
Abstract
The biological basis of the neurodegenerative movement disorder, Parkinson’s disease (PD), is still unclear despite it being ‘discovered’ over 200 years ago in Western Medicine. Based on current PD knowledge, there are widely varying theories as to its pathobiology. The aim of this article was to explore some of these different theories by summarizing the viewpoints of laboratory and clinician scientists in the PD field, on the biological basis of the disease. To achieve this aim, we posed this question to thirteen “PD experts” from six continents (for global representation) and collated their personal opinions into this article. The views were varied, ranging from toxin exposure as a PD trigger, to LRRK2 as a potential root cause, to toxic alpha-synuclein being the most important etiological contributor. Notably, there was also growing recognition that the definition of PD as a single disease should be reconsidered, perhaps each with its own unique pathobiology and treatment regimen.
Similar content being viewed by others

Introduction
Since the first description of Parkinson’s disease (PD) in Western Medicine over two centuries ago, significant progress has been made to better understand, diagnose and treat the disease1. Yet, despite this progress and years of research in the PD field, our understanding of its biological basis remains incomplete. Over the years, several different theories on the pathobiology of PD have emerged and evolved, with the general consensus being that the disease is complex with multiple factors (many still unknown) contributing to disease manifestation and progression.
The aim of this article was to explore some of these theories by asking PD clinician and laboratory scientists from around the globe to answer the question “What do you consider to be the starting point and the process that leads to the development of PD, and why?”. They could also comment on where future research efforts should be directed. Contributors were selected based on geographic spread, gender equity and diversity of their research interests. Overall, we had 12 contributions from six continents (Africa, Asia, Australia, Europe, North America, and South America). While authors sharing similar viewpoints were clustered together, their written and unedited views are presented here as a collection of their responses to the question.
Pathogenic sequence variants in conjunction with mitochondrial dysfunction and alpha-synuclein accumulation
Genetic studies that led to the identification of now well-established PD genes, including PRKN, PINK1, SNCA and LRRK22, have undoubtedly been key in elucidating the possible biological basis of the disease. One of our contributors highlights variants in these genes as an important contributing factor to the development of PD.
Carolyn M. Sue, MD, PhD, Australia
As with most complex diseases, there are numerous pathological processes that can contribute to the development of a final common clinical phenotype such as PD. Contributions from each process vary amongst affected individuals in the context of the individual’s predisposition to develop the disorder. In PD, some individual’s genetic background often encodes their risk of developing PD and for other individuals, their vulnerability to the pathological processes that may be triggered by disease process precipitants (e.g., toxin exposure).
Pathogenic mutations in causative PD genes are some of the strongest predisposing risk factors3, with biallelic mutations in some genes (e.g., PRKN, PINK1) being, by and large, fully penetrant. Mutations in other PD genes (e.g., GBA and LRRK2) may increase an individual’s risk, and are not associated with full penetrance. The extent by which aging, environmental factors or changes in lifestyle behaviors can modify disease onset in these mutation carriers is the subject of current research endeavors4.
Wherever an individual lies along this genetic risk spectrum, the two most important cellular processes that contribute to PD pathogenesis are mitochondrial dysfunction and the accumulation of aggregated alpha-synuclein. Each patient with PD may have varying degrees of disruption to each of these cellular pathways that when perturbed, interact to result in a cascade of events that accelerates cellular dysfunction and ultimately leads to the accumulation of toxic protein species such as aggregated alpha-synuclein, and cell death. Strong disruptors of mitochondrial function such as MPTP (or more accurately, MPP+) or rotenone, can cause PD, regardless of genetic status. Milder causes of mitochondrial dysfunction, such as biological aging, lead to more modest reductions in bioenergetic function and a cascade of cellular events including the generation and release of excessive reactive oxygen species, abnormal intraorganellar trafficking, inefficient protein clearance, impaired lysosomal function and the accumulation of alpha-synuclein, all of which can contribute and feed forward to the neurodegenerative process in PD5.
By contrast, aggregations of alpha-synuclein may result from over production of endogenous alpha-synuclein (e.g., SNCA triplications) that can overwhelm otherwise efficient protein clearance pathways. Protein clearance may be impaired due to mutations in GBA, be overloaded with increased exposure to alpha-synuclein or fail as a consequence of reduced bioenergetic function associated with the aging process. Mitochondrial dysfunction can lead to impaired protein clearance and the subsequent accumulation of aggregated alpha-synuclein. Completing the vicious cycle, aggregated alpha-synuclein impairs mitochondrial and lysosomal function to accelerate the neurodegenerative process6,7.
Leucine-rich repeat kinase 2 (lrrk2): a root cause?
As emphasized by one of our contributing authors below, LRRK28,9, has been repeatedly linked to PD since its discovery in the early 2000s, with its encoded protein acting as a notorious multitasker that may contribute to PD’s biological basis in multiple ways.
Matthew Farrer, PhD, USA
Increased LRRK2 kinase activity is the single most important causal factor as it is responsible for PD, whether sporadic10 or in families with a dominantly-inherited disease8,9. Importantly, LRRK2-parkinsonism is clinically indistinguishable from idiopathic late-onset PD11. Half of the genetically-identified LRRK2 individuals with PD that come to autopsy manifest midbrain Lewy body disease12 and satisfy a definitive pathologic diagnosis of PD13. Although there have been no formal epidemiologic studies, and the incidence, prevalence and penetrance figures cited in the literature are mostly clinic-based14, LRRK2 pathogenic substitutions appear to confer the highest individual genotypic/disease-penetrant and population-attributable risk of PD15. LRRK2 coding substitutions tend to be population specific; LRRK2 p.R1441G in the Euskera Basques16, p.G2019S in North African Berbers17,18 and Ashkenazi Jews19, and p.G2385R in South Eastern Asia20, define “patches” of PD on a world map. The majority of these mutant alleles originated from one or few founders, their frequencies evolutionarily increased by recent positive selection21. What that selective allelic advantage is remains to be discovered. In support, LRRK2 is highly expressed in cells of myeloid-lineage in the peripheral immune system and in brain-resident microglia22,23, the LRRK2 promoter contains interferon-gamma responsive elements24, and its level of expression is clearly responsive to inflammatory stimuli25. The locus has also been nominated in genome-wide association studies (GWAS) of idiopathic PD26, progressive supranuclear palsy27 and several chronic inflammatory disorders28,29. As the immune system mediates a host’s response to its environment, and PD has long been considered a multifactorial disorder30, it is attractive to hypothesize successive periods of inflammation and LRRK2 activation may explain the penetrance of LRRK2-parkinsonism, and such a mechanism may partially underlie the ontology and incidence of idiopathic PD that is steadily increasing. Hence, LRRK2 biology currently attracts much academic, philanthropic and pharmaceutical research interest and investment.
All LRRK2 mutations that cause PD activate its kinase activity, directly or indirectly, the most evident being p.G2019S that keeps the hinge of kinase “activation segment” ajar31. Mutations in other Mendelian genes for dominantly-inherited PD, such as vacuolar protein sorting 35 p.D620N32,33, also result in constitutive LRRK2 kinase activation34. Hence, LRRK2 kinase inhibitors are now in human clinical trials given their promise to halt disease progression. Nevertheless, how LRRK2 kinase dysfunction manifests in the selective vulnerability of dopaminergic neurons35, and what inter- (dopaminergic and/or non-cell autonomous) and intra-cellular events underlie their demise is unknown. LRRK2 forms a large, dimeric protein scaffold with several lysosomal targeting motifs36 and protein-interaction domains37. It is intimately associated with endosomal-lysosomal processes, cytoskeletal and vesicular trafficking38, and can phosphorylate multiple substrates including a subset of Rab GTPases39. Therefore, LRRK2’s functions are consequently pleiotropic.
While there is now much data and synthesis, the question of what biology is necessary and sufficient for the ontology of PD remains unanswered, and how can that be elucidated? One approach to an integrated and holistic understanding is to use conditional cre-loxP animal models40 that recapitulate mutant gene dysfunction. As PD is multifactorial, and a function of the relationship between neurons, glia, the immune system and the environment, then models should have these components. It will also be necessary to compare findings between models of different mutant genes implicated in PD to identify their biological “intersection”. While the magnitude of locus associations nominated through GWAS are too small and too pleiotropic to drive comparable neuroscience41, implicating the proteins encoded in a mechanistic “intersection” would be useful and an important validation.
Toxin exposure
Before the identification of genetic contributors such as LRRK2, PD was believed to be an archetypal “non-genetic” disease; a view that was supported by early observations that toxin exposure could cause a parkinsonism phenotype42. To date, toxin exposure is still considered an important but underappreciated (and understudied) contributor to PD that could give important insights into the disease’s pathobiology. This notion is also shared by two contributing authors whose opinions are provided below.
Marieke Dekker, MD, PhD, Tanzania
Genetic susceptibility in combination with exogenous causes, including toxins, are thought to have a cumulative impact on the brain. This results in pathogenic protein accumulation, low-grade inflammation and loss of dopamine-producing neurons. Current disease hypotheses are largely based on research done in high-income countries. Let us focus on Sub-Saharan Africa (SSA), where within a single generation’s time, life expectancy has increased by 10 years to 62 years43, which has implications for age-related disorders such as PD. The mean age of onset of PD in a Tanzanian community-based study from 2008 was 69.4 years. However, a number of recent, mostly hospital-based, studies in SSA report a trend towards a higher prevalence and a possible lower age of PD onset of 55–60 years44,45,46,47 (unpublished data).
Africans are more genetically diverse among themselves than they are with respect to other ethnicities48,49. Presently known PD genes do not seem to play a significant role in Southern and Western African PD patients47,49. Whole genome screening in SSA PD patients is ongoing but very limited47,50. It is unclear whether the lower life expectancy in SSA underestimates genetic factors. It also begs the question of whether the magnitude of genetic risk factors for PD is different in SSA, with perhaps a larger role for inflammation or environmental toxins.
Inflammation in PD has been widely studied but regarded as consequence rather than a cause. No infectious cause has been found. There is epidemiological and in-vitro evidence of some causal link between pesticide exposure and PD, especially in rural populations51. Most of SSA inhabitants rely on subsistence farming, and pesticide use is omnipresent. The longer life expectancy and a young median age of the population make for a large cohort of individuals in rural SSA, who are at risk for potential exposure to neurotoxic substances.
Population stratification by different African populations could enhance clinical neurological observations in PD. Eastern Africa still has exceptional rural non-sedentary populations such as the Tanzanian Hadzabe, one of the last hunter-gatherer tribes in the world, and the Maasai, semi-nomadic pastoralists. Local Tanzanian medical staff and residents, to the best of our knowledge, have never identified a case of PD in the Hadzabe. A largely similar observation applies to the much more numerous Maasai (numbered at ~1 million). Over 40 years of clinical practice in the area have identified just two cases of PD of Maasai origin, both patients being non-pastoralist and with higher education.unpublished data A common factor in the two tribes is a lack of occupational exposure to insecticides, whereas tribe-specific genetic risk factors are unknown. Although life expectancy is shorter in (semi)nomads, it may also imply a lower-than-average risk of PD in those individuals minimally exposed or unexposed to pesticides. It is anticipated that the frequency of PD in SSA is set to increase because of younger onset of disease, ageing, different risk genes, and toxic exposures particularly pesticides. To better understand this increase, large-scale and long-term studies on genetic predisposition and environmental toxin exposure in PD are needed. Also, inclusion of the world’s vanishing ethnic groups with a nomadic or non-pastoralist lifestyle may still help to increase understanding of what causes PD.
To learn a lesson from a neurological disease with a low frequency in persons of African ancestry living in Africa, multiple sclerosis (MS) is estimated to occur there at a rate of only 1–2/100,000 in contrast to 100/100,000 in high income countries52. A recently published study identified Epstein-Barr virus (EBV) infection in late teens or early twenties as an essential risk factor for the development of MS53. EBV infection typically occurs in early childhood in Africa54 and its causal role with malaria in the pathogenesis of Burkitt’s Lymphoma was among the first to demonstrate a role for viruses in malignancies. It is likely that the same environmental exposure to EBV infection in early childhood in Africa is providing protection against the later risk of developing MS. In a disease of unknown etiology, studies involving “the absence of” may just be as important as studies involving “the presence of”. Consequently, involving ethnic groups in Africa with a relative absence of PD may help increase our understanding of the causes of PD.
Artur F. S. Schuh, MD, PhD, Brazil
Much has been said about PD as being probably the fastest-growing neurological disorder, which led some authors to call it a pandemic55. The most common late-onset sporadic form of the disease is widely recognized as a result of an interaction between genetic and environmental factors, and the only way to change this pandemic is to study these many factors. Fortunately, in recent years, massive efforts have been made to understand the genetic factors driving the disease - efforts with the virtue of including previously underrepresented populations and the potential to point out new pathophysiological pathways and treatments. However, the genetic component may explain only around one-third of the disease risk26, and it is implausible that it is responsible for the alarming rising incidence observed in previous decades. Preventing new cases and slowing down this rising curve should be a priority, which can be achieved by also dissecting out environmental factors.
In this regard, there is strong evidence to implicate pesticides as a significant environmental factor associated with the disease, especially paraquat, rotenone, and organochlorines56,57,58. Overall, pesticides may increase the risk of PD and cause an earlier onset and premature death59,60,61. Among them, paraquat is one the most studied and is widely used as a herbicide in many crops worldwide62. Acute poisoning can cause death in humans, and chronic exposure has been associated with PD in epidemiology studies63,64,65,66. Paraquat undergoes redox cycling, producing an excess of oxidative and nitrosative stress, which harms the mitochondria and endoplasmic reticulum and causes apoptosis62,67. The molecule is similar to MPP+, the active compound of MPTP, which causes selective damage to substantia nigra42,68. Many pathophysiological processes usually associated with PD have been replicated by paraquat models, such as suppression of proteasomal degradation, phosphorylation of parkin, and alpha-synuclein modification and accumulation62,69. Interestingly, even developmental exposures seem to produce nigrostriatal toxicity later in life, which is boosted by combination with other pesticides70.
Even though the scientific community is aware of its risks, paraquat is still widely used in many countries. Pesticides are loosely regulated in Latin America, where the prevalence of PD seems even higher compared to the Global North and Asia. For example, Brazil is one of the biggest consumers of pesticides in the world, with hundreds of agrochemicals allowed here and forbidden in other countries. Studying populations from developing countries with significant exposure to pesticides, some banned in other countries, represents an excellent opportunity to understand their relationship with PD and to provide insights into disease pathogenesis. Also, efforts should be made to develop reliable biomarkers of chronic and long-term exposure to pesticides, considering it can happen many years before the onset of the disease. Finally, multi-omic population studies, combining genome and exposome (a measure of all the exposures of an individual over their entire lifetime from conception, and how these relate to health), may represent a significant step to disentangling the complex network of interactions leading to PD.
Toxic alpha-synuclein accumulation and spreading
Over 20 years ago, it was discovered that misfolded alpha-synuclein is the primary constituent of Lewy bodies71, and that pathogenic variants in the alpha-synuclein gene (SNCA) cause familial PD, thereby linking genetics to PD for the very first time72. These discoveries sparked years of further research demonstrating that alpha-synuclein accumulation is not only a hallmark of PD, but that it can also cause neurodegeneration, and forms part of PD’s pathobiology, as commented on below by two contributing authors. The authors comment on the link between alpha-synuclein and mitochondrial dysfunction, the spreading of alpha-synuclein in PD and highlight the utility of imaging tools to detect, better understand and monitor this spreading.
Nobutaka Hattori, MD, PhD, Japan
Most neurodegenerative diseases such as PD, Alzheimer’s disease and Progressive Supranuclear Palsy manifest protein toxicity as one of their critical pathogenic mechanisms, the details of which remain unclear. By systematically deconstructing the nature of toxic proteins, we aim to elucidate and illuminate some of the essential mechanisms of protein toxicity from which therapeutic insights may be drawn. In PD, alpha-synuclein as a potential prion-mimicking protein has been reported. Αlpha-synuclein can adopt a β-sheet rich structure that forms toxic oligomeric aggregates that accumulate within neurites in the central nervous system and the peripheral nervous system. Such alpha-synuclein can be secreted, taken up by neighboring cells (bodies, dendrites, or axons), and thus induce seeding and spread of the toxic oligomers. Suppose abnormal alpha-synuclein is generated in the intestinal tract and inflammation occurs in the intestinal tract73. In that case, it is thought that the seed may become elongated and enter the bloodstream or spread to the brain by ascending the vagus nerve or sympathetic nerves. Many people can eliminate the abnormal alpha-synuclein seed, but if they cannot do so, the seed potential is thought to increase and propagate. In fact, it has been reported that T cells that recognize synuclein peptides are more common in PD, making it highly likely that some immunological mechanism is at work.
Recently, CHCHD2 as a causative gene for hereditary PD has been identified74. CHCHD2 mutants result in reduced oxygen consumption and ATP production. Although CHCHD2 is localized to mitochondria and is known to be involved in the electron transfer system, brain pathology of individuals with the CHCHD2 T61I mutation has shown an accumulation of alpha-synuclein throughout the brain. In addition, the co-expression of alpha-synuclein with the CHCHD2 mutant in a Drosophila model resulted in increased alpha-synuclein accumulation and a shortened lifespan. In the knockout mouse model, expansion of alpha-synuclein and p62 has been observed. Therefore, a hypothetical scenario of the two possible series of molecular events is postulated intervening between either synaptic alpha-synuclein deposition leading to induction of mitochondrial dysfunction, or mitochondrial functional deficits leading to accumulation of alpha-synuclein at the synapse6. Notably, in both situations, Ca2+ rise and production of oxidative stress mediators are pivotally involved in the interconnection between mitochondrial impairment and alpha-synuclein synaptic pathology. Abnormal alpha-synuclein seed and mitochondrial dysfunction are closely related. In other words, the possibility that mitochondrial dysfunction may induce protein toxicity should also be considered.
Irena Rektorová, MD, PhD, Czech Republic
Based on a recent hypothesis75, alpha-synuclein in PD spreads in a prion-like fashion either from the gastrointestinal tract all the way to the brain following the Braak staging (so-called “body first” PD or dementia with Lewy bodies; DLB) with more malignant and symmetric course of the disease, or the brain pathology starts asymmetrically in the amygdala/limbic system and from there it spreads to the substantia nigra, locus coeruleus and other brain (cortical and subcortical) structures and the body via the autonomic nervous system (so-called “brain first” PD) with a presumably less malignant disease course. Our work is focused on studying which of these two options is the most plausible. We do this performing behavioral-magnetic resonance imaging (MRI)-immunohistochemical studies in several models of PD including a transgenic TNWT-61 mouse model overproducing human alpha-synuclein in the brain76,77, a toxic methamphetamine model showing massive loss of TH-stained cells in the striatum78 or a rotenone model that follows Braak staging with alpha-synuclein spreading from gut to the brain via the vagal dorsal motor nucleus (DMN)79.
We used MRI and the diffusion kurtosis imaging (DKI) method which precisely evaluates non-gaussian diffusion of water molecules in gray matter and is an indicator of the heterogeneous environment with restrictions to diffusion. We confirmed that DKI is a sensitive translational marker of alpha-synuclein and alpha-synuclein-induced brain pathology early in the course of the disease (in preclinical stages)79 as well as a dynamic marker for monitoring the spread of brain pathology in all of these animal models and in humans with PD of various cognitive subtypes77,79,80. Mean kurtosis values correlated with the amount of alpha-synuclein in the thalamus of TNWT-61 mice76. However, once alpha-synuclein reached the cortex and cognitive symptoms occurred, neurodegeneration (brain atrophy) was the major player on the scene78,79,80.
Another question still remains to be answered: How does alpha-synuclein spread in the brain? It has been hypothesized that brain atrophy progression in Lewy body diseases is shaped by (increased) connectivity and local vulnerability, with alpha-synuclein spreading trans-synaptically, via brain networks81. Synaptic plasticity can be studied by functional MRI (fMRI). Resting state fMRI is characterized by low-frequency BOLD (blood oxygen level dependent) fluctuations and various analytical methods can be used for studying large-scale brain networks connectivity. Temporal dynamics of large-scale brain networks alterations can be further assessed by dynamic fMRI82 and EEG microstates (MS); the latter representing transient, quasi-stable patterns of EEG that provide us with detailed information about spatial and temporal characteristics of EEG within well-described brain networks83. Hyperconnectivity/ hyperactivity of motor and particularly cognitive brain networks have been documented in early stages of PD with normal cognition84,85 as well as in prodromal stages of Alzheimer’s disease or frontotemporal dementia86 or prodromal DLB83. These alterations affect disease-specific vulnerable networks and are considered as compensatory mechanisms, however representing rather maladaptation. In line with this notion, we have demonstrated that increased occurrence of visual network EEG MS in prodromal DLB is associated with lower dominant alpha frequency (which is a supportive diagnostic biomarker of DLB)83. This network overactivity seems to decrease as the disease progresses86,87.
Taken together, our work indicates that diffusion MRI may detect early microstructural changes that reflect alpha-synuclein-induced brain changes already in preclinical stages of PD and can be used to monitor brain pathology spreading until major neurodegeneration occurs. We hypothesize that this brain pathology spreads via large-scale disease-specific brain networks that show hyperconnectivity (hyperactivity) in early disease stages.
A complex interaction of multiple factors
To date, the vast majority of PD research has been conducted based on the assumption that PD is a single disease88. However, the complex nature of PD has led some, including four contributing authors, to reconsider PD as multiple diseases (instead of a single entity), triggered by complex interactions of many factors that cumulatively push cells past their tipping point.
Mohamed Salama, MD, PhD, Egypt
PD is best described as a complex disease, of which the complexity is observed on different layers. The first layer of complexity can be seen in its nature; our understanding of PD has changed recently from being a localized neurologic disorder to accepting it as a systemic disease with multiple affections of various systems which reflects an advanced understanding of disease initiation and progression89. Another layer of complexity is expressed in the diverse clinical subtypes and even different phenotypes within each of these subtypes90. The new appreciation of PD as a mixture of different disorders motivated Farrow and colleagues to conceptualize what they called the “Parkinson’s Diseases Mountain Range model”88. It is generally believed that PD develops in response to the interaction of genetic and environmental factors, which has been proven previously in several works including research done by our group in Egypt91. The conventional patho-mechanistic model adopted for developing PD is the initial trigger (most probably a genetic risk) that increases the vulnerability to the effect of an environmental factor. This linear–chronological model, however, will not be able to accommodate the recent definition of PD as a diverse and complex mixture of disorders.
I believe that a “threshold model” could better embrace the different mechanisms for developing PD replacing the old (trigger and modifiers) linear one. In this model, the detrimental factor for developing the disease is the threshold after which cells will go into the cycle of degeneration. So, we could have different types of triggers that may lead to the development of the disease. These different triggers—that can work simultaneously—will build up stress on different systems until reaching the damage threshold, which usually does not start in the same time point in all systems, hence, developing the initial stage(s) of the disease. This could justify the earlier non-motor signs of PD that reflect early, non-midbrain damage. The idea of having a threshold, that once exceeded leads to damage, has been widely accepted as a mechanism for neurodegenerative disorders92.
We understand now that the disease initiation process is not following a single model. So, in one case, a pathogenic gene variant could interact with different environmental factors causing the disease (gene-environment interaction)93. In another case, different genetic variants could interact to cause the disease (gene–gene interaction)94, or different pollutants can interact to lead to the disease (environment-environment interaction)95. This can be even extended to involve more factors e.g., social stressors, metabolic diseases . etc. This model could allow us to adopt a more holistic – exposomic understanding of factors leading to PD instead of looking for a single interaction, which cannot be validated given the complex exposures a human being could have throughout their lifetime.
Given the diversity in causes, mechanisms and disease processes, it seems mandatory to adopt a more diverse stratification strategy for PD cases. This strategy should accept having different diseases within the spectrum of PD. This better understanding will be reflected in precision medicine approaches tailored to each specific sub-type’s features and needs.
George D. Mellick, PhD, Australia
The fundamental question regarding the biological basis for PD has led to tensions between geneticists and toxicologists, clinical “lumpers” and “splitters” and pathologists using very strict or more liberal diagnostic criteria. We now need to move beyond disciplinary boundaries to make progress. PD should be considered a syndromic spectrum of many different primary conditions. We know this because rare inheritable forms of Parkinsonism are genetically validated to involve different processes. Genetics has provided many critical clues to etiology since linkage studies in the Contursi kindred heralded the synuclein era72. Similarly, the “discovery” that toxins such as MPTP can induce Parkinsonism in humans and the identification of environmental risk factors demonstrate that non-genetic variables also have substantial impact. The truth is that every person living with PD has a unique expression of the dysfunction and precise balance of factors contributing to this idiosyncratic disease journey.
Excepting that there are unique combinations of contributing factors, there are also commonalities, which can inform our thinking and help to focus research efforts.
-
i.
PD is progressive and degenerative. Whilst death of dopaminergic neurons is an obvious consequence of the pathology, etiological investigations need to extend well beyond these cell types and brain tissue. The spread of synucleinopathies is important evidence that the initial problem(s) likely arise from areas outside the brain96,97.
-
ii.
PD involves the transformation of a “normal” homeostatic process past a tipping point towards a pathological spiral into cellular degeneration; a cascade of secondary and self-reinforcing sequalae such as neuroinflammation, abnormal protein aggregation and metabolic imbalance. Where precisely this is initiated remains elusive, and it may well differ from person to person. The fact that an identical genetic lesion can lead to different pathologies attests to this98.
-
iii.
The disease is triggered many years before symptoms of Parkinsonism become obvious and is ongoing in many people who never present with any symptoms99.
I believe that appreciating the commonalities while embracing each patient’s idiosyncrasies is key to understanding causal triggers and etiologically informed interventions.
Christine Klein, MD, Germany
When Bastiaan Bloem invited me to jointly write a Seminar “Parkinson’s Disease” for the Lancet in 2020, we both felt that the more appropriate title for this piece would have been “Parkinson’s Diseases”100, reflecting not only the plethora of known—mostly genetic—causes of clinical syndromes resembling idiopathic PD but also the fact that each person with PD suffers from his/her own PD not only in terms of disease expression but also cause and modifying factors. Clearly, genetics has been a key driver in our understanding of the etiology of PD and now as a contributor to first attempts at targeted treatment, all of this sparked by the discovery of alpha-synuclein mutations as the first established monogenic cause of PD 25 years ago72.
With additional forms of monogenic PD discovered in rapid succession, it quickly became obvious that there are multiple primary causes of PD101, which collectively explain almost 15% of PD(s)102. and Westenberger et al. in preparation Elegant functional work on shared pathways of the encoded proteins has led to the notion that multiple different pathogenic events converge on one (or more) final common pathway(s) resulting in dopaminergic neurodegeneration and, eventually, the clinical manifestation of PD.
After alpha-synuclein toxicity, mitochondrial dysfunction is probably the next best-established factor in the etiology of PD. Very recently, mitochondria were found to be directly connected to alpha-synuclein conversion from monomeric to oligomeric states in neurons with intracellular seeding events occurring preferentially at mitochondrial membranes where the mitochondrial lipid cardiolipin triggered the oligomerization of mutated alpha-synuclein103. However, it has also been demonstrated that “mitochondrial forms” of PD due to pathogenic variants in PRKN do not necessarily result in the formation of Lewy bodies, although typical Lewy body pathology has also been described in carriers of biallelic pathogenic PRKN variants104. To my mind, these findings do not contradict each other but rather support the notion that PD(s) is/are etiologically diverse.
Monogenic forms of PD, at least those that appear to be fully penetrant (such as PRKN pathogenic variants or alpha-synuclein triplications), raise the intriguing question as to when PD begins in carriers of such variants. In keeping with the recent biological classification of Huntington’s disease characterizing individuals for research purposes from birth starting at Stage 0 (individuals with a pathologically expanded repeat but without any detectable pathological change)105, one may postulate that there are “congenital” forms of PD, as well. This notion has major implications not only for our understanding of (some of) the etiology/ies of PD but also its preclinical phases, offering a much larger and much earlier window of potential intervention. A knock-in Huntington’s disease mouse model (HdhQ7/Q11) showed clear changes in cortical circuit physiology shortly after birth, which was “self-corrected” in the second week of life. Treating pups during the first week of life with an ampakine, thereby correcting the glutamatergic circuit defect, prevented the development of Huntington’s disease-like signs in these mice106. These findings suggest that functional abnormalities at the earliest stages of life can potentially be rescued and that treatment of (inherited) neurodegenerative diseases might have to start at the soonest possible time point.
Even with my geneticist’s hat on, I cannot reasonably postulate that “all PD” is genetic even though some of the ‘causal’ PD genes have been shown to play a role also in rare variant burden and even common variant risk107. Indeed, we have seen fascinating and independently confirmed developments also in the field of genetic risk factors and polygenic risk scores26,108,109 contributing to the etiology of PD. Furthermore, it is conceivable that pathogenic events occur and accumulate over an individual’s lifetime: For example, we have recently demonstrated a relationship between mitochondrial variant burden and development of PD in carriers of heterozygous PRKN and PINK1 pathogenic variants110.
Finally, while the field is preoccupied with finding causal and risk factors of PD, I would like to encourage the study and identification of protective and compensatory mechanisms, counteracting the development of PD and, thus, “interfering with” or even serving as a counterpart of its etiology. Some of the protective factors proposed include anti-inflammatory agents, antioxidants, calcium channel agonists, inhibitors of alpha-synuclein aggregation, neurotrophic factors and protective lifestyle factors, such as coffee drinking however this area of research is not yet well developed and requires further study111. Recently, “polygenic resilience” has been shown to reduce the penetrance of PD polygenic risk factors with a higher polygenic resilience score being associated with a lower risk for PD112. Intriguingly, also the effects of pathogenic variants causing monogenic PD can be mitigated by protective factors. For example, tobacco use and black tea consumption have independent but additive effects on delaying age of onset in carriers of pathogenic LRRK2 variants113.
A. Jon Stoessl, MD, Canada
More than 30 years ago, my mentor Donald Calne suggested that PD was a syndrome and not a single disease114. This has become increasingly obvious if for no other reason than the identification of several monogenic forms of PD, not all of which are associated with alpha-synuclein pathology. That said, and despite the recent failures of monoclonal antibodies targeting misfolded alpha-synuclein115,116, there can be little doubt that alpha-synuclein misfolding plays a critical role in a majority of PD, with numerous mechanisms postulated to lead to impaired clearance of misfolded protein. However, it may be worth asking whether PD is a synaptopathy that is associated in most cases with abnormal alpha-synuclein deposition, in others with impaired mitochondrial function, and perhaps in others with unknown underlying pathophysiology.
Some environmental contributors to PD may result in abnormal aggregation of alpha-synuclein (e.g., air pollution117; viral infection118). Regardless of which mechanism is proposed, hypotheses to date, including that of prion-like propagation of Lewy pathology, have for the most part failed to account for the (partial) neuronal selectivity of PD. Despite the undeniable importance of pathology in non-dopaminergic neurons, one cannot ignore the selective vulnerability of midbrain dopamine neurons and their consistent pattern of involvement, including asymmetry, even in genetic cases. High terminal arborization may contribute119, but any theory on cause must take into consideration the evidence that degeneration of dopaminergic neurons likely begins in nerve terminals, not in neuronal cell bodies35,120. Therefore, a focus in the future should be on changes that occur at the nigrostriatal terminal and those factors that may contribute not only to the vulnerability of dopamine neurons, but also on the somatotopic selectivity within that population. From that perspective, the role of corticostriatal activity has curiously received inadequate attention and deserves more121.
Three other areas deserve further attention in determining why people get PD. One is the role of infection and neuroinflammation. Alpha-Synuclein production and misfolding are triggered by infection118 and alpha-synuclein expression is required for Type 1 interferon responses122. It is thus of interest that exposure of mice to low levels of SARS-CoV-2 virus renders them susceptible to subtoxic doses of MPTP123. Mitochondria, whose dysfunction is the other key proposed culprit, particularly in recessively inherited forms of PD, are required for antigen presentation and that function is repressed by PINK1 and Parkin124. A second area that has been largely ignored is the contribution of developmental changes (see ref. 125 for a recent review) and stressors that occurred earlier in life (e.g. ref. 126) to the later appearance of selective neurodegeneration. In this respect, it is of great interest that in Huntington’s disease, while clinical manifestations do not occur until much later, there is evidence of extensive marked developmental pathology127. Finally, theories on PD have been excessively neuron-centric. It would be of great value to explore the contributions of other cells, particularly astrocytes128. Astrocytes play a major role in brain metabolism and synaptic function129 and are subject to mitochondrial oxidative phosphorylation defects in PD130. In addition to alterations in neurotransmitter reuptake and participating in the synaptopathy that may underlie PD, astrocytic dysfunction may contribute to impairment of glymphatic function and additional defects in clearance of misfolded protein. Astrocytes derived from induced pluripotent stem cells in patients with LRRK2 PD express increased alpha-synuclein, resulting in altered calcium homeostasis and increased cytokine release upon inflammatory stimulation131 and display altered morphology of extracellular vesicles and altered morphology and distribution of multi-vesicular bodies, with overaccumulation of phosphorylated alpha-synuclein and reduced trophic support and viability of dopamine neurons132. Astrocytes from LRRK2 knock-in mice have abnormal trafficking of glutamate transport133 and are impaired in their ability to internalize and degrade fibrillar alpha-synuclein134. Prevention of microglial-mediated conversion of astrocytes to the pro-inflammatory state by a GLP1R agonist is protective in both preformed fibril and human alpha-synuclein transgenic mouse models of PD135.
The microbiome–gut–brain axis and possible origin points
Ai Huey Tan, MD, PhD and Shen-Yang Lim, MD, Malaysia
The heterogeneity of PD continues to be a major conundrum in our field. Differences in the phenotypes at presentation (e.g., the presence/absence of prodromal features such as REM sleep behavior disorder [RBD], constipation and/or hyposmia, and the variability in age of onset and family history) and during the disease course (e.g., rapid vs. slow progression, differential response to dopaminergic medication, presence/absence of non-motor symptoms such as cognitive impairment and autonomic dysfunction which substantially impact clinical outcome) all suggest that there might be differences between individuals with PD, in the “starting point” (i.e., the anatomical location and triggers/causative factors), and in the pattern, speed and extent of spread of PD pathology.
With regards to differences in the starting anatomical location, there has been a growing body of literature on a central (i.e., “brain-first”) vs. peripheral (i.e., “body-first”) origin of PD pathology. The discovery, in the 1980s, of alpha-synuclein deposits in the enteric nervous system (ENS) of PD patients136, and subsequent observations that constipation and alpha-synuclein deposits in gastrointestinal biopsies can predate the diagnosis of PD by decades, as well as the marked peripheral autonomic nervous system imaging abnormalities with relative sparing of the nigrostriatal dopaminergic system in patients with prodromal PD (isolated RBD)137,138,139,140 have implicated the gut as a possible site of origin in PD. It has been postulated that insults acting on the gut could trigger the misfolding and aggregation of alpha-synuclein in the ENS, subsequently propagating into the brain via prion-like cell-to-cell transfer, with the vagus nerve serving as a conduit141. Imaging, neuropathological and animal/experimental studies have provided evidence both for and against this hypothesis, while the epidemiological association between full truncal vagotomy and PD risk remains inconclusive142. We can perhaps speculate that these mixed findings suggest that a single theory for PD origin is less likely, and central or peripheral degenerative processes may occur at varying timepoints and degrees in different patients. Although earlier autopsy studies by Braak et al. suggested a dual-hit hypothesis (i.e., simultaneous peripheral and central origin with entry points through the dorsal motor nucleus of the vagus [DMNV] and the olfactory bulb [OB], respectively)143, a recent re-analysis of post-mortem datasets revealed the lack of concomitant DMNV and OB pathology, suggesting that the pathologic process starts in either location, but rarely simultaneously144. Recent studies also indicate that, once begun, dysfunction and pathology can spread bidirectionally, from gut-to-brain, as well as from brain-to-gut145,146.
Besides anatomical heterogeneity, inter-individual differences in triggers/causative factors and pathogenic processes are likely to be at play. For example, there is convincing evidence that gastrointestinal inflammation occurs in PD147,148,149, at least in a subset of patients, which can be due to alterations in the composition and/or activities of the gut microbiome, as well as genetic variation (e.g., in LRRK2)142,150,151. In turn, an inflammatory gut environment is postulated to promote aggregation of alpha-synuclein in the ENS, as well as inducing gut hyperpermeability to toxins and other factors, with far-reaching effects on the brain (e.g., disrupting the blood-brain barrier and triggering a cascade of events culminating in neuroinflammation and neurodegeneration)142. Over the past decade, a swathe of studies in PD patients have been published reporting alterations in gut microbiome composition and function152,153,154 with, for example, enrichment of Enterobacteriaceae that express highly immune-stimulatory lipopolysaccharide (that activate Toll-like receptors) or produce curli (an amyloidogenic protein that can template α- synuclein aggregation in the gut)154,155; and depletion of bacteria producing beneficial/neuroprotective molecules such as short-chain fatty acids (deficits in which are linked to constipation, gut barrier dysfunction and inflammation)152,153,154. Corroborating these (largely correlative) human observations, animal models suggest that gut microbial-related factors can contribute crucially to PD-like pathogenesis142,155. However, definitive proof that they are causative in human PD will likely require large-scale community-based epidemiological studies with multiple sampling over many years to unravel the evolution of gut-related changes in PD. Whether and how lifestyle factors (caffeine intake, cigarette smoking, environmental toxin exposure) that modulate PD risk impact the gut microbiome is also ripe for study156.
Importantly, although gut inflammation and alpha-synuclein aggregation might be common events, we believe that PD ensues only when additional contributing factors, such as host genetic vulnerability or ageing, are present155,157,158, with ethno-geographic factors also playing a role153,159. Ultimately, a deeper understanding of potentially modifiable factors and events operating along the microbiome-gut-brain axis will open up new ways to prevent or change the course of this disabling disease142,160.
A combination of social, biological and environmental factors
René L. Vidal, PhD, Chile
Movement disorders (MDs) encompass heterogeneous nervous system conditions that cause either an excess of movement or a paucity of voluntary and involuntary movements161. The most prevalent MD, PD, affected over 6.1 million people in 2016, and the number of affected individuals have increased 2.4-fold from 1990 to 2016 and continues to grow162. In 2017, the economic burden of PD only in the USA was U$51.9 billion163. Most MDs feature a long prodromal phase, providing unique opportunities to identify significant risk factors for early detection and intervention, to ultimately improve patient care.
For unknown reasons, Chile, with its partially isolated environments combined with socioeconomic inequalities in rural and urban sectors, has the highest prevalence of PD in Latin America164. Ethnically, the Chilean population has an admixed genetic composition of European and South Amerindian ancestries, e.g., Aymaras and Mapuche. These unique ancestral combinations may be involved in the prevalence of MDs. For these reasons it is very relevant to dissect the social, environmental, and genetic factors associated with MDs such as PD in Chile or other South Amerindian populations.
During the past decade, epidemiological and demographic data from Chile, has been used to generate a unique and publicly accessible resource: a nationwide de-identified individual-level electronic health record database. In addition, we have access to clinical statistics (i.e., inpatient services) from the Ministry of Health through the Department of Health Statistics and Information (DHSI) and to environmental factor exposure data (i.e., registry of contaminants by geographic districts) through the Ministry of Environment and others. We have identified a population of more than 37,000 PD cases over the last 20 years who are mainly concentrated in overpopulated or industrialized regions, which once again demonstrates the impact of environmental factors on the development of this type of pathology. Other important epidemiological estimates such as regional disease prevalence, progressions (retrospectively), comorbidities, mortality, social determinants, and disease economic burden also arise as relevant factors involved in PD progression.
Moreover, it is important to consider how biological factors such as genomic variants, gene expression profiles, protein networks, among others associate with the phenotypes observed in individuals with PD. In this line, we recently investigated potential biomolecules in blood samples such as Insulin-like growth factor 2 (IGF2) and autophagic pathways which could play central roles in disease pathogenesis. Our preliminary work has shown a significant decrease in autophagy activity together with a drastic downregulation of IGF2 in peripheral blood mononuclear cell from Chilean persons with PD165.
Finally, in PD there are several subcategories related to severities and progressions, and its biological basis is largely unknown. For this reason, it is very important to validate the biological factors in human PD samples and in short-lived disease models at different times of disease progression, which may contribute to understanding the molecular basis associated with differing severity and progression of this disease. I believe that the combinations of biological factors such as genetic variations, protein modifications, autophagic dysfunction, alpha-synuclein accumulation and growth factor levels among others in blood samples, together with environmental and social factors could explain the heterogeneity of motor and nonmotor symptoms observed in PD patients.
Concluding remarks
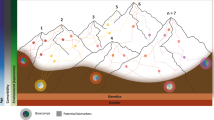
It is clear from the personal viewpoints above that there are a myriad of differing opinions on the biological basis of PD, as illustrated in Fig. 1. This shows that the PD experts are not in agreement with what exactly goes wrong for someone to develop the disorder. While some views overlap in terms of considering genetic factors, alpha-synuclein accumulation, exposure to mitochondrial toxins and neuroinflammation as key etiological drivers, we still do not know what exactly the cause of PD is. It should be noted that most of the views were from researchers with MD, PhD backgrounds which may have introduced a bias in the responses.

The gray box around all these factors represents the notion that multiple or all of these factors may collectively contribute to disease onset. Perspectives, future directions, and take-home messages related to these discussed factors are highlighted in green boxes.
We can consider that the etiology of the disorder is multifactorial and due to variances in individual genetic make-up, environment and lifestyle, is different in each person with PD. With the more or less highly penetrant monogenic or strong environmental causes, our etiologic understanding currently represents only the tip of the iceberg. Paradoxically, it will require both a lumper’s as well as a splitter’s approach to fully embrace the etiology of PD. Additional layers of omics studies, including exposomics, are warranted to further explore and potentially disentangle different etiologic contributions and their (epigenetic) changes over an individual’s lifetime. Consequently, this means that each person may have their own unique form of the disease.
From a translational perspective, with a view to more effective treatment or even cure, one may envision the development of a “cocktail” of drugs targeting alpha-synuclein accumulation, restoring GBA levels and mitochondrial function, and mitigating the effects of environmental exposures, with its “ingredients” possibly adjusted according to each individual’s etiological contributions in a personalized fashion.
Data availability
No datasets were generated or analyzed for this article.
References
Del Rey, N. L.-G. et al. Advances in Parkinson’s disease: 200 years later. Front. Neuroanat. 12, 113 (2018).
Cherian, A. & Divya, K. P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 120, 1297–1305 (2020).
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
San Luciano, M. et al. Nonsteroidal anti-inflammatory use and LRRK2 Parkinson’s disease penetrance. Mov. Disord. J. Mov. Disord. Soc. 35, 1755–1764 (2020).
Grünewald, A., Kumar, K. R. & Sue, C. M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 177, 73–93 (2019).
Zaltieri, M. et al. Mitochondrial dysfunction and α-synuclein synaptic pathology in Parkinson’s disease: who’s on first? Park. Dis. 2015, 108029 (2015).
Ganjam, G. K. et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 10, 865 (2019).
Zimprich, A. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004).
Paisán-Ruíz, C. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004).
Hulihan, M. M. et al. LRRK2 Gly2019Ser penetrance in Arab–Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 7, 591–594 (2008).
Aasly, J. O. et al. Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson’s disease: LRRK2 P.Asn1437His and Parkinson’s disease. Mov. Disord. 25, 2156–2163 (2010).
Ross, O. A. et al. Lrrk2 and Lewy body disease. Ann. Neurol. 59, 388–393 (2006).
Goedert, M., Spillantini, M. G., Del Tredici, K. & Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24 (2013).
Healy, D. G. et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590 (2008).
Ross, O. A. et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case–control study. Lancet Neurol. 10, 898–908 (2011).
Mata, I. F. et al. Lrrk2 R1441G-related Parkinson’s disease: evidence of a common founding event in the seventh century in Northern Spain. Neurogenetics 10, 347–353 (2009).
Ishihara, L. et al. Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch. Neurol. 63, 1250 (2006).
Lesage, S. et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N. Engl. J. Med. 354, 422–423 (2006).
Ozelius, L. J. et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 354, 424–425 (2006).
Farrer, M. J. et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinson. Relat. Disord. 13, 89–92 (2007).
Voight, B. F., Kudaravalli, S., Wen, X. & Pritchard, J. K. A map of recent positive selection in the human genome. PLoS Biol. 4, e72 (2006).
Hakimi, M. et al. Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural Transm. 118, 795–808 (2011).
Wallings, R. L. & Tansey, M. G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 47, 1581–1595 (2019).
Gardet, A. et al. LRRK2 is involved in the IFN-γ response and host response to pathogens. J. Immunol. 185, 5577–5585 (2010).
Shutinoski, B. et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl. Med. 11, eaas9292 (2019).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Jabbari, E. et al. Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol. 20, 107–116 (2021).
Li, Y. R. et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med. 21, 1018–1027 (2015).
Witoelar, A. et al. Genome-wide pleiotropy between Parkinson Disease And Autoimmune Diseases. JAMA Neurol. 74, 780 (2017).
Farrer, M. J. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat. Rev. Genet. 7, 306–318 (2006).
Alessi, D. R. & Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 360, 36–37 (2018).
The Austrian VPS-35 Investigators Team. et al. VPS35 Parkinson’s disease phenotype resembles the sporadic disease. J. Neural Transm. 121, 755–759 (2014).
Vilariño-Güell, C. et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167 (2011).
Mir, R. et al. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 475, 1861–1883 (2018).
Burke, R. E. & O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 246, 72–83 (2013).
Orenstein, S. J. et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406 (2013).
Deniston, C. K. et al. Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature 588, 344–349 (2020).
Bonet‐Ponce, L. & Cookson, M. R. LRRK2 recruitment, activity, and function in organelles. FEBS J. 289, 6871–6890 (2022).
Gomez, R. C., Wawro, P., Lis, P., Alessi, D. R. & Pfeffer, S. R. Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J. Cell Biol. 218, 4157–4170 (2019).
Tian, X. & Zhou, B. Strategies for site-specific recombination with high efficiency and precise spatiotemporal resolution. J. Biol. Chem. 296, 100509 (2021).
Crouch, D. J. M. & Bodmer, W. F. Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc. Natl Acad. Sci. 117, 18924–18933 (2020).
Langston, J. W., Ballard, P., Tetrud, J. W. & Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 (1983).
Gapminder. https://www.gapminder.org/.
Howlett, W. P. et al. Neurological disorders in Northern Tanzania: A 6-year prospective hospital-based case series. Afr. Health Sci. 22, 269–284 (2022).
Cilia, R. et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain J. Neurol. 137, 2731–2742 (2014).
Amod, F. H. & Bhigjee, A. I. Clinical series of Parkinson’s disease in KwaZulu-Natal, South Africa: Retrospective chart review. J. Neurol. Sci. 401, 62–65 (2019).
van Rensburg, Z. J. et al. The South African Parkinson’s disease study collection. Mov. Disord. J. Mov. Disord. Soc. 37, 230–232 (2022).
Dotchin, C. et al. The prevalence of Parkinson’s disease in rural Tanzania. Mov. Disord. J. Mov. Disord. Soc. 23, 1567–1672 (2008).
Dekker, M. C. J. et al. Parkinson’s disease research on the African continent: obstacles and opportunities. Front. Neurol. 11, 512 (2020).
Rizig, M. et al. The International Parkinson Disease Genomics Consortium Africa. Lancet Neurol. 20, 335 (2021).
Dorsey, E. R., Sherer, T., Okun, M. S. & Bloem, B. R. The emerging evidence of the parkinson pandemic. J. Park. Dis. 8, S3–S8 (2018).
Leray, E., Moreau, T., Fromont, A. & Edan, G. Epidemiology of multiple sclerosis. Rev. Neurol.172, 3–13 (2016).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Coleman, C. B. et al. Epstein-barr virus type 2 infects t cells in healthy Kenyan children. J. Infect. Dis. 216, 670–677 (2017).
Dorsey, E. R. & Bloem, B. R. The Parkinson pandemic-a call to action. JAMA Neurol. 75, 9–10 (2018).
De Miranda, B. R., Goldman, S. M., Miller, G. W., Greenamyre, J. T. & Dorsey, E. R. Preventing Parkinson’s disease: an environmental agenda. J. Park. Dis. 12, 45–68 (2022).
Ascherio, A. & Schwarzschild, M. A. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 15, 1257–1272 (2016).
Narayan, S., Liew, Z., Bronstein, J. M. & Ritz, B. Occupational pesticide use and Parkinson’s disease in the Parkinson Environment Gene (PEG) study. Environ. Int. 107, 266–273 (2017).
Schneider Medeiros, M. et al. Occupational pesticide exposure and the risk of death in patients with Parkinson’s disease: an observational study in southern Brazil. Environ. Health Glob. Access Sci. Source 19, 68 (2020).
Gamache, P.-L. et al. Exposure to pesticides and welding hastens the age-at-onset of Parkinson’s disease. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 46, 711–716 (2019).
Poortvliet, P. C., Gluch, A., Silburn, P. A. & Mellick, G. D. The Queensland Parkinson’s Project: an overview of 20 years of mortality from Parkinson’s disease. J. Mov. Disord. 14, 34–41 (2021).
See, W. Z. C., Naidu, R. & Tang, K. S. Cellular and molecular events leading to paraquat-induced apoptosis: mechanistic insights into parkinson’s disease pathophysiology. Mol. Neurobiol. 59, 3353–3369 (2022).
Tangamornsuksan, W. et al. Paraquat exposure and Parkinson’s disease: a systematic review and meta-analysis. Arch. Environ. Occup. Health 74, 225–238 (2019).
Tanner, C. M. et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 119, 866–872 (2011).
Tanner, C. M. et al. Occupation and risk of parkinsonism: a multicenter case-control study. Arch. Neurol. 66, 1106–1113 (2009).
Costello, S., Cockburn, M., Bronstein, J., Zhang, X. & Ritz, B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol. 169, 919–926 (2009).
Tsai, W.-T. A review on environmental exposure and health risks of herbicide paraquat. Toxicol. Environ. Chem. 95, 197–206 (2013).
Langston, J. W. The MPTP story. J. Park. Dis. 7, S11–S19 (2017).
Vellingiri, B. et al. Neurotoxicity of pesticides—a link to neurodegeneration. Ecotoxicol. Environ. Saf. 243, 113972 (2022).
Cory-Slechta, D. A., Thiruchelvam, M., Barlow, B. K. & Richfield, E. K. Developmental pesticide models of the Parkinson disease phenotype. Environ. Health Perspect. 113, 1263–1270 (2005).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Polymeropoulos, M. H. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997).
Houser, M. C. & Tansey, M. G. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Park. Dis. 3, 3 (2017).
Funayama, M. et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol. 14, 274–282 (2015).
Horsager, J. et al. Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain 143, 3077–3088 (2020).
Khairnar, A. et al. Late-stage α-synuclein accumulation in TNWT-61 mouse model of Parkinson’s disease detected by diffusion kurtosis imaging. J. Neurochem 136, 1259–1269 (2016).
Khairnar, A. et al. Early and progressive microstructural brain changes in mice overexpressing human α-Synuclein detected by diffusion kurtosis imaging. Brain Behav. Immun. 61, 197–208 (2017).
Arab, A. et al. Diffusion Kurtosis imaging detects microstructural changes in a methamphetamine-induced mouse model of Parkinson’s disease. Neurotox. Res 36, 724–735 (2019).
Khairnar, A. et al. Diffusion Kurtosis imaging detects the time-dependent progress of pathological changes in the oral rotenone mouse model of Parkinson’s disease. J. Neurochem 158, 779–797 (2021).
Sejnoha Minsterova, A. et al. Patterns of diffusion kurtosis changes in Parkinson’s disease subtypes. Parkinson. Relat. Disord. 81, 96–102 (2020).
Tremblay, C. et al. Brain atrophy progression in Parkinson’s disease is shaped by connectivity and local vulnerability. Brain Commun. 3, fcab269 (2021).
Mitterová, K. et al. Dynamic functional connectivity signifies the joint impact of dance intervention and cognitive reserve. Front Aging Neurosci. 13, 724094 (2021).
Lamoš, M., Morávková, I., Ondráček, D., Bočková, M. & Rektorová, I. Altered spatiotemporal dynamics of the resting brain in mild cognitive impairment with lewy bodies. Mov. Disord. 36, 2435–2440 (2021).
Anderkova, L., Barton, M. & Rektorova, I. Striato-cortical connections in Parkinson’s and Alzheimer’s diseases: relation to cognition. Mov. Disord. 32, 917–922 (2017).
Klobušiaková, P., Mareček, R., Fousek, J., Výtvarová, E. & Rektorová, I. Connectivity between brain networks dynamically reflects cognitive status of parkinson’s disease: a longitudinal study. J. Alzheimers Dis. 67, 3233–180834 (2019).
Bonanni, L. et al. Hyperconnectivity in dementia is early and focal and wanes with progression. Cereb. Cortex 31, 97–105 (2021).
Schumacher, J. et al. Dysfunctional brain dynamics and their origin in Lewy body dementia. Brain 142, 1767–1782 (2019).
Farrow, S. L., Cooper, A. A. & O’Sullivan, J. M. Redefining the hypotheses driving Parkinson’s diseases research. Npj Park. Dis. 8, 1–7 (2022).
Engelender, S. & Isacson, O. The threshold theory for Parkinson’s disease. Trends Neurosci. 40, 4–14 (2017).
Mou, L., Ding, W. & Fernandez-Funez, P. Open questions on the nature of Parkinson’s disease: from triggers to spreading pathology. J. Med. Genet. 57, 73–81 (2020).
Rösler, T. W. et al. K-variant BCHE and pesticide exposure: Gene-environment interactions in a case-control study of Parkinson’s disease in Egypt. Sci. Rep. 8, 16525 (2018).
Davey, G. P., Peuchen, S. & Clark, J. B. Energy thresholds in brain mitochondria. Potential Involv. Neurodegener. J. Biol. Chem. 273, 12753–12757 (1998).
Cannon, J. R. & Greenamyre, J. T. Gene-environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiol. Dis. 57, 38–46 (2013).
Gao, X. et al. Gene-gene interaction between FGF20 and MAOB in Parkinson disease. Ann. Hum. Genet. 72, 157–162 (2008).
Bellou, V., Belbasis, L., Tzoulaki, I., Evangelou, E. & Ioannidis, J. P. A. Environmental risk factors and Parkinson’s disease: an umbrella review of meta-analyses. Parkinson. Relat. Disord. 23, 1–9 (2016).
Olanow, C. W. & Prusiner, S. B. Is Parkinson’s disease a prion disorder? Proc. Natl Acad. Sci. USA 106, 12571–12572 (2009).
Luk, K. C. et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012).
Dächsel, J. C. & Farrer, M. J. LRRK2 and Parkinson disease. Arch. Neurol. 67, 542–547 (2010).
Siderowf, A. & Lang, A. E. Premotor Parkinson’s disease: concepts and definitions. Mov. Disord. J. Mov. Disord. Soc. 27, 608–616 (2012).
Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet 397, 2284–2303 (2021).
Lange, L. M. et al. Nomenclature of genetic movement disorders: recommendations of the international parkinson and movement disorder society task force—an update. Mov. Disord. J. Mov. Disord. Soc. 37, 905–935 (2022).
Skrahina, V. et al. The Rostock International Parkinson’s Disease (ROPAD) study: protocol and initial findings. Mov. Disord. J. Mov. Disord. Soc. 36, 1005–1010 (2021).
Choi, M. L. et al. Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat. Neurosci. 25, 1134–1148 (2022).
Pramstaller, P. P. et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann. Neurol. 58, 411–422 (2005).
Tabrizi, S. J. et al. A biological classification of Huntington’s disease: the Integrated Staging System. Lancet Neurol. 21, 632–644 (2022).
Braz, B. Y. et al. Treating early postnatal circuit defect delays Huntington’s disease onset and pathology in mice. Science 377, eabq5011 (2022).
Trinh, J. & Klein, C. Needle in a haystack: the common can inform the rare in restless legs syndrome. Ann. Neurol. 87, 172–174 (2020).
S, B.-C. et al. The genetic architecture of Parkinson Disease in Spain: characterizing population-specific risk, differential haplotype structures, and providing etiologic insight. Mov. Disord. J. Mov. Disord. Soc. 34, 1851–1863 (2019).
Koch, S. et al. Validity and prognostic value of a polygenic risk score for Parkinson’s disease. Genes 12, 1859 (2021).
Trinh, J. et al. Mitochondrial DNA heteroplasmy distinguishes disease manifestation in PINK1/PRKN-linked Parkinson’s disease. Brain J. Neurol. 7, awac464 (2022).
Sarkar, S., Raymick, J. & Imam, S. Neuroprotective and therapeutic strategies against parkinson’s disease: recent perspectives. Int. J. Mol. Sci. 17, 904 (2016).
Liu, H. et al. Polygenic resilience modulates the penetrance of parkinson disease genetic risk factors. Ann. Neurol. 92, 270–278 (2022).
T, L. et al. Age at onset of LRRK2 p.Gly2019Ser is related to environmental and lifestyle factors. Mov. Disord. J. Mov. Disord. Soc. 35, 1854–1858 (2020).
Calne, D. B. Is ‘Parkinson’s disease’ one disease? J. Neurol. Neurosurg. Psychiatry Suppl, 18–21 (1989).
Lang, A. E. et al. Trial of cinpanemab in early Parkinson’s disease. N. Engl. J. Med. 387, 408–420 (2022).
Pagano, G. et al. Trial of prasinezumab in early-stage Parkinson’s disease. N. Engl. J. Med. 387, 421–432 (2022).
Yuan, X. et al. Fine particulate matter triggers α-synuclein fibrillization and parkinson-like neurodegeneration. Mov. Disord. J. Mov. Disord. Soc. 37, 1817–1830 (2022).
Tulisiak, C. T., Mercado, G., Peelaerts, W., Brundin, L. & Brundin, P. Can infections trigger alpha-synucleinopathies? Prog. Mol. Biol. Transl. Sci. 168, 299–322 (2019).
Bolam, J. P. & Pissadaki, E. K. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov. Disord. J. Mov. Disord. Soc. 27, 1478–1483 (2012).
Kurowska, Z. et al. Is axonal degeneration a key early event in Parkinson’s disease? J. Park. Dis. 6, 703–707 (2016).
Foffani, G. & Obeso, J. A. A cortical pathogenic theory of Parkinson’s disease. Neuron 99, 1116–1128 (2018).
Monogue, B. et al. Alpha-synuclein supports type 1 interferon signalling in neurons and brain tissue. Brain J. Neurol. 145, 3622–3636 (2022).
Smeyne, R. J. et al. COVID-19 infection enhances susceptibility to oxidative stress-induced Parkinsonism. Mov. Disord. J. Mov. Disord. Soc. 37, 1394–1404 (2022).
Matheoud, D. et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 166, 314–327 (2016).
Del Rey, N. L.-G. & García-Cabezas, M. Á. Cytology, architecture, development, and connections of the primate striatum: Hints for human pathology. Neurobiol. Dis. 176, 105945 (2022).
Barer, Y., Chodick, G., Glaser Chodick, N. & Gurevich, T. Risk of Parkinson disease among adults with vs without posttraumatic stress disorder. JAMA Netw. Open 5, e2225445 (2022).
Barnat, M. et al. Huntington’s disease alters human neurodevelopment. Science 369, 787–793 (2020).
Brandebura, A. N., Paumier, A., Onur, T. S. & Allen, N. J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 24, 23–39 (2022).
Stoessl, A. J. Glucose utilization: still in the synapse. Nat. Neurosci. 20, 382–384 (2017).
Chen, C. et al. Astrocytic changes in mitochondrial oxidative phosphorylation protein levels in Parkinson’s disease. Mov. Disord. J. Mov. Disord. Soc. 37, 302–314 (2022).
Sonninen, T.-M. et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 10, 14474 (2020).
de Rus Jacquet, A. et al. The LRRK2 G2019S mutation alters astrocyte-to-neuron communication via extracellular vesicles and induces neuron atrophy in a human iPSC-derived model of Parkinson’s disease. eLife 10, e73062 (2021).
Iovino, L. et al. Trafficking of the glutamate transporter is impaired in LRRK2-related Parkinson’s disease. Acta Neuropathol. 144, 81–106 (2022).
Streubel-Gallasch, L. et al. Parkinson’s disease-associated LRRK2 interferes with astrocyte-mediated alpha-synuclein clearance. Mol. Neurobiol. 58, 3119–3140 (2021).
Yun, S. P. et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938 (2018).
Wakabayashi, K., Takahashi, H., Takeda, S., Ohama, E. & Ikuta, F. Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 76, 217–221 (1988).
Adams-Carr, K. L. et al. Constipation preceding Parkinson’s disease: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 87, 710–716 (2016).
Knudsen, K. et al. In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol. 17, 618–628 (2018).
Schrag, A., Horsfall, L., Walters, K., Noyce, A. & Petersen, I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 14, 57–64 (2015).
Shannon, K. M. et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease: colonic α-Synuclein in Parkinson’s disease. Mov. Disord. 27, 709–715 (2012).
Breen, D. P., Halliday, G. M. & Lang, A. E. Gut–brain axis and the spread of α‐synuclein pathology: vagal highway or dead end? Mov. Disord. 34, 307–316 (2019).
Tan, A. H., Lim, S. Y. & Lang, A. E. The microbiome–gut–brain axis in Parkinson disease—from basic research to the clinic. Nat. Rev. Neurol. 18, 476–495 (2022).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Borghammer, P. et al. A postmortem study suggests a revision of the dual-hit hypothesis of Parkinson’s disease. Npj Park. Dis. 8, 166 (2022).
Arotcarena, M.-L. et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 143, 1462–1475 (2020).
Leclair‐Visonneau, L., Neunlist, M., Derkinderen, P. & Lebouvier, T. The gut in Parkinson’s disease: bottom‐up, top‐down, or neither? Neurogastroenterol. Motil. 32, e13777 (2020).
Devos, D. et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 50, 42–48 (2013).
Hor, J. W. et al. Fecal calprotectin in Parkinson’s disease and multiple system atrophy. J. Mov. Disord. 15, 106–114 (2022).
Houser, M. C. et al. Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease: stool inflammatory profiles in PD patients. Mov. Disord. 33, 793–804 (2018).
Herrick, M. K. & Tansey, M. G. Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? Npj Park. Dis. 7, 26 (2021).
Lin, C. et al. Mild chronic colitis triggers Parkinsonism in LRRK2 mutant mice through activating TNF‐α pathway. Mov. Disord. 37, 745–757 (2022).
Tan, A. H. et al. Gut Microbial ecosystem in Parkinson disease: new clinicobiological insights from multi‐omics. Ann. Neurol. 89, 546–559 (2021).
Toh, T. S. et al. Gut microbiome in Parkinson’s disease: new insights from meta-analysis. Parkinson. Relat. Disord. 94, 1–9 (2022).
Wallen, Z. D. et al. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 13, 6958 (2022).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e12 (2016).
Derkinderen, P., Shannon, K. M. & Brundin, P. Gut feelings about smoking and coffee in Parkinson’s disease: smoking, coffee, and gut microbiota in PD. Mov. Disord. 29, 976–979 (2014).
Killinger, B. & Labrie, V. The appendix in Parkinson’s disease: from vestigial remnant to vital organ? J. Park. Dis. 9, S345–S358 (2019).
Matheoud, D. et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 571, 565–569 (2019).
Lim, S.-Y. et al. Parkinson’s disease in the Western Pacific Region. Lancet Neurol. 18, 865–879 (2019).
Tan, A. H. et al. Probiotics for constipation in Parkinson’s disease: a randomized placebo-controlled study. Neurology 10.1212/WNL.0000000000010998 https://doi.org/10.1212/WNL.0000000000010998 (2020).
ICD-10 Version:2019. https://icd.who.int/browse10/2019/en#/G20-G26.
Dorsey, E. R. et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953 (2018).
Yang, W. et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Park. Dis. 6, 15 (2020).
Leiva, A. M. et al. Parkinson’s disease in Chile: highest prevalence in Latin America. Rev. Med. Chil. 147, 535–536 (2019).
Sepúlveda, D. et al. Insulin-like growth factor 2 and autophagy gene expression alteration arise as potential biomarkers in Parkinson’s disease. Sci. Rep. 12, 2038 (2022).
Acknowledgements
We acknowledge the contribution of Prof. Howlett to Dr. Dekker’s section. A.C.M.-N. and S.B. are supported by the National Research Foundation of South Africa (Grant 129249). C.K. is supported by the DFG (FOR2488) and the MJFF (ASAP; GP2). C.M.S. is a National Health and Medical Research Council Practitioner Fellow (APP1136800) and is supported by National Health and Medical Research Council grants, MJFF (ASAP; GP2) and Shake it Up Australia. I.R. is supported by the Ministry of Health of the Czech Republic (Grant NU20–04–00294) and by the project LX22NPO5107 (MEYS) funded by European Union—Next Generation EU. M.S. is supported by the Bartlett Fund for Critical Challenges, the American University in Cairo (AUC). A.H.T. and S-Y.L. are supported by the University of Malaya Parkinson’s Disease and Movement Disorders Research Program (PV035–2017). R.L.V. is supported by Fondecyt 1191003.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
S.B., A.C.M.-N., and C.K. conceptualized the article. All authors contributed to writing the first draft, critical review and editing, and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.C.M.-N., S.B., C.M.S., M.C.J.D., G.D.M., A.H.T., R.L.V., M.J.F., M.S., and I.R. declare no competing interests. C.K. serves as a medical advisor to Centogene and Retromer therapeutics and received speakers’ honoraria from Desitin, for lecturing from the International Parkinson and Movement Disorder Society (MDS), a stipend as Deputy Editor of Movement Disorders and Science Advances, and royalties from Oxford University Press. A.J.S. chairs a DSMB for Neurocrine, serves on a DSMB for AskBio, serves as an advisor to Capsida and SioGene, and he receives a stipend as Editor-in-Chief of Movement Disorders. S-Y.L. serves as a consultant for the Michael J. Fox Foundation, serves on the Lundbeck International Neuroscience Foundation Editorial Board, serves on the Advisory Board for Eisai and received honoraria for lecturing from the International Parkinson and Movement Disorder Society (MDS), International Brain Research Organization (IBRO), Lundbeck, and Medtronic.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Müller-Nedebock, A.C., Dekker, M.C.J., Farrer, M.J. et al. Different pieces of the same puzzle: a multifaceted perspective on the complex biological basis of Parkinson’s disease. npj Parkinsons Dis. 9, 110 (2023). https://doi.org/10.1038/s41531-023-00535-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-023-00535-8
This article is cited by
-
Key Non-coding Variants in Three Neuroapoptosis and Neuroinflammation-Related LncRNAs Are Protectively Associated with Susceptibility to Parkinson’s Disease and Some of Its Clinical Features
Molecular Neurobiology (2024)
-
Clinicians’ viewpoints on current paradigms of care and research in Parkinson’s disease
Journal of Neural Transmission (2024)
