
Abstract
The relationship between APOE polymorphisms and Parkinson’s disease (PD) in black Africans has not been previously investigated. We evaluated the association between APOE polymorphic variability and self-declared cognition in 1100 Nigerians with PD and 1097 age-matched healthy controls. Cognition in PD was assessed using the single item cognition question (item 1.1) of the MDS-UPDRS. APOE genotype and allele frequencies did not differ between PD and controls (p > 0.05). No allelic or genotypic association was observed between APOE and age at onset of PD. In PD, APOE ε4/ε4 conferred a two-fold risk of cognitive impairment compared to one or no ε4 (HR: 2.09 (95% CI: 1.13–3.89; p = 0.02)), while APOE ε2 was associated with modest protection against cognitive impairment (HR: 0.41 (95% CI 0.19–0.99, p = 0.02)). Of 773 PD with motor phenotype and APOE characterized, tremor-dominant (TD) phenotype predominated significantly in ε2 carriers (87/135, 64.4%) compared to 22.2% in persons with postural instability/gait difficulty (PIGD) (30/135) and 13.3% in indeterminate (ID) (18/135, 13.3%) (p = 0.037). Although the frequency of the TD phenotype was highest in homozygous ε2 carriers (85.7%), the distribution of motor phenotypes across the six genotypes did not differ significantly (p = 0.18). Altogether, our findings support previous studies in other ethnicities, implying a role for APOE ε4 and ε2 as risk and protective factors, respectively, for cognitive impairment in PD.
Similar content being viewed by others


Introduction
The gene encoding apolipoprotein E (APOE), located on chromosome 19q13.2, has three commonly described polymorphic alleles (ε2, ε3, ε4) constituting six genotypes in humans (ε2/ε2, ε2/ ε3, ε2/ ε4, ε3/ ε3, ε3/ ε4, ε4/ ε4). Apolipoprotein E plays a vital role in lipid metabolism and has been linked to both vascular and neurodegenerative pathological processes. Allelic and genotypic variability in APOE have been extensively explored in Alzheimer’s disease (AD) and other neurodegenerative conditions including Parkinson’s disease (PD)1,2. Currently, variability in APOE is the strongest known common genetic risk factor for late onset AD, in which the APOE ε4 allele increases disease risk and lowers the age at disease onset, whereas the APOE ε2 allele confers a protective effect against AD2,3,4. In Northern European Ancestry individuals, homozygous carriers of APOE ε4 have up to a 12-fold increased risk for AD compared to non-carriers, whereas there is a weaker but significant effect for incident AD in persons of Yoruba ancestry in Nigeria1,5,6,7.
The relationship between APOE polymorphic variability and disease status and age at onset remains unclear for PD, with a significant number of studies yielding inconsistent results. Recent evidence from large genome-wide association studies (GWAS) of Northern European ancestry participants showed no convincing link between APOE genotype and PD disease status or age at onset8. However, recent meta-analyses combining data from cohorts of varied ethnicities including Europeans, Asians and Latin-Americans showed that the association between APOE genotype and PD risk could be ancestry dependent9. APOE ε4 but not ε2 was shown to be a consistent risk factor for PD in Asian populations but not in Northern European ancestry individuals and Latin-Americans8,9. APOE ε4 was also shown to be consistently associated with a higher incidence of cognitive decline in patients with PD from Northern European, Asian and Latino backgrounds9.
Few studies have examined the role of APOE (and specifically ε4) in neurodegeneration in Africans10,11,12. To date, the link between APOE and PD risk and age at onset or PD related cognitive impairment has not been explored in individuals of black African ancestry within or outside Africa. The interaction with motor phenotype is also unknown. The objective of this study was to examine the role of APOE polymorphisms in the genetic susceptibility to PD in Nigerians, and to interrogate possible interactions with age at onset, motor phenotype and cognitive status.
Results
Cohort characteristics
The study participants comprised of 1100 Nigerians with PD and 1097 cognitively normal age-matched controls of similar ancestry. Baseline characteristics are shown in Supplementary Table 1 and includes the sex distribution (female: PD—302 (27.5%), controls—382 (34.8%)), mean age at study (years) (PD: 64.6 ± 10.0, controls: 62.7 ± 9.0), mean age at onset of PD (59.6 ± 10.5 years) and median duration of PD (interquartile range (IQR)) 3.0 (3.0) years. A significantly higher proportion of controls in this study were female (p = 0.000). Among individuals with PD, mean age at study and mean age at onset did not differ by sex (p = 0.15, respectively). Disease duration, median PD stage (Hoehn and Yahr), median MDS UPDRS cognition score and the proportions with abnormal cognition (as defined, i.e. score of 0 or 1 on the MDS UPDRS cognition question) were also similar when compared by sex (p > 0.05 for all). In non-parametric (Spearman’s) correlation analysis, MDS UPDRS cognition score was significantly positively correlated with age at study (Rs = 0.145, p = 0.000), age at onset of PD (Rs = 0.094, p = 0.002) and duration of PD at study (Rs = 0.115, p = 0.000).
APOE allelic and genotypic frequency proportions in Nigerians with PD and controls
The allele frequencies of APOE in all participants (n = 2197) were: ε3 (58.9%), ε4 (29%) and ε2 (12.1%). The genotypic frequencies of APOE were as follows for homozygotes (ε3/ε3 (43.2%), ε4/ε4 (5.7%), ε2/ε2 (1.4%)) and heterozygotes (ε3/ε4 (33%), ε2/ε3 (12%), ε2/ε4 (4.7%)), respectively. As shown in Tables 1 and 2 and Supplementary Tables 2 and 3, there was no significant difference in allele (p = 0.17) or genotype frequencies (p = 0.56) in PD versus controls in this study. No sex differences were observed (p > 0.05). Tables 1 and 2 also compare the distribution of APOE alleles and genotypes in Nigerian PD patients and controls in this study to data from previous reports describing these frequencies in general populations from different ethnicities13,14,15.
Association between APOE and PD risk, age at onset and cognition status
The Odds ratios (95% CI) for the comparison between PD and controls for allele distribution (Supplementary Table 3) were as follows: ε2: 0.97 (0.87–1.08), p = 0.56; ε3: 1.10 (0.97–1.24), p = 0.15; ε4: 0.94 (0.97–1.03), p = 0.17. Supplementary Table 4 provides data on the association of ε2 and ε4 dose to disease status, demonstrating the lack of association with PD status (p>0.05 for all comparisons).
Supplementary Table 5 and Supplementary Fig. 1 explore the allelic and genotypic relationship of APOE to age at onset of PD. Genotypic and allelic genotypes in APOE did not influence age at onset of PD, and neither did ε2 or ε4 dose (data not shown; p > 0.05 for all iterations).
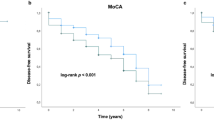
Table 3 provides data on the relationship of allelic and genotypic variability and ε2 and ε4 dose to cognition status in PD. Homozygosity for ε4 conferred a two-fold increased risk for cognitive impairment in PD (Hazards ratio 2.09 (95% CI 1.13–3.89), p = 0.02), whereas the presence of at least one ε2 allele reduced the likelihood of cognitive impairment (HR 0.41 (95% CI 0.19–0.88), p = 0.023). None of the 18 PD participants homozygous for ε2 had cognitive impairment.
Association between motor phenotype, cognition and APOE variability
773 persons with PD had both APOE genotype and motor phenotype data, and are included in this report. Motor phenotype was tremor dominant (TD) in 438 (56.7%), postural instability/gait difficulty (PIGD) in 244 (31.6%), and indeterminate (ID) in 91 (11.8%). Of these, self-declared abnormal cognition was present in a significantly higher number of PIGD (38/244, 15.6%), compared to TD (37/438, 8.4%) and ID (9/91, 9.9%), p = 0.02.
Considering the allele as the exposure, the frequency of motor phenotype varied significantly in ε2 carriers, being highest for TD (87, 64.4%) compared to PIGD (30, 22.2%) and ID (18, 13.3%), p = 0.037. There was no significant difference in distribution of motor phenotypes in ε3 and ε4 carriers (p = 0.59 and p = 0.97, respectively) (Supplementary table 7). The distribution of motor phenotypes per APOE genotype is also shown in Supplementary table 7, and did not vary significantly (p = 0.18), although the frequency of the TD phenotype was highest overall in homozygous ε2 carriers (12, 85.7%).
Discussion
This is the largest dataset from individuals of black African ancestry investigated to date describing genetic variability in APOE in the context of PD and providing a comparison to ethnically matched otherwise healthy subjects from the same geographical location. In addition, we provide further insight into the distribution of APOE in modern populations by adding to the existing data on the frequency of APOE alleles and genotypes from the healthy population in Nigeria. APOE allelic and genotypic frequencies for our entire cohort had a similar distribution to that reported in populations of African ancestry except for the ε4/ε4 genotype which was higher than the global average (5.7% in the present study versus 1.41% global) and higher than the average frequency in other black populations (3.44%)13,14,15. As has been described in other publications, the APOE ε3 was the most frequent allele, present in 59.9% of the healthy controls in this study (compared to the widely variable global range of 8.5–98.0% derived from populations across all continents)13, and within the range of rates reported from modern African populations (48–94%)13,14,15,16. The frequency of ε4 in our healthy controls (28.2%) is also mid-range of the typical rates (14.3–40.7%) for Africa, in which the highest frequencies are in Central Africans (from 29% in Fon to 40% in Aka pygmies)13. The least frequent allele was ε2 (present in 11.8%) and also coincides (though at the higher end of the range) with earlier reports from Africa (2.7–11.6%)13,17. The genotype distributions in our healthy controls (ε3/ε3 and ε3/ε4 most common) followed a similar trend with the most recent data for individuals of black ethnicity included in the systematic review by Qin et al.15.
Regarding the specific objectives of our study, we found no association between any specific APOE allele or genotype and the risk of PD in our population. Our findings corroborate previous observations in other populations, indicating that the distribution of APOE alleles (including specifically ε4 carrier rates), ε4 or ε2 allele dosage, and APOE genotypes are not significantly different between PD and controls18,19. We did not observe the significant over-representation of APOE ε2 carriers in PD reported in previous meta-analysis, and our observation is similar with the data presented by William-Gray et al. for the primary cohort of 528 PD and 512 controls, in which the frequency of ε2 was 8.3% for both PD and controls18,19,20.
We found a modest but significant (protective) association of the APOE ε2 allele with cognitive status in our cohort, with higher ε2 dosage conferring a lower (but small) risk of self-declared cognitive impairment in individuals with PD. On the other hand, the presence of the ε4 allele conferred a two-fold increased risk of abnormal cognition. These findings align with the postulate of a modest protective effect of ε2 and detrimental impact of ε4 on cognition in PD. Several studies and meta-analyses have demonstrated an over-representation of APOE ε4 carriers amongst individuals with PD cognitive impairment and dementia, although others have been equivocal or provided only modest evidence19,21,22,23,24. Studies including GWAS of neuropathologically confirmed PD have also strengthened the credibility of an association with APOE e4 carrier status by demonstrating a significant association with cognitive decline in PD25,26. The more appealing explanation of the effect of genetic variability on cognition in PD is that of a cumulative effect conferred by multiple common (often independently low risk) variants (polygenic risk) such as APOE ε4. A recent longitudinal genome-wide survival study not only confirmed the notion of an association between APOE and cognition in PD, but demonstrated a substantial aggregate association of polygenic progression scores (but not polygenic susceptibility scores) with dementia risk, and proposed diverging genetic architectures of cognitive disease progression and susceptibility27. The pathophysiologic basis for APOE ε4 and ε2 as risk and protective factors, respectively, for cognitive decline in neurodegenerative disorders have yet to be clearly elucidated although clues to biologically plausible mechanism are emerging28,29,30. APOE protein structure (and function) varies per allele due to the single amino acid substitutions that result in greater inherent stability in ε2, which also confers less domain specific interactions that deter neurodegenerative processes (such as amyloid-related damage, synaptic dysfunction, oxidation and inflammation) in contrast to ε428,29. Ultimately, the processes are likely complex, multiple/overlapping and reflect interactions with other genetic and environmental factors and aging.
Our findings regarding the higher frequency of abnormal cognition in the PIGD phenotype corroborate the previously documented association of this motor phenotype with a higher burden of abnormal cognition, greater risk for incident dementia in PD and a faster rate of cognitive decline31,32,33. The motor phenotype distribution in this study is similar to that previously described in our cohort overall (TD 56.5%, PIGD 31.4% and ID 12.1%, respectively)34. Although we consider our findings preliminary, the trend towards the less severe tremor-dominant motor phenotype in ε2 carriers may signify a protective effect on motor severity. However, this may also reflect the inherent heterogeneity of the genetics and pathophysiology of PD. Subtypes of PD may have different genetic predictors as demonstrated by Factor and colleagues who found that postural instability with falling (PIF) (proposed as a subtype of PIGD) was inversely associated with APOE ε4 (suggesting a protective effect), in contrast to the more severe variant of freezing of gait (FOG)35.
We acknowledge the limitations with respect to the measure of cognitive function utilized in this analysis, and understand the inherent challenge with specifically comparing our data with studies that have used more widely recommended and robust measures such as the Montreal Cognitive Assessment (MoCA) and other extensive cognition batteries36. The aspiration to provide an albeit exploratory impression of the relationship of APOE status to cognition in our population where no prior data exists, coupled with the precedence for the use of the MDS UPDRS single patient-reported cognition question in the absence of more robust assessments provided the rationale for this approach37. This single item reportedly is most strongly associated with visuospatial/executive function and delayed recall on the MoCA37. As alluded to by Mills and colleagues, the less complicated, more global patient-reported cognitive measure are externally valid and inherently useful. Our study is informative in that with only one question about global cognition, the patient had to sum his or her experiences and give a general response based on the degree of self-assessed severity while avoiding the distraction of more interrogative approaches. In addition, our observations are likely credible because the trend of association between APOE ε4 and cognitive impairment occurred despite the similarity in potential clinical confounders such as age at onset, duration of PD and age at study. Although the proportion of PD with cognitive impairment at the median duration of disease in this study using the single MDS UPDRS cognition screen is similar to previous studies employing more robust assessments of cognition, the interpretation of our findings must be cautious. A validation study of the MDS-UPDRS Part 1 for non-motor symptoms compared the single item cognition question to Addenbrooke’s Cognitive Examination (ACE), Scales for Outcome of Parkinson’s disease (SCOPA)-cognitive scale (SCOPACOG) and Frontal Assessment Battery (FAB) found a weak (though positive and statistically significant) correlation for all three cognitive scales (ACE, SCOPA-COG and FAB). The study alluded to the possibility of heterogeneity in cognitive profiles in PD, which makes demonstrating a stronger correlation with a single screening question difficult38. We acknowledge that our findings do not imply causality or define a mechanism for the cognitive impairment as no radiological, laboratory or neuropathological studies to exclude other underlying risk factors such as vascular or other co-existing neurodegenerative pathologies was conducted. In addition, the perception of cognitive status represents a combination of participant and/or caregiver’s report, and should be interpreted in this context. Our study is also limited by the draw-back of assessing cognitive profile derived from a static snapshot whereas cognitive decline is an inherently dynamic clinical variable which can occur with disease progression in neurodegenerative disorders such as PD.
In conclusion, our study provides an important dataset describing the association between APOE and PD in individuals of black African ancestry, demonstrating a lack of association with disease risk and age at onset and indicating a trend of association of cognitive impairment with APOE ε4 and protection by higher doses of APOE ε2.
Methods
Approval of the study protocol was obtained from the institutional health research ethics committees at all participating recruiting sites, the National Health Research Ethics Committee (NHREC) in Nigeria and the ethics committee of the University College London and the National Hospital for Neurology and Neurosurgery, London, United Kingdom. All participants provided written informed consent prior to inclusion in the study.
Participant recruitment and clinical assessments
A total of 1100 Nigerians with PD and 1097 healthy controls matched by age were included in this cohort study. All controls had an abridged neurological examination and were clinically assessed as cognitively normal based on the absence of any self-declared problems with memory, concentration, orientation or attention. We excluded 83 participants (31 controls and 52 PD participants) with incomplete genotyping data. The excluded participants did not differ from those included based on age at study (p = 0.998), male/female ratio (p = 0.98) or age at onset of PD (p = 0.56). Participants were recruited from an ongoing study being conducted by the Nigeria Parkinson’s Disease Research (NPDR) network in collaboration with the International Parkinson’s Disease Genomics Consortium Africa (IPDGC Africa)39,40. The NPDR includes participating sites from tertiary neurology clinics covering all 6 geopolitical regions in Nigeria39.
PD diagnosis was based on the United Kingdom Parkinson’s Disease Society Brain Bank (UKPDSBB) criteria41. Data available for analysis in this study include baseline demographics (age at study, sex, age at onset of PD, duration of PD (years), disease stage (Hoehn and Yahr) and patient-reported cognitive status. Cognitive status was clinically assessed in individuals with PD. No brain imaging or additional evaluation for aetiology of cognitive impairment was conducted. We used the Movement Disorder Society (MDS) Unified Parkinson’s Disease Rating Scale (UPDRS) (n = 988) or the earlier version of the UPDRS (n = 112) single item question on cognitive status (Part 1 item 1.1 of the instrument). The response is rated as 0: Normal: No cognitive impairment; (1) Slight: Impairment appreciated by patient or caregiver with no concrete interference with the patient’s ability to carry out normal activities and social interactions; (2) Mild: Clinically evident cognitive dysfunction, but only minimal interference with the patient’s ability to carry out normal activities and social interactions; (3) Moderate: Cognitive deficits interfere with but do not preclude the patient’s ability to carry out normal activities and social interactions and (4) Severe: Cognitive dysfunction precludes the patient’s ability to carry out normal activities and social interactions. The responses for the small sample of 113 with old UPDRS scores were recoded to the most approximate MDS UPDRS score (0→0, 1→1, 2→3 and 3 or 4→4). This convenience was adopted because the previously published formulae for calibration of data allows archival UPDRS Parts II and III data to be accurately transferred to MDS-UPDRS scores but are not accurate for Part I and IV scores42. In this study, cognition scores of 0 and 1 were interpreted as PD with normal cognition, whereas scores of 2–4 were regarded as abnormal cognition. Motor phenotype was determined using the method described by Stebbins et al.34,43. In summary, the phenotypes are computed using specified MDS UPDRS items for computing tremor score (Part II item 2.10 and Part III items 3.15–3.18 assessing postural, kinetic and rest tremor and rest tremor constancy) and postural instability/gait difficulty (PIGD) score (Part II items 2.12 (walking and balance), 2.13 (freezing), 3.10 (gait), 3.11 (freezing of gait) and 3.12 (postural instability)). The categorization of motor phenotype is based on the MDS-UPDRS TD/PIGD score, which is the mean of the tremor items divided by the mean of the PIGD items. Assignment of phenotype is based on the ratio obtained (TD ≥ 1.15, PIGD ≤ 0.90, indeterminate between 0.90 and 1.15)43.
APOE genotyping
DNA was extracted from saliva samples collected using DNA Genotek® saliva kits or from venous whole blood samples using standard protocols. APOE genetic variation was determined by genotyping two well established non-synonymous single nucleotide polymorphisms (SNPs): rs429358 and rs7412. The Kompetitive Allele-Specific Polymerase Chain Reaction (PCR) assay (KASP™, LGC Genomics. Herts, UK) was used to genotype both SNPs in 987 participants with PD and 1050 controls44. In addition, 113 samples from individuals with PD and 47 controls were genotyped using the Infinium® NeuroChip Consortium Array (Illumina, San Diego, CA, USA)45,46. NeuroChip array description and validation of its ability to accurately identify APOE genotype calls compared to standard Taqman genotyping is well established47. Quality control assessments for the arrays were carried out using PLINK version 1.9 and genotype calls of the rs429358 and rs7412 SNPs were extracted to define the APOE alleles45,46. SNPs genotypes were assessed for Hardy-Weinberg equilibrium (HWE) using Fisher exact test.
Data analyses
Cohort characteristics are expressed as counts (%), mean ± SD or medians and compared between groups (PD and controls) using two-tailed X2 test for categorical variables (or) analysis of variance (ANOVA) or non-parametric alternative for continuous variables as relevant. Frequency proportions in percent of APOE alleles (ε2, ε3, ε4) and genotypes (ε2/ε2, ε2/ ε3, ε2/ ε4, ε3/ ε3, ε3/ ε4, ε4/ ε4) were calculated and compared to published data in subjects from other populations. Logistic regression was used to analyze the association between APOE and PD risk, and cognitive performance (in individuals with PD with normal cognition versus PD with impaired cognition). The differences in the frequencies of genotypes and allele between persons with PD and controls, and Hardy-Weinberg equilibrium (HWE) was tested using the Pearson’s Chi-square test. SNP rs429358 was in HWE for both control and cases (p = 0.76 and p = 0.40, respectively). For SNP rs7412, HWE was preserved in controls (p = 0.56) but not in (cases p = 0.03) (See Supplementary Fig. 1). Cox proportional hazards regression was used to investigate the influence of APOE on age of onset of PD. For all analyses, PD cases and (or) controls were used as the dependent variable and the relevant APOE allele, genotype and ε4 dose as the independent variables, adjusting where relevant for sex, age at onset or at study in PD and age at recruitment for controls. APOE ε4 dose was defined as 0 dose = ε2/ε2, ε2/ ε3 and ε3/ ε3, 1 dose = ε3/ ε4 and 2 doses = ε4/ ε4. Genotype ε2/ε4 was excluded from the analysis because ε2 is considered protective and ε4 is considered a risk variant. Data were analyzed using Stata/MP version 16.0 statistical software (Stata Corporation, College Station, TX: StataCorp LLC).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The dataset analyzed during the current study is available from the corresponding author upon reasonable request (e.g. reproducibility of research). Sharing restrictions will be applied to sensitive data to preserve the privacy of participants.
References
Reitz, C., Brayne, C. & Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 7, 137–152 (2011).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Jun, G. R. et al. Protein phosphatase 2A and complement component 4 are linked to the protective effect of APOE ɛ2 for Alzheimer’s disease. Alzheimers Dement. https://doi.org/10.1002/alz.12607 (2022).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Farrer, L. A. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997).
Serrano-Pozo, A., Das, S. & Hyman, B. T. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 20, 68–80 (2021).
Hendrie, H. C. et al. APOE ε4 and the risk for Alzheimer disease and cognitive decline in African Americans and Yoruba. Int. Psychogeriatr. 26, 977–985 (2014).
Sun, R., Yang, S., Zheng, B., Liu, J. & Ma, X. Apolipoprotein E Polymorphisms and Parkinson disease with or without dementia: a meta-analysis including 6453 participants. J. Geriatr. Psychiatry Neurol. 32, 3–15 (2019).
Li, J., Luo, J., Liu, L., Fu, H. & Tang, L. The genetic association between apolipoprotein E gene polymorphism and Parkinson disease: a meta-analysis of 47 studies. Medicine 97, e12884 (2018).
Sawyer, K., Sachs-Ericsson, N., Preacher, K. J. & Blazer, D. G. Racial differences in the influence of the APOE epsilon 4 allele on cognitive decline in a sample of community-dwelling older adults. Gerontology 55, 32–40 (2009).
Knopman, D. S., Mosley, T. H., Catellier, D. J., Coker, L. H. & Atherosclerosis Risk in Communities Study Brain MRI Study. Fourteen-year longitudinal study of vascular risk factors, APOE genotype, and cognition: the ARIC MRI Study. Alzheimers Dement. 5, 207–214 (2009).
Akinyemi, R. O. et al. Dementia in Africa: current evidence, knowledge gaps, and future directions. Alzheimers Dement. 18, 790–809 (2022).
Abondio, P. et al. The genetic variability of APOE in different human populations and its implications for longevity. Genes (Basel) 10, 222 (2019).
Corbo, R. M. & Scacchi, R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann. Hum. Genet. 63, 301–310 (1999).
Qin, W. et al. Race-Related Association between APOE genotype and Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimers Dis. 83, 897–906 (2021).
Eisenberg, D. T., Kuzawa, C. W. & Hayes, M. G. Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history. Am. J. Phys. Anthropol. 143, 100–111 (2010).
Kamboh, M. I., Sepehrnia, B. & Ferrell, R. E. Genetic studies of human apolipoproteins. VI. Common polymorphism of apolipoprotein E in blacks. Dis. Markers 7, 49–55 (1989).
Federoff, M., Jimenez-Rolando, B., Nalls, M. A. & Singleton, A. B. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol. Dis. 46, 389–392 (2012).
Williams-Gray, C. H. et al. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J. Neurol. 256, 493–498 (2009).
Huang, X., Chen, P. C. & Poole, C. APOE-ε2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology 62, 2198–2202 (2004).
Gonzalez-Latapi, P., Bayram, E., Litvan, I. & Marras, C. Cognitive impairment in Parkinson’s disease: epidemiology, clinical profile, protective and risk factors. Behav. Sci. (Basel) 11, 74 (2021).
D’Souza, T. & Rajkumar, A. P. Systematic review of genetic variants associated with cognitive impairment and depressive symptoms in Parkinson’s disease. Acta Neuropsychiatr. 32, 10–22 (2020).
Mata, I. F. et al. Large-scale exploratory genetic analysis of cognitive impairment in Parkinson’s disease. Neurobiol. Aging 56, 211.e1–211.e7 (2017).
Mata, I. F. et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 71, 1405–1412 (2014).
Tan, M. M. X. et al. Genome-wide association studies of cognitive and motor progression in Parkinson’s disease. Mov. Disord. 36, 424–433 (2021).
Tunold, J. A. et al. APOE and MAPT are associated with dementia in Neuropathologically confirmed Parkinson’s disease. Front. Neurol. 12, 631145 (2021).
Liu, G. et al. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat. Genet. 53, 787–793 (2021).
Suri, S., Heise, V., Trachtenberg, A. J. & Mackay, C. E. The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE ɛ2. Neurosci. Biobehav Rev. 37, 2878–2886 (2013).
Szwedo, A. A. et al. GBA and APOE impact cognitive decline in parkinson’s disease: a 10-year population-based study. Mov. Disord. 37, 1016–1027 (2022).
Dilliott, A. A. et al. Association of apolipoprotein E variation with cognitive impairment across multiple neurodegenerative diagnoses. Neurobiol. Aging 105, 378.e1–378.e9 (2021).
Michels, J. et al. Long-term cognitive decline related to the motor phenotype in Parkinson’s disease. J. Parkinsons Dis. 12, 905–916 (2022).
Arie, L., Herman, T., Shema-Shiratzky, S., Giladi, N. & Hausdorff, J. M. Do cognition and other non-motor symptoms decline similarly among patients with Parkinson’s disease motor subtypes? Findings from a 5-year prospective study. J. Neurol. 264, 2149–2157 (2017).
Burn, D. J. et al. Motor subtype and cognitive decline in Parkinson’s disease, Parkinson’s disease with dementia, and dementia with Lewy bodies. J. Neurol. Neurosurg. Psychiatry 77, 585–589 (2006).
Ojo, O. O. et al. A cross-sectional comprehensive assessment of the profile and burden of non-motor symptoms in relation to motor phenotype in the Nigeria Parkinson Disease Registry cohort. Mov. Disord. Clin. Pract. 8, 1206–1215 (2021).
Factor, S. A. et al. Postural instability/gait disturbance in Parkinson’s disease has distinct subtypes: an exploratory analysis. J. Neurol. Neurosurg. Psychiatry 82, 564–568 (2011).
Skorvanek, M. et al. Global scales for cognitive screening in Parkinson’s disease: critique and recommendations. Mov. Disord. 33, 208–218 (2018).
Mills, K. A. et al. Cognitive impairment in Parkinson’s disease: association between patient-reported and clinically measured outcomes. Parkinsonism Relat. Disord. 33, 107–114 (2016).
Gallagher, D. A., Goetz, C. G., Stebbins, G., Lees, A. J. & Schrag, A. Validation of the MDS-UPDRS Part I for nonmotor symptoms in Parkinson’s disease. Mov. Disord. 27, 79–83 (2012).
Ojo, O. O. et al. The Nigeria Parkinson Disease Registry: process, profile, and prospects of a collaborative project. Mov. Disord. 35, 1315–1322 (2020).
Rizig, M. et al. The International Parkinson Disease Genomics Consortium Africa. Lancet Neurol. 20, 335 (2021).
Hughes, A. J., Daniel, S. E., Kilford, L. & Lees, A. J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 55, 181–184 (1992).
Goetz, C. G., Stebbins, G. T. & Tilley, B. C. Calibration of Unified Parkinson’s Disease Rating Scale scores to Movement Disorder Society-Unified Parkinson’s Disease Rating Scale cores. Mov. Disord. 27, 1239–1242 (2012).
Stebbins, G. T. et al. How to identify tremor dominant and postural instability/gait difficulty groups with the movement disorder society unified Parkinson’s disease rating scale: comparison with the unified Parkinson’s disease rating scale. Mov. Disord. 28, 668–670 (2013).
Zhang, L. et al. Apolipoprotein E polymorphisms contribute to statin response in Chinese ASCVD patients with dyslipidemia. Lipids Health Dis. 18, 129 (2019).
Sabir, M. S. et al. Assessment of APOE in atypical parkinsonism syndromes. Neurobiol. Dis. 127, 142–146 (2019).
Hannon, E. et al. Genetic risk for Alzheimer’s disease influences neuropathology via multiple biological pathways. Brain Commun. 2, fcaa167 (2020).
Blauwendraat, C. et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol. Aging 57, 247.e9–247.e13 (2017).
Acknowledgements
We would like to thank the patients, their care partners and the controls for their participation. We acknowledge Dr. David Curtis of the UCL Genetics Institute, University College London, London, United Kingdom for his advice regarding data analysis. Funding support was provided by the Michael J Fox Foundation Genetic Diversity in Parkinson’s Disease 2019 (Grant ID:17483) awarded to N.O., M.R., H.H., J.H. and A.S., and the Tertiary Education Trust Fund (TETFund) National Research Fund (NRF) Intervention 2019 (Batch 6) (N.O.).
Author information
Authors and Affiliations
Contributions
Conceptualization and design: N.O., O. Okunoye, O. Ojo, M.R. Data acquisition, analysis or interpretation: All authors Drafting of manuscript: N.O., O. Okunoye, O. Ojo, M.R. Critical revision of manuscript for intellectual content: All authors Statistical analysis: N.O., O. Okunoye, O. Ojo, M.R. Obtained funding: N.O., M.R., H.H., J.H., A.S. Administrative, technical or material support: N.O., O. Ojo, M.R. Supervision: N.O., O. Ojo, M.R. Supervision of genotyping: M.R., D.H. Data management: N.O., O. Okunoye, O. Ojo, M.R.
Corresponding author
Ethics declarations
Competing interests
A.S. is an editor for npj Parkinson’s Disease. A.S. was not involved in the journal’s review of, or decisions related to, this manuscript. R.A. is supported by the following grants: US NIH/NHGRI (U01HG010273) and the UK Royal Society /African Academy of Sciences (FLR/R1/191813). M.R. received funding from the University College London Grand challenges Small Grants (Award ID:177813 and the Michael J Fox Foundation Genetic Diversity in Parkinson’s Disease 2019 (Grant ID:17483). H.H. is supported by the Michael J Fox Foundation Genetic Diversity in Parkinson’s Disease (Grant ID: 17483). N.U.O. is supported by the Michael J Fox Foundation Genetic Diversity in Parkinson’s Disease 2019 (Grant ID:17483) and the TETFund National Research Fund (NRF) 2019. S.B.-C., C.B. and A.S. are supported by the Intramural Research Program, National Institute on Aging, National Institutes of Health and US Department of Health and Human Services project ZO1 AG000949 and all these authors declare no non-financial competing interests. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okubadejo, N.U., Okunoye, O., Ojo, O.O. et al. APOE E4 is associated with impaired self-declared cognition but not disease risk or age of onset in Nigerians with Parkinson’s disease. npj Parkinsons Dis. 8, 155 (2022). https://doi.org/10.1038/s41531-022-00411-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-022-00411-x
This article is cited by
-
Carboxyl truncation of α-synuclein occurs early and is influenced by human APOE genotype in transgenic mouse models of α-synuclein pathogenesis
Acta Neuropathologica Communications (2023)
