
Abstract
The pathogenesis and clinical heterogeneity of Parkinson’s disease (PD) have been evaluated from molecular, pathophysiological, and clinical perspectives. High-throughput proteomic analysis of cerebrospinal fluid (CSF) opened new opportunities for scrutinizing this heterogeneity. To date, this is the most comprehensive CSF-based proteomics profiling study in PD with 569 patients (350 idiopathic patients, 65 GBA + mutation carriers and 154 LRRK2 + mutation carriers), 534 controls, and 4135 proteins analyzed. Combining CSF aptamer-based proteomics with genetics we determined protein quantitative trait loci (pQTLs). Analyses of pQTLs together with summary statistics from the largest PD genome wide association study (GWAS) identified 68 potential causal proteins by Mendelian randomization. The top causal protein, GPNMB, was previously reported to be upregulated in the substantia nigra of PD patients. We also compared the CSF proteomes of patients and controls. Proteome differences between GBA + patients and unaffected GBA + controls suggest degeneration of dopaminergic neurons, altered dopamine metabolism and increased brain inflammation. In the LRRK2 + subcohort we found dysregulated lysosomal degradation, altered alpha-synuclein processing, and neurotransmission. Proteome differences between idiopathic patients and controls suggest increased neuroinflammation, mitochondrial dysfunction/oxidative stress, altered iron metabolism and potential neuroprotection mediated by vasoactive substances. Finally, we used proteomic data to stratify idiopathic patients into “endotypes”. The identified endotypes show differences in cognitive and motor disease progression based on previously reported protein-based risk scores.Our findings not only contribute to the identification of new therapeutic targets but also to shape personalized medicine in CNS neurodegeneration.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder worlwide1. Parkinson’s disease patients experience selective degeneration and loss of dopaminergic neurons in the substantia nigra pars compacta. In most cases, PD is classified as idiopathic, but a growing set of genetic variants increase PD risk or accelerate its onset. Many of the identified genes are involved either in mitochondrial or endo-lysosomal biology2. The two most common PD risk genes are leucine rich kinase 2 (LRRK2) and glucosidase beta acid (GBA). Mutations in these genes are linked to ~10% of sporadic cases and up to 30% in specific ethnic subgroups and familial disease3. Some of the genetic variants in LRRK2 and GBA have been associated with specific clinical phenotypes4,5. For both genes, the pathological mutations are thought to exacerbate the toxicity of alpha-synuclein, which—in an aggregated form—contributes to neuronal death and amplifies the neuroinflammatory response6,7,8,9.
Clinical heterogeneity of PD has motivated many disease stratification efforts. Some of those have focused on clinical variables, mostly hypothesis-driven, while others have focused on molecular data, mostly hypothesis-free10. While carriers of specific mutations may present specific clinical phenotypes4,5, patient strata defined by clinical or molecular variables are not easily linked to specific PD risk mutations11.
Proteins hold great potential as predictors, causal biomarkers and surrogates of disease progression and/or stratification. However, the biological and pathophysiological complexity of PD, the difficulties of collecting standardized biological samples (especially cerebrospinal fluid; CSF) from large cohorts throughout the course of disease, and the technical limitations of high-throughput proteomic analyses hamper the identification of biomarkers at a proteomic level. The multicenter Parkinson Progression Marker Initiative (PPMI) was initiated to overcome some of these limitations, particularly in terms of number of samples and clinical data12. In this collaboration, 1103 baseline (not longitudinal) CSF samples from patients and control participants with known status of LRRK2 and GBA pathogenic variants were analyzed using the SomaScan aptamer-based proteomics platform13. In parallel we also looked at the whole genome sequencing data from 804 patients out of the 1103, after quality control (QC). To our knowledge, this is, to date, the largest proteomic and genetic data set for interrogating causal proteins for PD in a neurologically relevant biofluid. Previously reported characterizations of the proteome in CSF of PD paved the way to identify potential biomarkers and help to better understand the etiology of the disease14,15. The conclusions reported in those studies were extracted from smaller patient populations. A recent study16 has performed a CSF proteomic profiling of over 1700 proteins using mass spectrometry in the PPMI subcohort of LRRK2 + patients and a subset of PPMI idiopathic patients as well as in an independent set (Harvard Biomarker Study). The overlaps with our findings are considered in the discussion section.
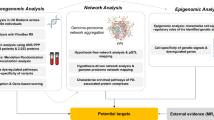
The main goals of this study are summarized in Fig. 1: i. Parkinson’s disease causal protein identification using mendelian randomization based on proteomics and genetics, ii. identification of differences between PD patients and controls within and between subcohorts (LRRK2+, GBA+ and idiopathic), and iii. hypothesis-free stratification of idiopathic PD patients into clinically relevant endotypes and then compared by means of protein-based risk scores reflecting cognitive and motor progression17.

1103 subjects were analyzed with SomaScan for 4135 unique proteins in CSF. The comparison between GBA + PD patients and GBA + unaffected controls (UC) retrieved six differentially expressed (DE) proteins. The comparison between LRRK2 + PD patients and LRRK2 + UC retrieved seven DE proteins. The comparison between idiopathic PD patients and HC non-mutation carriers retrieved 23 DE proteins. Patients and controls were also combined and compared, which retrieved 122 DE proteins. Idiopathic PD patients were further analyzed, and two endotypes were identified based on CSF proteomics. 1264 subjects were sequenced genome wide to detect a total of 9743041 SNPs. For the 804 patients that had both genomic and proteomic data, a pQTL analysis was performed that identified 744 unique proteins with a significant cis-pQTL. The pQTLs combined with a meta GWAS for PD performed by Nalls et al.70, led to the proposal of 68 unique CSF proteins presumed to be causal for PD.
Results
Causal analysis
Our analysis reported significant cis-proteomic quantitative trait loci (pQTLs) for 856 Slow Off-rate Modified Aptamers (SOMAmers)—corresponding to 744 unique proteins (Supplementary Table 1). From these 856 cis-pQTLs, we identified statistically significant evidence for causation for 68 proteins in CSF (Table 2). Out of those proteins, GPNMB, FCGR2A and FCGR2B also had a strong colocalization signal (see Methods), indicating the same single nucleotide polymorphism (SNP) is both associated with protein level and PD risk (Fig. 2). The full tables with nominal P ≤ 0.05 are included in Supplementary Tables 2 and 3 for less or more stringent clumping, respectively.

A Locus visualization of GPNMB pQTL hits suggest a strong association between GPNMB SOMAmer levels and its cis-SNPs. Colors indicate the linkage disequilibrium (LD) correlation of other SNPs with chr7:23294144 (rs858275). B Locus visualization of GWAS hits in the GPNMB locus for the risk of developing PD. The y-axis is the −log10 nominal p value of the GWAS results.
Differential protein expression in subcohorts of Parkinson’s disease patients
To identify proteins differentially expressed in PD, we compared SOMAmers in each of the subcohorts (GBA+, LRRK2+ and idiopathic) to their corresponding controls. Our statistical analyses revealed six differentially expressed SOMAmers for GBA + patients, seven SOMAmers for LRRK2 + patients and 23 SOMAmers for idiopathic patients. Directionality of the change and adjusted P values for each of these markers are reported in Table 3.
For each subcohort, several identified markers confirmed previously reported proteins dysregulated in PD. Interestingly, there was little overlap between proteins dysregulated in GBA+, LRRK2+ and idiopathic subcohorts (SEMG2 and DLK1 were shared by GBA+ and the idiopathic subcohort) though in each list there is a high percentage (4/6 in GBA+, 4/7 in LRRK2+ and 10/23 in the idiopathic subcohort) of markers previously reported in relation to PD (Table 3).
Only one protein, CTSB, passed the FDR significance threshold for the interaction between disease status and mutation status. As shown in Table 3, CTSB was also differentially expressed in the LRRK2 + subcohort.
Additional analysis comparing all patient subcohorts with all controls, using subcohort membership (no mutation/GBA+/LRRK2+) and treatment status (yes/no) as additional covariates resulted in 129 SOMAmers, tagging 122 distinct proteins, passing false discovery rate (FDR) correction (Supplementary Table 4).
Identification of subtypes of idiopathic Parkinson’s disease patients
Identification of endotypes
To determine if distinct endotypes were present in the idiopathic subcohort we performed a network analysis on the CSF proteome of idiopathic patients (Weighted Gene Co-expression Network Analysis (WGCNA) block-wise automatic module detection; soft threshold = 11; unsigned TOM, minimum module size = 500, Pearson correlation type). Two modules of co-expressed proteins were identified. They comprised 889 and 600 SOMAmers, respectively. Applying consensus clustering on these two protein modules split the idiopathic subcohort (350 patients) in proteome-based patient endotypes 1 (185 patients) and 2 (165 patients) (Fig. 3A). As seen in the tracking plot (Fig. 3B) these endotypes suffer only negligible changes as the number of modeled subclasses increases. Moreover, a predictive model for the endotypes was built based on clinical parameters, avoiding the re-use of the same proteomic data involved in the definition of the endotypes. Patients with CSF phospho-tau ≥11 pg/mL (as measured with the Elecsys® assay) were enriched for endotype 1, and patients with CSF phospho-tau <11 pg/mL were enriched for endotype 2 (Fig. 3C). The model accuracy in the training (244 patients) and in the independent test (106 patients) sets, was 0.82 and 0.73, respectively. The estimated area under the curve for the test set was 0.77.

A Heatmap of z-scores of the protein values as measured with SomaScan, corresponding to the two modules identified using Weighted Gene Co-expression Network Analysis (WGCNA). The proteins in these modules are used for cluster analysis using Consensus Clustering, which retrieves two clusters (endotypes) of idiopathic PD patients. Patients are shown in the x-axis, separated by endotype, while proteins are shown in the y-axis, separated by module. B Tracking plot depicting how the idiopathic PD patients are assigned to specific endotypes by Consensus Clustering as the number of potential endotypes increases. C Partition tree predicting endotype membership of the idiopathic PD patients based on clinical variables only. One node suffices to separate patients into endotypes based on phospho-tau levels (p-tau) in CSF as measured with a clinical assay, the cut-off being 11 pg/mL.
Endotypes did not significantly differ in age or sex. There were neither significant differences in caudate, striatum or putamen thickness, nor in UPDRS score parts II and III or MoCA scores. Significant differences were found for UPDRS score part I (higher for endotype 2; P = 0.044), as well as CSF levels of amyloid beta (lower for endotype 2; P = 1.95 × 10−15), phospho-tau (lower for endotype 2; P = 1.25 × 10−15), total tau (lower for endotype 2; P < 2 × 10−16) and alpha-synuclein (lower for endotype 2; P < 2 × 10−16). Endotypes also showed significant differences in “non-genetic Parkinson’s Disease-associated Proteomic Scores” (higher ngPD-ProS for endotype 2; P = 0.034).
Proteins differentially expressed in endotypes (CSF SomaScan)
To identify the unique proteins significantly dysregulated in each endotype, a linear model was used to identify the differences between each of these two endotypes and the healthy controls to the idiopathic subcohort. For endotype 1, five markers were significantly different compared to the control group (CNTFR, LPO, MMP10, RIPK2, and VEGFA). LPO, RIPK2 and VEGFA were also part of the differences between healthy controls and the whole idiopathic group (see above). Endotype 2, however, showed 200 differentially expressed SOMAmers, 197 unique proteins (see Supplementary Table 5). Among those proteins, AK1, CCL14, FRZB, GPI, HAMP, LPO, NETO1, PTPRR, RAB31, RELT, RIPK2, ROBO3, RSPO4, SHANK1, SPINK9, VEGFA, and VIP were dysregulated for the whole idiopathic group as compared to healthy controls. 155 SOMAmers (i.e., 153 unique proteins; see Supplementary Table 5) were differentially expressed between endotypes.
Discussion
Independently of its etiology (genetic or idiopathic), multiple cellular and metabolic alterations (e.g., iron metabolism), inflammation, and oxidative stress underly neurodegeneration in PD. In this regard, the identification of causal proteins not only enhances its understanding, but also assists the search for druggable targets. This study identified 68 CSF proteins as causal and GPNMB, FCGR2A, FCGR2B and CTSB were among the top ones.
GPNMB—expressed by myeloid cells—is found at high levels in the substantia nigra of PD patients18. Recently, GPNMB has been demonstrated in a cellular model to be necessary and sufficient for the internalization of alpha-synuclein fibrils in the neurons via a functional interaction which would in turn lead to the development of PD. Complementary to that finding, higher GPNMB levels are associated with higher disease risk and higher disease severity19. It has also been suggested that GPNMB modulates immune response20,21 having primarily a protective role21,22,23. The anti-inflammatory role of GPNMB is mediated by the CD44 non-kinase transmembrane glycoprotein24, which has been found to be augmented in PD patients using mass spectrometry in a combination of cohorts from a subset of the PPMI cohort and an independent data set16. CTSB cleaves alpha-synuclein fibrils with the potential for decreasing alpha-synuclein aggregation25. Both the CTSB locus26 and a specific genetic variant of CTSB have been found to be associated with the risk of developing PD27. FCGR2A and FCGR2B are involved in phagocytosis and modulate inflammatory responses28. Supporting our findings, independent reports suggest that GPNMB, CTSB and FCGR2A are causal to PD19,29,30,31. Alpha-synuclein aggregates bind to the microglia’s FCGR2B receptor, which leads to inhibition of the microglia’s phagocytic activity and indirectly enhances neurodegeneration32. In neurons, the FCGR2B receptor may mediate cell-to-cell propagation of alpha-synuclein and in the formation of intracellular Lewy bodies33. Both FCGR2A and FCGR2B require binding to IgG-specific immune complexes to trigger their specific signaling cascades. The SomaScan panel used here does not distinguish between IgG isotypes. The identification and quantification of those isotypes in CSF could provide additional evidence to support or exclude FCGR2A and FCGR2B as being functionally linked to PD.
Out of the identified 68 causal proteins, only two reappear in the analysis of subcohorts: i.e., ARSA and CTSB. But while the former analysis is focused on casual effects on the whole patients’ group, the latter does not distinguish between cause and consequence and is subcohort-specific.
GBA and LRRK2 are enzymes involved in ceramide metabolism and lysosomal functions including the removal of aggregated alpha-synuclein. Genetic variants of the GBA and LRRK2 genes are known risk factors for the development of PD and dementia associated with accumulation of Lewy bodies34,35,36. To better understand the differences between diseased and unaffected mutation carriers we compared their CSF proteomes (see Table 3).
Protein differences between GBA + patients and unaffected GBA + controls are related to brain dopaminergic neurons (DLK1), dopamine metabolism (CALCA, GCH1) and inflammation effector cells (IL17A). All these proteins have been previously linked to PD, though not specifically to GBA+. DLK1 is coupled to both tyrosine hydroxylase expression and neurotrophic signaling37. Significantly lower levels of DLK1 in GBA + patients suggest increased degeneration of dopaminergic neurons. GCH1 and CALCA are involved in nigrostriatal dopamine synthesis38, and dopamine release and metabolism39,40, respectively. We found both elevated in GBA + patients. GCH1 variants contribute to the risk and earlier age-at-onset of PD41. IL17A was also augmented in the CSF of GBA + PD patients. It has been recently proposed that elevated IL17A plays a key role in neurodegenerative diseases42. Moreover, one of the inflammatory mechanisms proposed for neurodegeneration in PD involves Th17 cells43. Taking the above observations together, we hypothesize that GBA + induced loss of nigral dopaminergic neurons44 may trigger the neuroinflammatory events that lead to increased release of cytokines such as IL17A. IL17A in CSF maybe secreted by either Th17 brain resident cells and/or by Th17 cells infiltrating through the blood brain barrier45. Also, it cannot be excluded that IL17A could be also released by CD8+, lymphoid cells, NKTs and/or microglia.
Among the CSF proteins differentially expressed between LRRK2 + patients and unaffected LRRK2 + controls, several stood out for their relevance in PD: ARSA, SMPD1, CTSB and TENM4. ARSA (causal protein, see causal analysis) is a lysosomal chaperone that prevents alpha-synuclein aggregation, secretion and cell-to-cell propagation46. We found ARSA elevated in LRRK2 + patients, which could be interpreted as a protection mechanism to prevent the formation of alpha-synuclein aggregates. CTSB (causal protein, see causal analysis) and SMPD1 play important roles in PD autophagy and lysosomal degradation processes47. CTSB and SMPD1 genetic variants are known to be associated with PD risk47. In this study, higher CTSB and SMPD1 levels in LRRK2 + patients indicate dysregulation of autophagy-endolysosomal pathway and potentially increased macroautophagy48,49. Brain TENM4 is involved in axon guidance and myelination50. In our study, it was significantly reduced in LRRK2 + patients. Loss-of-function and missense variants in TENM4 are associated with early onset PD and essential tremor, a potential risk factor for developing PD51,52,53,54,55,56,57,58,59,60.
Worldwide, ~90% of PD cases are idiopathic. Proteome differences between idiopathic patients and controls comprise not only markers of inflammation, but also of mitochondrial dysfunction/oxidative stress, iron metabolism and other pathological processes (see Table 3). The AK1 kinase is expressed by neurons and astrocytes52. At advanced PD stages, AK1 is downregulated in the substantia nigra probably due to mitochondrial dysfunction and dopaminergic neuronal death52. The observed elevation of CSF AK1 levels may be associated with PD progression stages, frontal cortex primary alteration or compensation of altered purine metabolism52. SHANK may be regulated by the mitochondrial kinase PINK1, for which variants are known to be causal for PD53. It has been reported that knockdown in neurons of PINK decreases PSD95 and SHANK153. SHANK1 was decreased in idiopathic patients suggesting impaired synaptic plasticity. TXN promotes cell proliferation, protection against oxidative stress and anti-apoptotic functions in the brain, which makes it a good candidate for a neurodegeneration marker. Here we find TXN decreased in PD patients. Iron dysregulation is associated with oxidative stress and lipid peroxidation54. It has been proposed that LPO—heme peroxidase—in the substantia nigra is involved in neurodegeneration55. Lower LPO levels in idiopathic patients found here contrasts with previously reported elevated CSF LPO levels55. The high CSF levels of HAMP could help explaining this discrepancy. HAMP reduces iron accumulation and neuroinflammation by decreasing mitochondrial dysfunction, oxidative stress, and ultimately dopamine neuronal loss56. Moreover, HAMP overexpression—as seen here—promotes alpha-synuclein clearance through autophagy57. It has been also reported that dopamine and levodopa reduce LPO levels55. Given that the idiopathic patients recruited were drug-naïve early PD patients, dopamine levels in this subpopulation may have helped maintaining low levels of CSF LPO. The glucose metabolism enzyme GPI was elevated in idiopathic patients and may be protective. Its overexpression in dopaminergic neurons protects against alpha-synuclein-induced neurotoxicity58. As seen in GBA + patients, reduced DLK1 levels in idiopathic patients may suggest neurodegeneration. Lower levels of RAB31, a small GTPase involved in exosome biogenesis59 and potentially in alpha-synuclein spreading in PD60 were observed, too. RIPK2, a LRRK2 substrate, is lower in idiopathic patients, which matches the fact that LRRK2 deficiency leads to reduced activation of RIPK261. The neurotrophic factor VEGFA is neuroprotective and has genetic variant associated with PD risk62. VIP enhances striatal plasticity and prevents dopaminergic cell loss in parkinsonian rats63. VEGFA and VIP at lower levels in idiopathic patients may reflect ongoing neurodegeneration.
Disease heterogeneity challenges the development of disease modifying therapies. In this study, we used proteomic data to stratify idiopathic patients into two clinically relevant endotypes.
The endotype robustness is confirmed by the high patient cluster stability (Fig. 3B) and the good performance of the endotype predictive model (see Fig. 3C). The fact that CSF phospho-tau levels sufficed to predict endotypes, suggests that targeted assessment of CSF proteins may be appropriate for idiopathic patient stratification in a clinical setting.
The lack of differences in the CSF levels of causal proteins, suggests that endotype molecular differences may be downstream from causal effects and affect the specific characteristics of PD phenotypes rather than PD risk. Pathway analysis of module proteins suggests inflammatory activity in endotype 2 (increased levels of proteins related to Th1, Th2, Th17 and macrophage activation pathways, as well as IL17 and other cytokines’ signaling pathways). In contrast, endotype 1 seems to have elevated levels of proteins related to synaptic connectivity and signaling, axonal guidance, and neurotransmission (Supplementary Fig. 1). Examples thereof could be glial fibrillary acidic protein, alpha-synuclein, microtubule associated protein tau, apolipoprotein E, 14-3-3 proteins (i.e., YWHAG, YWHAB, YWHAQ, YWHAZ). These results may reflect a higher neuroinflammatory activity for endotype 2, while activity for endotype 1 would be pointing to a compensation of the neurodegeneration process, as supported by the fact that endotype 1 shows less differences to controls while having enhanced levels of proteins related to neuronal function. Since endotypes showed differential progression as reflected by their ngPD-ProS, understanding their molecular differences might assist the design of personalized therapy approaches.
It is worth noting that, while in Tsukita et al.17 the ngPD-ProS perform similarly for both the idiopathic and the combined genetic subcohorts, here we see little overlap in the differentially expressed proteins between the idiopathic and each of the genetic subcohorts separately. This apparent discrepancy could just be reflecting a common CSF proteomic signature rising from the combination of genetic subcohorts even though each genetic subcohort may have different etiology. Supporting this hypothesis, when we compared the proteomes of the combined genetic subcohorts (GBA+ and LRRK2+) with their respective controls (data not shown) we found seven proteins shared with the 55 in the ngPD-ProS (i.e., DLK1, LPO, NEFH, RIPK2, SEMG2, VIP and LRFN2). These seven proteins intersected almost completely with the ten proteins that overlapped between the 55 proteins in the ngPD-ProS and the proteins differentially expressed between idiopathic patients and controls (i.e., DLK1, LPO, NEFH, RIPK2, SEMG2, VIP, CCL14, HAMP, RSPO4, and TXN). This similarity between the idiopathic subcohort and the combination of the genetic subcohorts could help explaining the apparent discrepancy mentioned above.
In summary, we: (i) identified causal proteins for PD, (ii) assessed CSF proteome differences in PD patients of genetic and idiopathic etiology, and (iii) stratified idiopathic patients into robust subtypes. Our findings not only contribute to the identification of new therapeutic targets but also to shaping personalized medicine in CNS neurodegeneration.
Methods
The clinical data and samples used in this study were obtained from the PPMI (http://www.ppmi-info.org/data) on October 1, 2020. PPMI samples were collected under a standardized protocol over 33 centers and includes clinical and imaging data as well as plasma and CSF samples. Study protocol and manuals are available online (http://www.ppmi-info.org/study-design).
Separate subcohorts of patients with PD and their respective controls were enrolled following inclusion and exclusion criteria64. One subcohort is comprised of recently diagnosed, drug-naïve, idiopathic PD patients and healthy controls, while the second and third subcohorts are comprised of PD patients, carriers of a severe GBA or LRRK2 mutation, either PD patients or unaffected controls. Parkinson’s disease patients from the genetic subcohorts had a longer disease duration, were partially under PD medication (n = 203), were over-represented for individuals of Ashkenazi Jewish descent and differed by sex distribution from the idiopathic PD patients (higher proportion of men among idiopathic). The study was approved by the Institutional Review Board at each site, and participants provided written informed consent.
Genetic testing was done by the centralized PPMI genetic testing core. Non-manifesting carriers received pre-testing and post-testing genetic counselling by phone from certified genetic counsellors at the University of Indiana or site-qualified personnel. The mutations considered severe in the LRRK2 genetic testing battery included G2019S and R1441G. GBA genetic testing included five mutations considered severe: N370S (tested for all participants), and L483P, L444P, IVS2 + 1, and 84GG (tested for a subset of participants). Dual mutation carriers (LRRK2 and GBA) were considered as LRRK2 carriers for simplicity (n = 1).
Six patients were diagnosed as idiopathic PD at enrolment but were re-classified during follow-up (two patients were diagnosed as multiple system atrophy and four patients did not have a final diagnose but PD had been excluded). These patients were removed from the analysis. Four patients were initially diagnosed as genetic PD, but the diagnose changed to prodromal during follow up. These patients were considered as unaffected controls in their corresponding genetic subcohort (Five LRRK2 + unaffected controls and one GBA + unaffected control).
One subject originally classified as healthy, but later shown to have an unclear health status, was removed from the analysis. Subjects recruited into the subcohort of idiopathic PD patients and healthy controls but identified as carriers of a severe GBA or LRRK2 mutation, were moved to the corresponding genetic subcohort (GBA n = 15; LRRK2 n = 7).
The genetic screening also detected GBA mutations of unknown or moderate risk: A459P, E365K, T408M. Carriers of these mutations were removed from analysis (n = 38).
Finally, ten carriers of a mutation in SCNA (eight PD patients and two unaffected controls), were also removed due to lack of statistical power for analysis.
The original data set was comprised of 1190 samples out of which 32 samples were pools, which were discarded for this study, and as described above, additional six PD patients and one healthy control were removed due to change in diagnose, 38 subjects were removed due to non-severe GBA mutations, and 10 patients were removed for being carriers of a mutation in SCNA. Hence, the final data set used for analysis was comprised of 1103 proteomic samples divided into three subcohorts: no mutation (idiopathic PD patients and healthy controls with no severe mutation in GBA or LRRK2), GBA + (PD patient carriers of severe GBA mutations and unaffected controls carrying the same mutations) and LRRK2 + (PD patients, carriers of severe LRRK2 mutations and unaffected controls carrying the same mutations). The exact composition is summarized in Table 1.
We make an explicit distinction between “healthy” and “unaffected” controls, because there is evidence of prodromal pathophysiology in LRRK2 + and GBA + controls when compared to healthy controls that are non-mutation carriers65.
Proteomics
Proteomic profiling was performed using SomaScan® in a platform version that is proprietary to Novartis and includes 4785 SOMAmers® (Slow Off-rate Modified Aptamers) targeting 4135 human proteins. SOMAmer levels were determined and standardized at SomaLogic Inc. (Boulder, US) including hybridization normalization (controls for variability in the readout of individual microarrays), plate scaling (accounts for plate-by-plate variation), median signal normalization (controls for total signal differences between individual samples) and calibration (removes the variation between assay runs within and across experiments).
Relative fluorescence units are transformed to log2 scale, normalized to the median separately by dilution level across all plates. Finally, the data set is adjusted for batch effects between plates using an empirical Bayes method as implemented in the R package sva v3.40.066.
It should be noted that SomaScan does not discriminate protein isoforms or post-translational modifications. In turn it is to date the technology that allows quantifying the largest number of proteins in a single run, thus reasonably covering most biological pathways.
Genetics
PPMI whole genome sequencing results were lifted over to hg19 coordinates. Biallelic SNPs on autosomes were extracted. Standard GWAS QC was applied at both individual and SNP level. 22 patients with outlying heterozygosity and 93 patients with high identity-by-descent were excluded after QC. 306031 SNPs were removed due to missing genotype and 35907596 SNPs removed due to minor allele count less than 20. Finally, 9743041 variants and 1264 subjects passed QC.
pQTL calculation
Among the 1264 subjects who passed QC for genetics and the 1103 who passed QC for proteomics, 804 subjects overlapped. Protein expression values were ranked and inverse normal transformed. For pQTL calculation, each protein level was regressed with each independent genetic variant (SNP; Major Allele Frequency (MAF) > 0.05), adjusted for age, sex, subcohort, protein principal components 1–4 and genetic principal components 1–10 using R package MatrixEQTL v2.367. Cis-pQTLs were defined as SNPs located inside the ±1 Mb region flanking the gene that encodes the given protein. A genome-wide threshold of P < 5 × 10-8 defined a significant cis-pQTL.
Causal analysis
The two-sample mendelian randomization method implemented in the R package TwoSampleMR v0.5.668,69 was applied to find causal proteins for PD in CSF. Although PPMI is a well-controlled study with genetic data, to avoid weak instrumental variable bias and take the advantage of a larger PD GWAS we relied on the meta-analysis from Nalls et al.70, which includes 17 datasets with PD cases ranging from 363 to 33674 individuals, and healthy controls ranging from 165 to 449056 individuals. Instrumental variables were selected for each SNP with MAF > 0.05, F-statistics larger than 10 and significant cis-pQTL P < 5 × 10−8. Shared SNPs in both cis-pQTL and PD GWAS were harmonized and then clumped using a linkage disequilibrium (LD) threshold of either r2 < 0.01 or r2 < 0.3. The more stringent LD threshold of r2 < 0.01 resulted in only one instrumental variable for most proteins, therefore the results we present are from the less stringent threshold of r2 < 0.3. The Wald ratio was used when only one instrument survived clumping, while the inverse variance weighted meta-analysis method was used when more than one instrumented SNP was available. Horizontal pleiotropy was tested using the R package MRPRESSO v1.071.
Colocalization probability was calculated using the R package coloc v5.1.072. Default priors of p1 = 10−4, p2 = 10−4, and p12 = 10−5 were used, where p1 is the prior probability of a SNP being associated with PD, p2 is the prior probability of a SNP being associated with CSF pQTL, and p12 is the prior probability of a SNP being associated with both PD and CSF pQTL. We considered PPH4 > 0.75 as strong evidence for colocalization. PPH4 is the posterior probability of one shared SNP being associated with both PD and CSF pQTL.
Whether mendelian randomization is enough to prove biological causality or not, is debatable. To facilitate readability, we will use the term “causal” as a simplification of “consistent with a potential causal effect”.
Clinical variables
The clinical assessment battery is described on the PPMI website (http://www.ppmi-info.org). Parkinson’s disease status was assessed with the Unified Parkinson’s Disease Rating Scale in the revised version published by the Movement Disorder Society (MDS-UPDRS) scores 1, 2, and 373. Cognitive testing comprised screening with the Montreal Cognitive Assessment (MoCA)74. High resolution xy-weighted 3 tesla MRI was available for 545 PD patients and 177 controls. Caudate, putamen and striatum thicknesses were calculated as the arithmetical mean between the values from the right and left brain hemispheres.
Cerebrospinal fluid was collected using standardized lumbar puncture procedures. Sample handling, shipment and storage were carried out as described in the PPMI biologics manual (http://ppmi-info.org). Besides the SomaScan analysis, data from immunoassay kits were also used for measuring CSF total amyloid-beta 1-42, total tau and phospho-tau (p-tau 181) and alpha-synuclein protein75,76. Briefly, for amyloid-beta 1-42 and total tau, standards, controls, and CSF samples were analyzed in duplicate with research-use-only Fujirebio-Innogenetics INNO-BIA AlzBio3 immunoassay kit–based reagents from Innogenetics Inc. The result was defined as the arithmetic mean of the calculated concentration of duplicates.
The amount of alpha-synuclein in CSF was analyzed by using a commercially available enzyme-linked immunosorbent assay kit from Covance. The concentration of alpha-synuclein was measured using standard alpha-synuclein curves (range, 6.1–1500 pg/mL using reconstituted stock) with 4-parameter curve fitting. The antibodies used in the kit do not cross react with beta-synuclein or gamma-synuclein.
Phospho-tau was measured with the Elecsys® assay run on the fully automated Roche Cobas® system.
Use of medications for PD was recorded at each visit after baseline assessment. For simplicity, we used this as a binary variable (medication present/absent).
Protein risk scores
The PD protein risk scores used in this study were taken from Tsukita et al.17. They were defined as “non-genetic Parkinson’s Disease-associated Proteomic Score (ngPD-ProS).
Statistical analysis
We first compared the protein profiles of PD patients with controls within each subcohort (idiopathic, GBA+ and LRRK2+ ). A linear regression model was applied using the Bioconductor R package limma v3.48.377. The model included the following covariates: age, sex, study center and proteomic principal components 1–4. The genetic subcohorts also included levodopa treatment (yes/no) as a covariate in the model to exclude treatment effects. This was skipped for idiopathic patients as they were drug-naïve.
In addition, a linear model with an interaction term was tested. The interaction term was between the disease status (case /control) and the mutation status (no mutation/GBA+/LRRK2+). The covariates were the same as in the models above.
Comparisons of clinical variables between endotypes of idiopathic patients were performed using a chi-squared test (two-sided) for categorical variables or a generalized linear model adjusted for age and sex for quantitative variables. To test differences in age between endotypes a Mann Whitney U-test (two-sided) was used.
Predictive modeling for the idiopathic classes was performed using a partition tree with pruning as implemented in the R package rpart v4.1.1678. The model was defined on a training set (70% of the idiopathic patients) and tested on an independent test set (30% of the idiopathic patients). Ten-fold cross validation was used for pruning.
All p values were adjusted for multiple testing using false discovery rate (FDR). Our SomaScan v3 -based findings were indirectly validated by assessing whether SOMAmers measure the correct protein or not. For this purpose, sera proteomes from heart failure patients (n = 88) were assessed using both SomaScan v3 and Olink®, a high-throughput proteomics technology based on dual antibody target recognition. The values of 1329 SOMAmers overlapped with those from 804 unique Olink® assays (there is some SOMAmer redundancy in SomaScan technology). The Pearson’s correlation coefficient as well as the p value79 were calculated for each overlap (see Tables 2 and 3). It should be noted that a high correlation suggests that the SOMAmer is correctly measuring the correct protein. However, the opposite is not necessarily true. A poor correlation does not automatically discard the result, since the two platforms may target different isoforms or post-translationally modified proteins. Moreover, they could just be measuring semiquantitative protein abundances beyond the linear fraction of their dynamic range.
Cluster analysis
Network analysis of the CSF proteome of idiopathic patients was carried out using the R package WGCNA v1.70.380. (WGCNA parameters: block-wise automatic module detection; soft threshold = 11; unsigned TOM, minimum module size = 500, Pearson correlation). Co-expressed proteins were grouped into modules of correlated proteins. Qiagen Ingenuity Pathway Analysis (IPA; July 2022 release) assisted unsupervised pathway analysis of proteins in modules.
Consensus clustering as implemented in the R package ConsensusClusterPlus v1.56.081. used the SOMAmer modules to identify idiopathic patient subclasses. To avoid confounders being responsible for the differences between patient subclasses, network analysis was performed on the SOMAmer residuals of a linear regression on age, sex and study center.
Heatmaps were generated using the R package Heatplus v3.0.082.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data used for this study is publicly available in the PPMI web page https://www.ppmi-info.org/access-data-specimens/download-data. The free access requires registration. The clinical data snapshot used here is kept under the tab “Archived PPMI data” » “Publication Associated Archives” » “2022-0001 Serrano-Fernandez: Parkinson’s Disease Proteogenomics (Version: 2022-05-18)”. The proteomic data is available under “Biospecimen” » “Proteomic Analysis” » “Project 151 Identification of proteins & protein networks & pQTL analysis in CSF x of 7 (Batch Corrected)” (7 files in total). The original adat files are also available under “Biospecimen” » “Proteomic Analysis” » “Project 151 Identification of proteins & protein networks & pQTL analysis in CSF - ADAT files”.
References
Group, G. B. D. N. D. C. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 16, 877–897 (2017).
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
Bonifati, V. Genetics of Parkinson’s disease-state of the art, 2013. Parkinsonism Relat. Disord. 20, S23–S28 (2014).
Kestenbaum, M. & Alcalay, R. N. Clinical Features of LRRK2 Carriers with Parkinson’s Disease. Adv. Neurobiol. 14, 31–48 (2017).
Thaler, A. et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat. Disord. 55, 45–49 (2018).
Behl, T. et al. Cross-talks among GBA mutations, glucocerebrosidase, and alpha-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: a comprehensive review. Transl. Neurodegener. 10, 4 (2021).
Ginns, E. I. et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet Metab. 111, 152–162 (2014).
Kozina, E., Byrne, M. & Smeyne, R. J. Mutant LRRK2 in lymphocytes regulates neurodegeneration via IL-6 in an inflammatory model of Parkinson’s disease. NPJ Parkinsons Dis. 8, 24 (2022).
Ho, D. H. et al. LRRK2 Inhibition Mitigates the Neuroinflammation Caused by TLR2-Specific alpha-Synuclein and Alleviates Neuroinflammation-Derived Dopaminergic Neuronal Loss. Cells 11, 861 (2022).
Qian, E. & Huang, Y. Subtyping of Parkinson’s Disease - Where Are We Up To? Aging Dis. 10, 1130–1139 (2019).
Ma, L. Y., Chan, P., Gu, Z. Q., Li, F. F. & Feng, T. Heterogeneity among patients with Parkinson’s disease: cluster analysis and genetic association. J. Neurol. Sci. 351, 41–45 (2015).
Parkinson Progression Marker, I. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Gold, L. et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5, e15004 (2010).
Rotunno, M. S. et al. Cerebrospinal fluid proteomics implicates the granin family in Parkinson’s disease. Sci. Rep. 10, 2479 (2020).
Marques, T. M. et al. Identification of cerebrospinal fluid biomarkers for parkinsonism using a proteomics approach. NPJ Parkinsons Dis. 7, 107 (2021).
Karayel, O. et al. Proteome profiling of cerebrospinal fluid reveals biomarker candidates for Parkinson’s disease. Cell Rep. Med. 3, 100661 (2022).
Tsukita, K. et al. Changes in the cerebrospinal fluid proteome precede and stratify the course of Parkinson’s Disease. medRxiv, 2022.2006.2008.22276035. https://doi.org/10.1101/2022.06.08.22276035 (2022).
Moloney, E. B., Moskites, A., Ferrari, E. J., Isacson, O. & Hallett, P. J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 120, 1–11 (2018).
Diaz-Ortiz, M. E. et al. GPNMB confers risk for Parkinson’s disease through interaction with alpha-synuclein. Science 377, eabk0637 (2022).
Ripoll, V. M., Irvine, K. M., Ravasi, T., Sweet, M. J. & Hume, D. A. Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J. Immunol. 178, 6557–6566 (2007).
Chung, J. S., Sato, K., Dougherty, I. I., Cruz, P. D. Jr & Ariizumi, K. DC-HIL is a negative regulator of T lymphocyte activation. Blood 109, 4320–4327 (2007).
Nagahara, Y. et al. Glycoprotein nonmetastatic melanoma protein B ameliorates skeletal muscle lesions in a SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neurosci. Res. 93, 1552–1566 (2015).
Nagahara, Y. et al. GPNMB ameliorates mutant TDP-43-induced motor neuron cell death. J. Neurosci. Res. 95, 1647–1665 (2017).
Saade, M., Araujo de Souza, G., Scavone, C. & Kinoshita, P. F. The Role of GPNMB in Inflammation. Front. Immunol. 12, 674739 (2021).
McGlinchey, R. P. & Lee, J. C. Cysteine cathepsins are essential in lysosomal degradation of alpha-synuclein. Proc. Natl Acad. Sci. USA 112, 9322–9327 (2015).
Farrow, S. L. et al. Establishing gene regulatory networks from Parkinson’s disease risk loci. Brain 145, 2422–2435 (2022).
Milanowski, L. M. et al. Cathepsin B p.Gly284Val Variant in Parkinson’s Disease Pathogenesis. Int J. Mol. Sci. 23, 7086 (2022).
Bournazos, S., Gupta, A. & Ravetch, J. V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 20, 633–643 (2020).
Kia, D. A. et al. Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study, Expression, and Epigenetic Data Sets. JAMA Neurol. 78, 464–472 (2021).
Storm, C. S. et al. Finding genetically-supported drug targets for Parkinson’s disease using Mendelian randomization of the druggable genome. Nat. Commun. 12, 7342 (2021).
Schilder, B. M. & Raj, T. Fine-mapping of Parkinson’s disease susceptibility loci identifies putative causal variants. Hum. Mol. Genet. 31, 888–900 (2022).
Choi, Y. R. et al. FcgammaRIIB mediates the inhibitory effect of aggregated alpha-synuclein on microglial phagocytosis. Neurobiol. Dis. 83, 90–99 (2015).
Choi, Y. R. et al. Prion-like Propagation of alpha-Synuclein Is Regulated by the FcgammaRIIB-SHP-1/2 Signaling Pathway in Neurons. Cell Rep. 22, 136–148 (2018).
Bastien, J., Menon, S., Messa, M. & Nyfeler, B. Molecular targets and approaches to restore autophagy and lysosomal capacity in neurodegenerative disorders. Mol. Asp. Med. 82, 101018 (2021).
Pang, S. Y. et al. LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl. Neurodegener. 11, 5 (2022).
Rivero-Rios, P., Romo-Lozano, M., Fasiczka, R., Naaldijk, Y. & Hilfiker, S. LRRK2-Related Parkinson’s Disease Due to Altered Endolysosomal Biology With Variable Lewy Body Pathology: A Hypothesis. Front. Neurosci. 14, 556 (2020).
Bossers, K. et al. Analysis of gene expression in Parkinson’s disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol. 19, 91–107 (2009).
Kapatos, G. The neurobiology of tetrahydrobiopterin biosynthesis: a model for regulation of GTP cyclohydrolase I gene transcription within nigrostriatal dopamine neurons. IUBMB Life 65, 323–333 (2013).
Deutch, A. Y. & Roth, R. H. Calcitonin gene-related peptide in the ventral tegmental area: selective modulation of prefrontal cortical dopamine metabolism. Neurosci. Lett. 74, 169–174 (1987).
Drumheller, A., Menard, D., Fournier, A. & Jolicoeur, F. B. Neurochemical effects of CGRP. Ann. N. Y Acad. Sci. 657, 546–548 (1992).
Pan, H. X. et al. GCH1 variants contribute to the risk and earlier age-at-onset of Parkinson’s disease: a two-cohort case-control study. Transl. Neurodegener. 9, 31 (2020).
Chen, J., Liu, X. & Zhong, Y. Interleukin-17A: The Key Cytokine in Neurodegenerative Diseases. Front Aging Neurosci. 12, 566922 (2020).
Bolte, A. C. & Lukens, J. R. Th17 Cells in Parkinson’s Disease: The Bane of the Midbrain. Cell Stem Cell 23, 5–6 (2018).
Reza, S., Ugorski, M. & Suchanski, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 31, 1416–1434 (2021).
Liu, Z., Huang, Y., Cao, B. B., Qiu, Y. H. & Peng, Y. P. Th17 Cells Induce Dopaminergic Neuronal Death via LFA-1/ICAM-1 Interaction in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 54, 7762–7776 (2017).
Angelopoulou, E., Paudel, Y. N., Villa, C. & Piperi, C. Arylsulfatase A (ASA) in Parkinson’s Disease: From Pathogenesis to Biomarker Potential. Brain Sci. 10, 713 (2020).
Navarro-Romero, A., Montpeyo, M. & Martinez-Vicente, M. The Emerging Role of the Lysosome in Parkinson’s Disease. Cells 9, 2399 (2020).
Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 45, 155–162 (2017).
Plowey, E. D., Cherra, S. J. 3rd, Liu, Y. J. & Chu, C. T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105, 1048–1056 (2008).
Hor, H. et al. Missense mutations in TENM4, a regulator of axon guidance and central myelination, cause essential tremor. Hum. Mol. Genet. 24, 5677–5686 (2015).
Algarni, M. & Fasano, A. The overlap between Essential tremor and Parkinson disease. Parkinsonism Relat. Disord. 46, S101–S104 (2018).
Garcia-Esparcia, P., Hernandez-Ortega, K., Ansoleaga, B., Carmona, M. & Ferrer, I. Purine metabolism gene deregulation in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 41, 926–940 (2015).
Hernandez, C. J., Baez-Becerra, C., Contreras-Zarate, M. J., Arboleda, H. & Arboleda, G. PINK1 Silencing Modifies Dendritic Spine Dynamics of Mouse Hippocampal Neurons. J. Mol. Neurosci. 69, 570–579 (2019).
Ndayisaba, A., Kaindlstorfer, C. & Wenning, G. K. Iron in Neurodegeneration - Cause or Consequence? Front. Neurosci. 13, 180 (2019).
Fernandez-Espejo, E., Rodriguez de Fonseca, F., Suarez, J. & Martin de Pablos, A. Cerebrospinal fluid lactoperoxidase level is enhanced in idiopathic Parkinson’s disease, and correlates with levodopa equivalent daily dose. Brain Res. 1761, 147411 (2021).
Liang, T., Qian, Z. M., Mu, M. D., Yung, W. H. & Ke, Y. Brain Hepcidin Suppresses Major Pathologies in Experimental Parkinsonism. iScience 23, 101284 (2020).
Urrutia, P. J., Borquez, D. A. & Nunez, M. T. Inflaming the Brain with Iron. Antioxid. (Basel) 10, 61 (2021).
Knight, A. L. et al. The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson’s models. Cell Metab. 20, 145–157 (2014).
Wei, D. et al. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 31, 157–177 (2021).
Kumar, R. et al. FGF2 Affects Parkinson’s Disease-Associated Molecular Networks Through Exosomal Rab8b/Rab31. Front. Genet. 11, 572058 (2020).
Yan, R. & Liu, Z. LRRK2 enhances Nod1/2-mediated inflammatory cytokine production by promoting Rip2 phosphorylation. Protein Cell 8, 55–66 (2017).
Wu, Y. et al. Association of VEGF gene polymorphisms with sporadic Parkinson’s disease in Chinese Han population. Neurol. Sci. 37, 1923–1929 (2016).
Korkmaz, O., Ay, H., Ulupinar, E. & Tuncel, N. Vasoactive intestinal peptide enhances striatal plasticity and prevents dopaminergic cell loss in Parkinsonian rats. J. Mol. Neurosci. 48, 565–573 (2012).
Marek, K. et al. The Parkinson’s progression markers initiative (PPMI) - establishing a PD biomarker cohort. Ann. Clin. Transl. Neurol. 5, 1460–1477 (2018).
Simuni, T. et al. Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol. 19, 71–80 (2020).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Shabalin, A. A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 13, e1007081 (2017).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet 50, 693–698 (2018).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014).
Goetz, C. G. et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 23, 2129–2170 (2008).
Nasreddine, Z. S. et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 53, 695–699 (2005).
Kang, J. H. et al. Association of cerebrospinal fluid beta-amyloid 1-42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 70, 1277–1287 (2013).
Mollenhauer, B. et al. Longitudinal analyses of cerebrospinal fluid alpha-Synuclein in prodromal and early Parkinson’s disease. Mov. Disord. 34, 1354–1364 (2019).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Therneau, T., et al. Rpart: Recursive Partitioning and Regression Trees, R Package Version 4.1-15. (2019).
Best, D. J. & Roberts, D. E. Algorithm AS 89: The Upper Tail Probabilities of Spearman’s Rho. J. R. Stat. Soc. Ser. C. (Appl. Stat.) 24, 377–379 (1975).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010).
Heatplus: Heatmaps with row and/or column covariates and colored clusters. R package (Bioconductor, https://github.com/alexploner/Heatplus, 2015).
Svenningsson, P., Palhagen, S. & Mathe, A. A. Neuropeptide Y and Calcitonin Gene-Related Peptide in Cerebrospinal Fluid in Parkinson’s Disease with Comorbid Depression versus Patients with Major Depressive Disorder. Front Psychiatry 8, 102 (2017).
Pu, J. L. et al. Parkinson’s Disease in Teneurin Transmembrane Protein 4 (TENM4) Mutation Carriers. Front Genet 11, 598064 (2020).
Liang, D. et al. Rare variant analysis of essential tremor-associated genes in early-onset Parkinson’s disease. Ann Clin Transl Neurol 8, 119–125 (2021).
Acknowledgements
The authors would like to thank Rose Case (Indiana University Genetics Biobank, Indiana University School of Medicine, Indianapolis, USA) for her key role in sample management and Myung Shin (Genome and Biomarker Sciences. Merck Exploratory Science Center. Cambridge, USA), Karla Gonzalez (Department of Medical and Molecular Genetics, Indiana University School of Medicine. Indianapolis, USA) and Faraz Faghri (Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health. Bethesda, USA) for constructive feedback. The study was funded by the Novartis Institutes for Biomedical Research and Merck. Protein measurements were performed at SomaLogic.
Author information
Authors and Affiliations
Contributions
Study conception and design: F.E., B.B., S.K., P.S.F., M.M., S.J.H.; Sample organization and data upload: Mary.D., V.S., M.M., P.S.F., K.C.; Conceived, designed and performed the analysis: S.K., P.S.F., L.Z., J.J., B.P.; Interpretation of results: S.K., P.S.F., L.Z., J.J., B.M., S.L., J.D.L., J.M., N.R., D.S., O.F., D.G., Mohammed.D, A.K., J.L., M.R., T.F., H.I., A.S., K.M.; Draft paper preparation: S.K., P.S.F., L.Z., B.M., F.E., M.M. All authors reviewed the results and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors report no competing interests. A.S. is an editor for npj Parkinson’s Disease. A.S. was not involved in the journal’s review of, or decisions related to, this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaiser, S., Zhang, L., Mollenhauer, B. et al. A proteogenomic view of Parkinson’s disease causality and heterogeneity. npj Parkinsons Dis. 9, 24 (2023). https://doi.org/10.1038/s41531-023-00461-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-023-00461-9
This article is cited by
-
Proteogenomic network analysis reveals dysregulated mechanisms and potential mediators in Parkinson’s disease
Nature Communications (2024)
-
Paradigm shift required for translational research on the brain
Experimental & Molecular Medicine (2024)
-
Machine learning and deep learning approach to Parkinson’s disease detection: present state-of-the-art and a bibliometric review
Multimedia Tools and Applications (2024)
-
The Molecular Impact of Glucosylceramidase Beta 1 (Gba1) in Parkinson’s Disease: a New Genetic State of the Art
Molecular Neurobiology (2024)
-
Proteome wide association studies of LRRK2 variants identify novel causal and druggable proteins for Parkinson’s disease
npj Parkinson's Disease (2023)
