11084 (2017).
Roth Flach, R. J. et al. Endothelial protein kinase MAP4K4 promotes vascular inflammation and atherosclerosis. Nat. Commun. 6, 8995 (2015).
Chuang, H. C. et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat. Commun. 5, 4602 (2014).
Meng, Z. et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 6, 8357 (2015).
Weinberger, F., Mannhardt, I. & Eschenhagen, T. Engineering cardiac muscle tissue: A maturating field of research. Circ. Res. 120, 1487–1500 (2017).
Ibanez, B. et al. Effect of early metoprolol on infarct size in ST-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: the Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial. Circulation 128, 1495–1503 (2013).
Pizarro, G. et al. Long-term benefit of early pre-reperfusion metoprolol administration in patients with acute myocardial infarction: results from the METOCARD-CNIC trial (Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction). J. Am. Coll. Cardiol. 63, 2356–2362 (2014).
Friese, A. et al. The convergence of stem cell technologies and phenotypic drug discovery. Cell Chem. Biol. 26, 1050–1066 (2019).
Kirby, R. J. et al. Discovery of novel small-molecule inducers of heme oxygenase-1 that protect human iPSC-derived cardiomyocytes from oxidative stress. J. Pharm. Exp. Ther. 364, 87–96 (2018).
Kane, A. et al. Cardioprotective Inhibitors of Reperfusion Injury (National Center for Biotechnology Information, 2010).
Drawnel, F. M. et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 9, 810–821 (2014).
Ng, K. M. et al. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci. Rep. 8, 14872 (2018).
Chong, J. J. et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510, 273–277 (2014).
Chen, H., Zhang, A. & Wu, J. C. Harnessing cell pluripotency for cardiovascular regenerative medicine. Nat. Biomed. Eng. 2, 392–398 (2018).
Li, M. & Izpisua Belmonte, J. C. Mending a faltering heart. Circ. Res. 118, 344–351 (2016).
Heallen, T. R., Kadow, Z. A., Kim, J. H., Wang, J. & Martin, J. F. Stimulating cardiogenesis as a treatment for heart failure. Circ. Res. 124, 1647–1657 (2019).
Mills, R. J. et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl Acad. Sci. USA 114, E8372–E8381 (2017).
Mills, R. J. et al. Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell 24, 895–907.e896 (2019).
Kim, C. et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110 (2013).
Horikoshi, Y. et al. Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells 8, 1095 (2019).
Parikh, S. S. et al. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 121, 1323–1330 (2017).
Garbern, J. C. et al. Inhibition of mTOR signaling enhances maturation of cardiomyocytes derived from human induced pluripotent stem cells via p53-induced quiescence. Circulation 141, 285–300 (2019).
Lemoine, M. D. et al. Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci. Rep. 7, 5464 (2017).
Tiburcy, M. et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation 135, 1832–1847 (2017).
Wang, G. et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623 (2014).
Ronaldson-Bouchard, K. et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243 (2018).
Zhao, Y. et al. A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell 176, 913–927 (2019).
MacQueen, L. A. et al. A tissue-engineered scale model of the heart ventricle. Nat. Biomed. Eng. 2, 930–941 (2018).
Feric, N. T. & Radisic, M. Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv. Drug Deliv. Rev. 96, 110–134 (2016).
Chen, T. & Vunjak-Novakovic, G. In vitro models of ischemia-reperfusion injury. Regen. Eng. Transl. Med. 4, 142–153 (2018).
Hidalgo, A. et al. Modelling ischemia-reperfusion injury (IRI) in vitro using metabolically matured induced pluripotent stem cell-derived cardiomyocytes. APL Bioeng. 2, 026102 (2018).
Li, M. et al. Overexpression of KCNJ2 in induced pluripotent stem cell-derived cardiomyocytes for the assessment of QT-prolonging drugs. J. Pharm. Sci. 134, 75–85 (2017).
Giacomelli, E. et al. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 144, 1008–1017 (2017).
Feric, N. T. et al. Engineered cardiac tissues generated in the Biowire II: a platform for human-based drug discovery. Toxicol. Sci. 172, 89–97 (2019).
Weng, Z. et al. A simple, cost-effective but highly efficient system for deriving ventricular cardiomyocytes from human pluripotent stem cells. Stem Cells Dev. 23, 1704–1716 (2014).
Lee, J. H., Protze, S. I., Laksman, Z., Backx, P. H. & Keller, G. M. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell 21, 179–194.e174 (2017).
Devalla, H. D. et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 7, 394–410 (2015).
Liang, W. et al. Canonical Wnt signaling promotes pacemaker cell specification of cardiac mesodermal cells derived from mouse and human embryonic stem cells. Stem Cells (2019). https://doi.org/10.1002/stem.3106. [Epub ahead of print].
Ebert, A. D. et al. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci. Transl. Med. 6, 255ra130 (2014).
Doherty, K. R. et al. Structural and functional screening in human induced-pluripotent stem cell-derived cardiomyocytes accurately identifies cardiotoxicity of multiple drug types. Toxicol. Appl. Pharm. 285, 51–60 (2015).
Ieda, M. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010).
Hashimoto, H. et al. Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers. Cell Stem Cell 25, 69–86 (2019).
Mertens, J., Reid, D., Lau, S., Kim, Y. & Gage, F. H. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu Rev. Genet. 52, 271–293 (2018).
US Food and Drug Administration. Novel Drug Approvals for 2019, https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2019 (2019).
Pointon, A. et al. High-throughput imaging of cardiac microtissues for the assessment of cardiac contraction during drug discovery. Toxicol. Sci. 155, 444–457 (2017).
Acknowledgements
This work of the authors is supported by Wellcome Trust Seeding Drug Discovery awards (WT10638, WT205256) and Apollo Therapeutics.
Author information
Authors and Affiliations
Contributions
P.G. and M.D.S. wrote and edited this review.
Corresponding author
Ethics declarations
Competing interests
M.D.S. declares patent applications relating to MAP4K4 as a druggable target in human cardiac muscle cell death (UK Patent Applications 1716867.5, WO/2019/073253; 1819839.0; 1905472.5; 1905476.6). P.G. has no conflict of interest to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Golforoush, P., Schneider, M.D. Intensive care for human hearts in pluripotent stem cell models. npj Regen Med 5, 4 (2020). https://doi.org/10.1038/s41536-020-0090-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41536-020-0090-7
This article is cited by
-
Preclinical trial of a MAP4K4 inhibitor to reduce infarct size in the pig: does cardioprotection in human stem cell-derived myocytes predict success in large mammals?
Basic Research in Cardiology (2021)
-
Selective protection of human cardiomyocytes from anthracycline cardiotoxicity by small molecule inhibitors of MAP4K4
Scientific Reports (2020)
