
Abstract
Parkinson’s disease (PD) guidelines lack clear criteria for genetic evaluation. We assessed the yield and rationale of genetic testing for PD in a routine clinical setting on a multicenter cohort of 149 early-onset and familial patients by exome sequencing and semi-quantitative multiplex ligation-dependent probe amplification of evidence-based PD-associated gene panel. We show that genetic testing for PD should be considered for both early-onset and familial patients alike, and a clinical yield of about 10% in the Caucasian population can be expected.
Similar content being viewed by others
Parkinson’s disease (PD) is a common extrapyramidal disorder with an onset around 65 years of age. Onset before age 50 is considered early-onset PD (EOPD), and ~15% of PD patients are familial (FPD). PD is a multifactorial disease, and an estimated 5–10% can be contributed to monogenic causes. Whereas patients with EOPD and FPD are generally considered to be at increased risk for monogenic genetic predisposition there is limited evidence on the clinical use of comprehensive genetic testing in these populations. Predominant pathogenic and likely pathogenic variants (P/LP) in PD genes are population and phenotype specific, and identifying those patients remains a diagnostic challenge1. Additionally, copy-number variants (CNV) may represent potential missing heritability2, and their presence should be examined in genes where known P/LP CNVs have been identified, such as SNCA, as well as in patients showing heterozygous SNV variants in AR PD genes such as PARK2 and PINK3.
The latest recommendations by EFNS/MDS-ES from 2013 regarding PD genetic testing precede accessibility of next-generation sequencing (NGS) in routine diagnostics and suggest testing should be individually decided and performed only for a few select genes4. In the last decade availability of NGS technology has enabled research of PD genetics to expand to many potential genes as well as rare variants5,6,7,8,9. However, the selection of the target gene panel is critical for effective translation into clinical practice, as clear clinical validity of an evaluated gene is required to classify variants according to the ACMG criteria for diagnosing monogenic disorders10. Furthermore, clinical reporting of variants must consider the proposed inheritance model for each PD-gene11.
The majority of previous genetic studies of PD looked at many target genes and reported several rare variants and candidate PD genes, with yields varying from 7.5% to as much as 43.5%1,5,6,7,8,9,12,13,14,15. The higher reported yields should be examined carefully for clinical validity, as many of these genes do not satisfy the stringency of ACMG criteria for reporting, with clinically reportable results being limited to evidence-based PD-associated genes only. Indeed, to the best of our knowledge, there is only one study that focused on the causality of detected variants, like our own study, and reported a 7.5% clinical yield9. This highlights the difference between actual reportable variants compared to other studies that reported variants based on their P/LP prediction alone.
This lack of clear clinical guidelines on which patients warrant testing, and no consensus on the exact genes to test for PD, currently translates to a lack of genetic testing in routine clinical practice. Indeed, a survey from 2019 reported that 41% of experienced PD physicians did not perform genetic testing in the prior year and more than 80% referred less than 11 patients in that period16. Given the relatively high cost and low yield of genetic testing in PD, clinicians need to have clear data on which they can base their testing decisions.
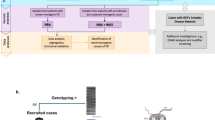
Therefore, we aimed to evaluate the yield and rationale of genetic testing for PD in the routine clinical setting by testing EOPD and FPD patients, by using exome sequencing (ES) of a 35 evidence-based PD-associated gene panel, as evaluated by expert groups ClinGen17 and Genomics England18, followed by the complementary multiplex ligation-dependent probe amplification (MLPA) method. We hypothesize that routine genetic testing of PD should include only clearly disease-associated genes, and only variants consistent with the inheritance model of each gene are clinically relevant and represent true yield.
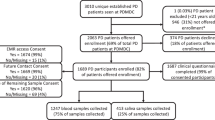
A total of 149 EOPD and FPD patients of Slavic ethnicity were included in the study (Table 1). Using ES, we detected a genetic contributor in 15 patients (10.1%), of which 14 had P/LP variants in GBA, and one had two P/LP variants in PARK2, an SNV and an exon 5 deletion detected using MLPA (Tables 1 and 2). No difference in the yield of causative P/LP variants was observed between the EOPD and FPD groups (X2 (1, N = 15) = 0.04, p = 0.85) (Table 1).
The involvement of GBA P/LP variants in familial PD and their penetrance is complex19,20,21,22, and our results showing P/LP variants in GBA to be the main reportable findings in our PD patients are in line with previous studies of PD patients of European ancestry5,7 as well as international studies23.
We identified the majority of causative P/LP using ES, however, MLPA revealed an additional 3 CNV in PARK2 (Supplementary information), one of which revealed a compound heterozygosity, leading to the genetic diagnosis in the patient. Therefore, to comprehensively address molecular pathology in the PD-associated genes, ES should be followed by MLPA as a complementary method. The two additionally detected heterozygous P/LP copy-number variants (CNV) in PARK2 detected by MLPA were classified as low-risk variants or risk-factors with incomplete penetrance, as no additional SNV P/LP in PARK2 were found in these two patients (Supplementary information).
Finally, variants of uncertain significance (VUS) in PD-associated genes were identified in 22 patients using ES (GBA in 8, LRRK2 in 5, ATP13A2 in 2, and ATP1A3, CSF1R, FTL, PLA2G6, SNCA, TUBB4A, and VPS35 each in one patient) (Supplementary information). At the moment, these results do not constitute clinically reportable findings, but since their role in PD pathogenesis may be resolved in the future, annual re-interpretation of such findings is advisable.
To conclude, our study identifies that genetic testing for PD should be considered in EOPD and FPD patients alike. Furthermore, a clear clinical testing focus should remain on a comprehensive set of validated/curated genes, and there is currently no rationale to test and classify variants in other genes in the clinical setting. By using these recommendations, a clinical yield of about 10% in the Caucasian population can be expected using ES and MLPA combined.
Methods
Our study cohort included 149 patients with PD, consecutively referred for routine genetic testing at the Clinical Institute of Genomic Medicine (CIGM)(Slovenia), and patients from Neurology departments Rijeka (Croatia) and Belgrade (Serbia), from January 2014 to October 2021.
All medical procedures in the study were performed in accordance with the ethical standards of the 1964 Helsinki Declaration and its later amendments or comparable ethical standards and national regulations of Slovenia, Croatia and Serbia.Written informed consent for genetic testing was obtained from the patients during their clinical appointment in the Slovenian, Croatian or Serbian language, granting the Clinical Institute of Genomic Medicine, where all the genetic testing was performed, rights to publish the findings of genetic testing in de-identified form in scientific literature. The informed consent statement was prepared according to National review board guidelines and approved by the Institutional Ethics Board at the University Medical Centre Ljubljana, Slovenia, and subsequently translated and approved for use by the Institutional Boards of Faculty of Medicine Rijeka, Croatia, and Faculty of Medicine, Belgrade, Serbia.
The inclusion criteria were confirmed clinical PD diagnosis based on United Kingdom Parkinson’s Disease Society Brain Bank clinical diagnostic criteria24, and either sporadic EOPD (<50 years) or familial PD (FPD) with at least one affected 1st-degree relative. Exclusion criteria were inconsistent clinical presentation, Parkinson-plus syndromes, idiopathic sporadic late-onset PD, or insufficient clinical information.
Gene panel analysis
ES was performed at CIGM using standardized protocols in use at the time of processing. In brief, we performed ES capture using TruSight One, TruSight Exome, Nextera Coding Exome, and IDT Exome capture kits (Illumina, San Diego, CA), Agilent SureSelect Human All Exon v2, v5, v6, and v7 capture kits (Agilent Technologies, Santa Clara, CA), as well as Twist Library Preparation Kit (Twist Bioscience, San Francisco, CA). Sequencing was performed on Illumina sequencing platforms in either 2 × 100 or 2 × 150 paired-end sequencing mode.
Initially, sequencing data analysis and variant interpretation were performed as previously described25,26. Archived raw data from all samples were re-analyzed using the most current software and annotation databases using a gene panel with ClinGen validated PD-associated genes17 (https://clinicalgenome.org/affiliation/40079/) and Genomics England panel for Parkinson’s disease and complex parkinsonism Version 1.718 (https://panelapp.genomicsengland.co.uk/panels/39/). Genes where tandem repeat expansions were previously reported as pathogenic have not been analyzed due to NGS limitations. Our final panel included the following genes (genes included in both panels are indicated in bold): ATP13A2, ATP1A3, C19orf12, CSF1R, DCTN1, DNAJC6, FBXO7, FTL, GBA, GCH1, GRN, LRRK2, LYST, MAPT, OPA3, PANK2, PARK7, PINK1, PLA2G6, PARK2, PRKRA, PTRHD1, RAB39B, SLC30A10, SLC39A14, SLC6A3, SNCA, SPG11, SPR, SYNJ1, TH, TUBB4A, VPS13A, VPS35, and WDR45.
Identified variants were classified according to the ACMG and AMP 2015 joint consensus recommendation10. The evidence support level was additionally weighted according to the ACGS recommendations where applicable27. Pathogenic and likely pathogenic (P/LP) variants in patients consistent with the inheritance model of individual gene disorders were considered clinically relevant.
Multiplex ligation-dependent probe amplification analysis
The semi-quantitative Multiplex Ligation-dependent Probe Amplification (MLPA) SALSA MLPA Probemixes P051 and P052 Parkinson mix assay (MRC Holland, Amsterdam, The Netherlands) were used for the detection of deletions or duplications in SNCA, PARK2, UCHL1, PINK1, PARK7, ATP13A2, LRRK2, and GCH1 genes.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. Reported P/LP variants’ ClinVar database accession numbers (https://www.ncbi.nlm.nih.gov/clinvar/)28 are available in Supplementary information. The raw ES datasets generated during and analyzed during the current study are available from the corresponding author B.P. on reasonable request.
References
Farlow, J. et al. Parkinson Disease Overview. In Genereviews® (University of Washington, Seattle, 1993).
La Cognata, V., Morello, G., D’Agata, V. & Cavallaro, S. Copy number variability in Parkinson’s disease: assembling the puzzle through a systems biology approach. Hum. Genet. 136, 13–37 (2017).
Jankovic, M. Z. et al. Identification of mutations in the PARK2 gene in Serbian patients with Parkinson’s disease. J. Neurol. Sci. 393, 27–30 (2018).
Berardelli, A. et al. EFNS/MDS-ES recommendations for the diagnosis of Parkinson’s disease. Eur. J. Neurol. 20, 16–34 (2013).
Schormair, B. et al. Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin. Genet. 93, 603–612 (2018).
Lin, C.-H. et al. A clinical and genetic study of early-onset and familial parkinsonism in Taiwan: An integrated approach combining gene dosage analysis and next-generation sequencing. Mov. Disord. 34, 506–515 (2019).
Trinh, J. et al. Utility and implications of exome sequencing in early-onset Parkinson’s disease. Mov. Disord. 34, 133–137 (2019).
Jiang, Y. et al. Parkin is the most common causative gene in a cohort of mainland Chinese patients with sporadic early-onset Parkinson’s disease. Brain Behav. 10, e01765 (2020).
Li, N. et al. Whole-exome sequencing in early-onset Parkinson’s disease among ethnic Chinese. Neurobiol. Aging 90, 150.e5–150.e11 (2020).
The ACMG Laboratory Quality Assurance Committee. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Singleton, A. B., Farrer, M. J. & Bonifati, V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov. Disord. 28, 14–23 (2013).
Youn, J. et al. Genetic variants of PARK genes in Korean patients with early-onset Parkinson’s disease. Neurobiol. Aging 75, 224.e9–224.e15 (2019).
Siitonen, A. et al. Genetics of early onset Parkinson’s disease in Finland: exome sequencing and genome-wide association study. Neurobiol. Aging 53, 195.e7–195.e10 (2017).
Yemni, E. A. et al. Integrated analysis of whole exome sequencing and copy number evaluation in Parkinson’s Disease. Sci. Rep. 9, 3344 (2019).
Gaare, J. J. et al. Meta-analysis of whole-exome sequencing data from two independent cohorts finds no evidence for rare variant enrichment in Parkinson disease associated loci. PLoS ONE 15, e0239824 (2020).
Alcalay, R. N. et al. Genetic testing for Parkinson disease: current practice, knowledge, and attitudes among US and Canadian movement disorders specialists. Genet. Med. 22, 574–580 (2020).
Rehm, H. L. et al. ClinGen — The Clinical Genome Resource. N. Engl. J. Med. 372, 2235–2242 (2015).
Martin, A. R. et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 51, 1560–1565 (2019).
Nichols, W. C. et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72, 310–316 (2009).
Neumann, J. et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794 (2009).
Anheim, M. et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78, 417–420 (2012).
Menozzi, E. & Schapira, A. H. V. Exploring the genotype–phenotype correlation in GBA-Parkinson disease: clinical aspects, biomarkers, and potential modifiers. Front. Neurol. 12, 694764 (2021).
Skrahina, V. et al. The Rostock International Parkinson’s Disease (ROPAD) Study: protocol and initial findings. Mov. Disord. J. Mov. Disord. Soc. 36, 1005–1010 (2021).
Gibb, W. R. & Lees, A. J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 51, 745–752 (1988).
Bergant, G. et al. Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: a retrospective survey in 1,059 cases. Genet. Med. 20, 303–312 (2018).
Ales, M. et al. Phenotype-driven gene target definition in clinical genome-wide sequencing data interpretation. Genet. Med. 18, 1102–1110 (2016).
Ellard, S. et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020 (ACGS, 2020).
Landrum, M. J. et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 48, D835–D844 (2020).
Acknowledgements
This work was supported in part by the Croatian Science Foundation (Grant IP-2019-04-7276), University of Rijeka (Grant uniri-biomed-18-1981353), Ministry of Education, Science and Technological Development of the Republic of Serbia, (Grant 175090 to V.S.K.), and the Slovenian Research Agency (Grants J3-9280 and V3-1911).
Author information
Authors and Affiliations
Contributions
A.K. and V.R. contributed equally to this work and are co-first authors. All authors contributed substantially to the study. V.V. and B.P. conceptualized the research project. A.K., V.R., G.B., A.M., and B.P. organized and A.K., V.R., G.B., D.G., D.F., A.M., N.T., E.P., M.B., M.J., M.S., V.S.K., I.N., Z.P., M.R., V.V., and B.P. implemented the project. A.K., A.M., and B.P. designed the analyses, A.K., V.R., G.B., and A.M. performed the statistical analyses, and D.G., D.F., A.M., N.T., E.P., M.B., M.J., M.S., V.S.K., I.N., Z.P., M.R., V.V., and B.P. critically reviewed the data. A.K., V.R., and G.B. wrote the first draft of the manuscript, and G.B., D.G., D.F., A.M., N.T., E.P., M.B., M.J., M.S., V.S.K., I.N., Z.P., M.R., V.V., and B.P. reviewed and critically corrected the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kovanda, A., Rački, V., Bergant, G. et al. A multicenter study of genetic testing for Parkinson’s disease in the clinical setting. npj Parkinsons Dis. 8, 149 (2022). https://doi.org/10.1038/s41531-022-00408-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-022-00408-6
This article is cited by
-
Deciphering the role of a SINE-VNTR-Alu retrotransposon polymorphism as a biomarker of Parkinson’s disease progression
Scientific Reports (2024)
-
Clinical Outcome of Hypertrophic Cardiomyopathy in Probands with the Founder Variant c.913_914del in MYBPC3: A Slovenian Cohort Study
Journal of Cardiovascular Translational Research (2024)
-
GBA-associated Parkinson’s disease in Hungary: clinical features and genetic insights
Neurological Sciences (2024)
