
Abstract
There is great heterogeneity in both the clinical presentation and rate of disease progression among patients with Parkinson’s disease (PD). This can pose prognostic difficulties in a clinical setting, and a greater understanding of the risk factors that contribute to modify disease course is of clear importance for optimizing patient care and clinical trial design. Genetic variants in SNCA are an established risk factor for PD and are candidates to modify disease presentation and progression. This systematic review aimed to summarize all available primary research reporting the association of SNCA polymorphisms with features of PD. We systematically searched PubMed and Web of Science, from inception to 1 June 2020, for studies evaluating the association of common SNCA variants with age at onset (AAO) or any clinical feature attributed to PD in patients with idiopathic PD. Fifty-eight studies were included in the review that investigated the association between SNCA polymorphisms and a broad range of outcomes, including motor and cognitive impairment, sleep disorders, mental health, hyposmia, or AAO. The most reproducible findings were with the REP1 polymorphism or rs356219 and an earlier AAO, but no clear associations were identified with an SNCA polymorphism and any individual clinical outcome. The results of this comprehensive summary suggest that, while there is evidence that genetic variance in the SNCA region may have a small impact on clinical outcomes in PD, the mechanisms underlying the association of SNCA polymorphisms with PD risk may not be a major factor driving clinical heterogeneity in PD.
Similar content being viewed by others

Introduction
There is great heterogeneity in both the clinical presentation and the rate of disease progression between patients with Parkinson’s disease (PD)1,2. While PD is characterized by motor symptoms such as resting tremor, bradykinesia, rigidity, and postural instability, patients also experience a variety of non-motor symptoms, including cognitive impairment and dementia, sleep disorders, depression, anxiety, hallucinations, and hyposmia3. These non-motor symptoms complicate the clinical management of the disorder and are significant determinants of poor quality of life for patients and their caregivers. The observed heterogeneity can also pose prognostic difficulties in a clinical setting, and a greater understanding of the risk factors that contribute to modify disease course is of importance in terms of informing patients more accurately how they may be affected by the disease and developing personalized medicine strategies for patient management. Further, there is increased interest in targeting patients with a higher risk of specific disease outcomes in tailored clinical trials.
A logical first step in seeking genetic modifiers for progression in PD is to examine well-established PD susceptibility genes. These include SNCA, the gene encoding the α-synuclein protein that constitutes the major protein component of Lewy bodies (LBs)4. The abnormal aggregation of α-synuclein has a primary role in the formation of the LBs and other α-synuclein pathological aggregates and is regarded as a critical step in the molecular pathogenesis of PD5. In familial cases of PD, specific mutations or copy number variations (CNVs) of SNCA have been shown to cause increased production of α-synuclein and this is correlated with increasing disease severity6,7. Such mutations and CNVs of SNCA are rare in the general PD population, but candidate gene studies and genome-wide association studies (GWAS) have established the SNCA locus as a risk factor for idiopathic PD8,9 and accordingly as a promising candidate for disease modification.
The analysis of common polymorphisms in the SNCA region with clinical outcomes or age at PD onset has received much attention, but the use of different clinical scales, populations, and study designs complicates the comparison of the results, and no clear disease-modifying polymorphisms have been identified so far. For a better understanding of the impact of SNCA genetic polymorphisms on the progression of disease, a comprehensive overview of the associations between clinical evaluations and SNCA genotypes in PD is necessary. This systematic review aims to summarize and compare all available primary literature that has evaluated the association between SNCA polymorphisms and any PD clinical outcome. The review is limited to the analysis of patients with idiopathic PD, but given the broad symptomatology accredited to PD, no restrictions were placed on the clinical outcome measures evaluated in the studies. The potential contribution of SNCA polymorphisms to the clinical manifestations of PD is summarized. Further, the implications of the results for our understanding on the pathophysiology of PD and the design of future studies are discussed.
Results
Characteristics of included studies
The search strategy yielded 597 unique studies (Fig. 1) of which 58 were found to be eligible for inclusion. Of the 58 studies included in the review, 24 evaluated PD age at onset (AAO; median size 832, range 81–28,568 participants) and 41 evaluated clinical features of PD (median size 330, range 50–2011 participants), and predominantly included patients of European (Europe, Australia, or North America; n = 37) or Asian ancestry (n = 24). Only one study population was from South America (Brazil). All studies used defined diagnostic criteria for the diagnosis of PD (Table 1) as specified in the inclusion criteria for the review, though notably one study additionally incorporated data from a commercial genotyping project (23andMe) in which the diagnosis of PD was self-reported10. Assessment of bias in each study revealed that those that stood out as having the lowest number of sources of potential bias replicated the findings in independent cohorts, properly accounted for multiple testing, or were based on population-based cohorts (Supplementary Table 1).

Asterisk (*): eight further articles were retrieved from a manual search of the 50 evaluated papers’ references (n = 5) and from the wider PD literature (n = 3). iPD idiopathic Parkinson’s disease, SNP single-nucleotide polymorphism.
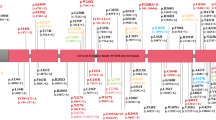
A total of 51 different SNCA genetic polymorphisms were investigated, comprising 50 single-nucleotide polymorphisms (SNPs) and variations in the complex microsatellite D4S3481 known as REP1, located approximately 10 kb upstream of the translational start of SNCA. The location and population frequency (European and East Asian) of each polymorphism is provided (Supplementary Table 2). Of the 51 polymorphisms studied, 6 were reported to be significantly associated with AAO and 10 were reported to be significantly associated with a clinical outcome (Fig. 2 and Supplementary Table 3), defined by a p value of <0.05, unless the authors of the study specifically reported that the association was not significant after adjustment for multiple testing or covariates. A summary of all of the SNPs investigated and not shown to be significantly associated with any clinical outcome analyzed is given in Supplementary Fig. 1 and Supplementary Table 3.

a Schematic of SNCA showing the location of each polymorphism. Exons are indicated by boxes. Chromosome 4 positions according to GRCh 37. Adapted from the output generated by LDLink83. b The number of studies reporting a significant association of a clinical outcome with the polymorphism indicated. c The number of studies reporting no significant association between a clinical outcome with the polymorphism indicated. In both b and c, the analysis of clinical outcomes is separated according to a cross-sectional (cs) or longitudinal (long) study design. A single study can be recorded more than once if both designs were included in the manuscript or if the study reported both significant and non-significant associations for a SNP in the same category. Full details of the SNPs and the outcomes in each category are given in Supplementary Tables 2 and 3. Asterisk (*): the unadjusted p value was <0.05 but the authors reported that the association did not remain significant after adjustment for multiple testing or covariates. Dagger (†): the study identified the association in a discovery cohort and replicated the finding in an independent cohort. AAO age at onset, cs cross-sectional, H&Y Hoehn and Yahr, long longitudinal.
The association of SNCA polymorphisms with age at PD onset
One of the distinguishing features of autosomal-dominant PD caused by several SNCA gene mutations or multiplications is an early AAO, and this has led to an extensive investigation of common SNCA polymorphisms as modifiers of AAO in idiopathic PD. We identified 24 studies that assessed the role of SNCA polymorphisms on modifying AAO and 6 out of the 40 studied SNPs were associated with differences (Fig. 2 and Supplementary Table 3).
Seven small studies, including patients of European (n = 6)11,12,13,14,15,16 and Asian (n = 2)16,17 ancestry, did not find evidence that the SNCA REP1-risk genotype impacts AAO in PD (Fig. 2 and Supplementary Table 3). In contrast, three studies showed a significant association between REP1 and an earlier AAO18,19,20. Further, a meta-analysis on data from 4190 cases from 15 sites involved in the Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium, including some of the previously cited studies, found higher REP1 scores (defined here as the sum of allele score, with each 259-, 261-, or 263-bp allele contributing 0, 1, or 2 points, respectively) to be associated with an earlier AAO (hazard ratio (HR) = 1.06; p = 0.01)21.
Five studies took a broader approach to identify genetic AAO modifiers by analyzing the association of SNPs across many genes previously associated with the risk of sporadic PD, including polymorphisms in the SNCA region10,11,22,23,24. In a German study of 21 SNPs associated with PD risk, AAO was assessed in 1396 patients with PD and rs356219 from the SNCA region was shown to significantly contribute to earlier AAO (rs356219-G, p = 0.001)23. These data are consistent with reports from both a Chinese population of 685 cases (rs356219-G dominant model, p = 0.02)25 and a Spanish population of 898 cases (rs356219-G log-additive model, p = 0.0034)24, each of which reported that carriers of the rs356219-G allele had a significantly younger AAO than carriers of rs356219-A allele. Notably, the results from the Spanish population were extended to identify a three-loci epistatic combination of rs356219 with RPTOR rs11868112 and RPS6KA2 rs6456121 that was associated with AAO in both the discovery cohort (n = 898; odds ratio (OR) 2.89; p < 0.0001) and a replication sample from the International Parkinson Disease Genomics Consortium (IPDGC; n = 4170; OR 1.56; p = 0.046–0.047). The most recent AAO study was also the largest study to be identified in this review, achieved by combining cases from the IPDGC with participants from the 23andMe research data set, which consisted of customers of the personal genetics company (23andMe, Inc.) who self-reported diagnosis and their age of PD diagnosis10: Blauwendraat et al. took two approaches, first performing a GWAS for AAO10. The top SNP associated with PD AAO in this GWAS was rs356203, resulting in an earlier AAO by approximately 0.6 years (p_meta = 1.90E−12). Interestingly, rs356203 has previously been shown to be associated with an increased risk of PD8 but has not been investigated for an association with other clinical outcomes in PD. Second, the authors analyzed an updated set of 44 PD-risk SNPs and identified variants in six different genes to be significantly associated with AAO, including the SNCA SNP rs365182 (p = 1.12 × 109)10.
Motor outcomes in PD
PD is primarily a movement disorder and accordingly motor signs and symptoms were the most frequent clinical outcomes evaluated. Of the 22 studies analyzing a total of 16 polymorphisms, 9 reported a significant association between a motor-related outcome and 4 SNPs26,27,28,29,30,31 or the REP1 polymorphism32,33,34 (Fig. 2 and Supplementary Table 3).
In cross-sectional analyses, significant associations of SNCA polymorphisms with Unified Parkinson’s Disease Rating Scale (UPDRS) scores were found in two Asian populations for REP1–263 allele carriers (UPDRS III, β 3.921; p = 0.026)34 and rs11931074 (UPDRS II, standardized coefficient (SC) −0.083; p = 0.035; UPDRS III, SC −0.140; p ≤ 0.001)28. However, these results were not replicated by other studies (Fig. 2 and Supplementary Table 3)27,35,36, including a large study from the Chinese National Consortium on Neurodegenerative Diseases (CNCPD; n = 2011)36. Cooper et al. showed an association between rs356182 and tremor-dominant (TD) vs. postural-instability gait disorder motor subtypes (corrected p = 0.04), which they subsequently replicated in an independent multi-center cohort (p = 0.002). In both the discovery and replication cohorts, the authors found that the rs356182-GG genotype was associated with a more TD phenotype29. Cooper et al. built on these findings and showed that rs356182 also predicted slower progression of motor impairment, with the annual rate of increase in UPDRS III scores found to be approximately 1 point per year less in rs356182-GG carriers compared to those with the AG or AA genotypes (p = 0.01)29. Further evidence that SNCA polymorphisms predict the rate of motor symptom decline in PD was reported in work from the Parkinson Environment and Genes cohort, a prospective, population-based study of patients in California, USA. In this study, the risk of faster decline of motor function (defined by a change of >5 points per year in UPDRS III score) was increased fourfold in carriers of the REP1–263 allele (OR 4.03; p = 0.004)32.
Few studies have addressed a possible association between SNCA polymorphisms and the development of motor complications related to dopaminergic treatment (Supplementary Table 3) and replication studies are lacking. The development of motor fluctuations and levodopa-induced dyskinesias (LIDs) up to 13 years from diagnosis was evaluated in patients from the prospective population-based Cambridgeshire Parkinson’s Incidence from GP to Neurologist (CamPaIGN) cohort. Carriers of rs356219-A were at increased risk of developing motor fluctuations compared to the rs356219-GG genotype (adjusted HR 1.902; p = 0.039), while no difference was observed in the development of LIDs between the two groups (unadjusted p = 0.907)26. Two studies assessed wearing off (WO): in an Italian study, carriers of the REP-263 allele were found to have a higher risk of WO compared to noncarriers at 10 years from disease onset (65.33% vs. 53.02%; log-rank test p = 0.028), although this result was not significant in multivariate Cox regression analysis18. In a separate Chinese study, rs11931074 was not found to be associated with WO (p = 0.520) in 724 patients with a comparably short disease duration (rs11931074-T carriers 4.61 ± 4.42 years; rs11931074-GG carriers 4.78 ± 3.97 years)28. Given the higher risk of motor problems with increasing disease duration, additional studies of more advanced PD will be needed to validate these results.
Measures of global PD severity and disability
The Hoehn and Yahr (H&Y) scale provides an overall assessment of severity of PD based on clinical features and functional disability and, since its introduction, has remained the most widely used scale to describe the general severity of PD. Fourteen studies including a total of 31 different polymorphisms used the H&Y scale to measure PD severity (Supplementary Table 3), but the means by which the scale was used varied widely. For example, Davis et al. report no significant association between rs356219 or rs11931074 and the annual change in H&Y stage22, while in other studies these same SNPs were shown to be significantly associated with either a longer time to a 0.5-point increase in H&Y stage (rs356219; HR 0.20; p = 0.005)37 or a longer time to reach H&Y stage ≥3 (mild-to-moderate disability) (rs11931074; HR 0.43; p = 0.03)31. Similarly, REP1 score was reported to be associated with a longer time to reach H&Y stage ≥4 (severe disability) (HR 0.87; p = 0.046)38, while reports on the association of the REP1 polymorphism with H&Y stage at the time of examination are conflicting34,36. Shi et al. (CNCPD, n = 2011) reported no associations with H&Y stage in an exploratory study of 9 genetic variants from 6 different genes that included REP136, while, in a smaller study (n = 172), Ng et al., found an association between REP1–263 and H&Y stage at examination (β 0.231; p = 0.008)34. These studies also vary greatly in the rigor of their design, but variability in the application of the H&Y scale makes a direct comparison of the results particularly challenging.
Cognitive impairment and the development of dementia in PD
Cognitive decline is an important aspect of PD as it brings a substantial additional burden for the patient, caregivers, and the healthcare system. In 8 of the 21 studies that assessed the association between 36 SNCA polymorphisms and cognition, 4 SNPs27,31,35,37,39 and the REP1 polymorphism18,34,38 were shown to be significantly associated with measures of cognitive impairment or the diagnosis of dementia (Fig. 2 and Supplementary Table 3). Among these, rs356219 was the most frequently studied SNP. In the most comprehensive assessment of cognitive performance and rs356219, Mata et al. analyzed global cognitive function using the Montreal Cognitive Assessment (MoCA), and memory; attention and executive function; language processing; and visuospatial skills in 1079 patients with PD of European descent and found no significant association between rs356219 and any outcome40. Two small studies did show a significant association of rs356219 with global cognitive impairment, with conflicting results: in a Chinese explorative study of 50 patients, the rs356219-G allele was significantly associated with an 18% decreased risk of cognitive decline (p = 0.006), measured by the time to a 1-point decrease on the MoCA scale37. Conversely, a Brazilian study of 105 patients found that both rs356219 heterozygotes (rs356219-GA; OR 4.74; p = 0.021) and homozygotes (rs356219-GG; OR 5.74; p = 0.014) had significantly increased risk of cognitive impairment as defined by an education-corrected Mini-Mental State Examination (MMSE) cut-off35. Given Mata’s findings and the small number of participants and events observed in the latter two studies, there is currently limited support for a role of rs356219 in modifying cognitive impairment.
The association of rs356219 and dementia in PD has been studied in three independent populations. The rs356219-G allele was found to be associated with an increased risk of PD dementia (PDD; p = 0.01) in a Chinese population of 291 patients39. In contrast, studies of two European populations of patients with PD from the UK (CamPaIGN)41,42 and Spain43 found no link between rs356219 and the risk or development of dementia. All four studies used clinical diagnostic criteria for PDD (Supplementary Table 3). The differences in findings could be due to type I errors or reflect variance in ethnic backgrounds or study design, such as the inclusion of incident patients vs. patients from specialist units.
One of the largest studies to explore the impact of SNCA polymorphisms on cognitive decline analyzed the role of REP1 and 19 additional haplotype-tagging SNPs in 922 PD patients from the Molecular Epidemiology of Parkinson’s Disease study. Participants were assessed by telephone interview, either directly with the cases (Modified Telephone Interview for Cognitive Status) or via proxy (Alzheimer’s Disease Dementia Screening Interview)38. The group of patients with higher REP1 scores (linked with highest risk of PD) were shown to have reduced risk of developing cognitive impairment (HR 0.81; p = 0.0017)38. However, this protective effect of REP1 on cognitive decline was not replicated by an Italian study of 426 PD patients which showed that carriers of the REP1–263 allele (similarly linked with PD risk) showed significantly increased risk of dementia compared to carriers of shorter alleles (HR 3.03; p < 0.001)18 or by a Singapore study which showed that REP1–263 allele carriers were associated with lower MMSE scores (β –1.46; p = 0.010)34. While these later results are more aligned with the predicted effect of REP1 on disease outcomes, it is difficult to directly compare the results because of the scales used to asses cognition and the handling of the REP1 genotype are different in all three studies.
Other non-motor symptoms in PD
Anxiety and depression are two of the most common mental health symptoms that affect people with PD. Seven studies from China (n = 6) and Brazil (n = 1) looked at depression or anxiety and a total of 11 SNCA polymorphisms (Supplementary Table 3). Only two studies reported a significant association: first, in a small (n = 105) Brazilian population, the rs2583988-TT genotype was found to significantly reduce the risk of depression, defined using the Beck Depression Inventory (OR 0.21; p = 0.046)35, and second, a large study in China (n = 1047) found that a REP1 “risk allele” (in this study defined by the copy number of a CA repeat) was associated with a decreased risk for the presence of mild-to-marked depression defined using the Hamilton Rating Scale for depression (OR 0.54; p = 0.049)44. Neither of these findings has been replicated.
As PD progresses, many patients will develop psychotic symptoms, primarily hallucinations, which is considered an important clinical milestone in the course of PD. However, only two studies reported the association between SNCA and the incidence of psychosis in PD (Supplementary Table 3). The first study found no evidence for an association with the SNCA REP1 alleles and psychotic symptoms measured by item 2 of the UPDRS part I (“benign hallucinations with insight retained” or worse) in the multi-center NeuroGenetics Research Consortium45. In the second study, with patients recruited in Italy, REP1–263 allele carriers were shown to have a 2.69-fold higher risk of developing visual hallucinations over the course of disease (p = 0.001) recorded from retrospective review of clinical records18.
Sleep disorders, including excessive daytime sleepiness and rapid eye movement (REM) sleep behavior disorder (RBD), usually increase in frequency over the course of PD. RBD is a parasomnia strongly linked to synucleinopathies and characterized by the absence of muscle atonia during REM sleep46. In a study of 325 patients with PD from Norway47, possible RBD (pRBD) was assessed using the RBD screening questionnaire (history of dream enactment without polysomnographically confirmed REM sleep without atonia). pRBD was significantly associated with the SNCA SNP rs3756063 (OR 1.44; p = 0.018) but not rs356165 or rs2245801. This result was replicated for rs3756063 in 382 patients from the Parkinson’s Progression Markers Initiative (PPMI) cohort with marginal significance (OR 1.35; one-sided p = 0.036)47. Recently, a large study analyzed the effect of four other SNCA SNPs, selected for their association with risk of PD or isolated RBD, in the Norwegian and PPMI cohorts and an additional cohort from McGill University48. Meta-analysis of the three cohorts identified a significant association of rs287004 with a reduced incidence of pRBD in PD (OR = 0.76; p = 0.009)48.
Two of the studies identified in this systematic review49,50 diagnosed RBD using the “gold standard” set by the International Classification of Sleep Disorder-II criteria51. These studies did not identify an association of RBD with multiple SNCA genetic polymorphisms in Chinese49 or Italian50 populations (Supplementary Table 3), but given their relatively small size (n = 5750 and 15249), as typified by studies of clinically defined RBD using overnight polysomnography, larger studies will be needed to characterize the impact of SNCA polymorphisms in formally diagnosed RBD in PD.
Hyposmia, identified as reduced sensitivity to odor, is a common non-motor symptom of PD and can antedate the typical motor symptoms by several years52. However, only three small studies identified in this review have explored the association of SNCA polymorphisms with hyposmia, each using the 16-item odor identification “Sniffin’ Sticks” (SS-16) tool to measure hyposmia in PD. Of these, 1 Chinese study of 50 patients followed for 30 months suggests that of the six SNPs selected, rs894278-G (HR 0.47; p = 0.029) and rs356219-G (HR 0.32; p = 0.021) were associated with slower olfactory impairment37. Another Chinese study of 218 patients report that the rs11931074-TT genotype increased the risk of hyposmia more than 3-fold compared with the GG genotype (crude OR 3.41; p = 0.011) but did not find any significant associations for rs89427853. The third study (n = 295) found no associations for SS-16 outcome measures and REP1 in a population from the Netherlands (Supplementary Table 3)54.
Discussion
In this systematic review, we provide a comprehensive summary of available data regarding the association of SNCA polymorphisms and clinical outcomes or AAO in patients with idiopathic PD. In total, 58 studies were identified that analyzed not only the association of 51 different SNCA polymorphisms with a broad range of outcomes, most commonly addressing AAO or motor impairment that is the core feature of PD, but also a broad range of non-motor symptoms, including cognitive impairment, sleep, mental health, and psychosis. Despite the large number of studies that have been conducted, the only associations that were replicated by independent studies were between SNCA polymorphisms and AAO, and no robust associations were identified for any of the clinical outcomes considered. This can not only be attributed to the lack of consistency in study designs and methodological shortcomings, but may also indicate that common SNCA variants are not major drivers of the heterogeneity in clinical presentation and progression that is observed in PD.
For the majority of polymorphisms (n = 39; 76%) identified in this review, no significant associations were reported with a clinical outcome or AAO, although it is notable that 31 of these SNPs were only included in 1 or 2 studies (Supplementary Fig. 1 and Supplementary Table 3). At the other extreme, the most frequently analyzed polymorphisms rs356219 and REP1 were each included in 21 studies. These two polymorphisms were among the very first genetic polymorphisms studied as modifiers of PD risk16,55,56 and are common in populations of both European and Asian ancestry, which may contribute to their popularity. The only replicated association for both rs356219 and REP1 was with AAO (rs356219, n = 323,24,25; REP1, n = 418,19,20,21). Clinical outcomes were included in 17 of the studies analyzing rs356219 and 11 of the studies analyzing REP1. Multiple significant associations, spanning the spectrum of the clinical outcomes identified in this review, were reported for both rs356219 and REP1, but notably none were replicated in independent populations (Fig. 2 and Supplementary Table 3). Therefore, the results warrant cautious interpretation. The possible reasons for the lack of robust associations are discussed below.
While the inclusion criteria pre-specified idiopathic PD diagnosed using defined diagnostic criteria, no limitations were put on other aspects of the study design, and the review revealed a striking heterogeneity in the design of the study cohorts, sample size, and the choice of the SNCA polymorphisms and the outcome measures, each of which can diminish the capacity to compare the effects of SNCA variants among the different studies. Most striking was the large heterogeneity in the outcome measures used to assess the severity of symptoms and disease progression, with >30 different clinical scales or questionnaires identified in the review, many of which were assessed only once. Adding to the heterogeneity, even when studies used the most common PD rating scales, the way in which these scales were applied varied greatly. This was not only exemplified in the analysis of H&Y staging (see “Results”) but was also notable in the application of the UPDRS and MMSE (see outcomes in Supplementary Table 3). There were also considerable differences in the demographic and clinical characteristics of the populations studied. Most were recruited from hospitals or specialized clinics and very few were population-based cohorts, which can limit the generalizability of the results. Further, most cohorts were limited to Caucasian or Asian populations, and ethnic differences can preclude the generalization of the results in genetic studies.
Increasing evidence suggests that variants in SNCA modify the risk for developing PD by altering the levels of α-synuclein and promoting the abnormal aggregation into LBs. In this model, small changes in α-synuclein expression may, over many decades, predispose to PD and different SNCA variants can contribute to this by altering the transcriptional regulation of α-synuclein expression. Of the polymorphisms identified in this review, the most compelling evidence for a mechanistic role in α-synuclein regulation is found for REP1 and rs35621957,58,59,60. These common genetic variants have been correlated with higher α-synuclein expression in vitro58 or with elevated brain or peripheral levels of α-synuclein in vivo57,59 and it is a biologically plausible hypothesis that these same mechanisms could impact the heterogeneity that is observed in PD. The results of this review support a role for these variants in modifying AAO; however, we did not find any strong evidence for a link to disease outcomes. Indeed, some studies even suggest the opposite findings, with those SNPs increasing PD risk being linked to fewer or milder symptoms, with examples found for REP138, rs35621926, rs1193107430,39, and rs35618229. These observations could reflect the shortage of well-designed replication studies but may also indicate that SNCA is an initiator of PD risk but not of the development of specific symptoms or changes in the rates of disease progression. This hypothesis is in line with a recent study combining data from 13 longitudinal PD cohorts, which found that a genetic risk score based on 31 PD-risk SNPs was inversely associated with AAO but not with a comprehensive battery of clinical outcomes61.
Several limitations must be considered in interpreting the results presented in this review. Despite the large number of studies identified, the heterogeneity in study design meant that it was not possible to compare the effect sizes among the studies or perform a meta-analysis. Further, we only reported the effects of multiple testing if the authors themselves included this as part of their analysis plans, although the assessment of potential sources of bias served to highlight some studies in which this was a weakness in the design (Supplementary Table 1). The review inclusion criteria may have missed some works, especially those that included negative findings related to SNCA as data not shown, and this highlights the plausible existence of positive reporting bias. Finally, the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) statement was used as a guide to ensure a rigorous review process; however, we note that the protocol for our review was not published before the search was initiated. The strengths of our review include the broad inclusion criteria, which captured studies of any clinical outcome associated with PD and including any SNCA polymorphism. Furthermore, several aspects of the design of each study were evaluated using pre-determined criteria in order to provide an assessment of the sources of bias in the studies.
To clarify the role of genetic variation in the SNCA region in PD heterogeneity, further studies are required. This review has highlighted the need for well-designed studies with standardized cohort designs, optimally recruiting patients early in the disease and following them prospectively using comprehensive batteries of clinical scales to measure disease severity and progression. Further, approaches to harmonize how disease progression is measured should be prioritized. One explanation for the broad range of tools used is that we currently lack clinical tools that assess PD progression in an accurate and meaningful way across the spectrum of disease stages. A new approach was recently adopted in a GWAS of cognitive and motor progression in which multiple assessments were combined in a data-driven principal components analysis to derive scores for composite, motor, and cognitive progression62. Alternatively, biomarkers may prove to be more sensitive measures of disease progression, with less susceptibility to the effects of subjectivity, medication, and placebo. Clearly, further efforts to improve phenotypic measures and define disease progression should be pursued.
There was a notable scarcity of population-based incident cases representative of the general PD population, which may lead to bias if the associations between polymorphisms and clinical features vary by age. The mean age of PD onset of the study cohorts reported in the studies was 58.8 (±5.3) years and the mean age at examination was 63.7 (±4.5) years. This is typical of general research studies, which are generally unrepresentative of the population age distribution of PD63. Finally, it is plausible that the effect sizes of SNCA polymorphisms will be small, and therefore large studies are needed to detect small effect sizes, as was demonstrated by the AAO GWAS of Blauwendraat et al.10. This can result in a trade-off between sample size and data quality, but there is an increasing drive for world-leading PD cohorts to collaborate or for journals to require the deposition of original data in public databases, and these initiatives will provide excellent platforms to further probe the role of SNCA variants in PD progression.
The lack of robust findings casts doubts on the relevance of SNCA SNPs in modifying disease course in PD and suggests that the genetics of PD risk and progression are largely separate. However, methodological aspects may explain some of the discrepancies in the effects of polymorphisms across the different studies. Most significant associations identified in this review found that SNCA polymorphisms had minor effects on the clinical outcomes and so, even if validated, these findings are not currently relevant in a clinical setting. However, given that α-synuclein is a major drug target, they have the potential to be used as a genetic risk stratification tool to reduce variability in clinical trials, and so further studies using large, well-designed cohorts are warranted.
Methods
Systematic review strategy
The review process was conducted according to the PRISMA statement64. A systematic review of literature was performed to summarize all available primary research reporting the association of SNCA polymorphisms with clinical features of PD. The following inclusion criteria were used: subjects were patients with PD diagnosed according to defined diagnostic criteria (i.e., UKBB, Gelb, Bower or other clearly defined criteria that were endorsed by a movement disorder expert (G.A.)), genetic polymorphisms were within the SNCA gene region, outcomes were any clinical feature attributed to PD or AAO, the association of SNCA polymorphisms with outcomes was assessed statistically, and the source was primary literature in English. Full exclusion criteria are listed in Supplementary Table 4.
The searches were performed on June 15, 2020 by the first and last author using two databases (PubMed and Web of Science) and the following search terms: Parkinson* AND SNCA AND (variant* OR polymorphism* OR SNP* OR GWAS OR genome-wide) (Supplementary Table 5). Five hundred and ninety-seven unique publications were identified, of which 431 were excluded for clearly not meeting the inclusion and exclusion criteria based on reading the title and abstract (Supplementary Fig. 2). The remaining 166 publications were read in full by the first and last author. Cases of diverging opinion about an article’s eligibility based on the inclusion/exclusion criteria were discussed at both stages. Fifty publications were found to be eligible for this review (Fig. 1). Eight further articles were retrieved from a manual search of the 50 evaluated papers’ references (n = 5) and from the wider PD literature (n = 3) (identified in Table 1). For the final 58 publications, the publication year, country of study, cohort name or main study site, criteria for diagnosis of PD, number of patients, AAO, age at examination, disease duration, number of patients followed longitudinally, length of follow-up, SNCA polymorphisms, clinical features analyzed, and result of association analyses (p value of best model) were retrieved, as available. An overview of the papers included in this review (Table 1) and of the SNCA polymorphisms and outcomes assessed (Supplementary Tables 2 and 3) are provided. The risk of bias for each study was assessed by the first and last author using a simple traffic light system to grade potential sources of bias as low level (green), of some concern (yellow), or high level (red). A third author (J.L.) resolved cases of diverging assessment. The factors taken into consideration were sample representativeness, adequate PD definition, adequate assessment of outcomes, multiple comparisons tests, pre-specified plan, and replication in an independent cohort (Supplementary Table 1).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this systematic review are provided in the Supplementary files.
References
Aarsland, D. et al. Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 13, 217–231 (2017).
Williams-Gray, C. H. et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 132, 2958–2969 (2009).
Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 318–324 (2016).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Goedert, M., Jakes, R. & Spillantini, M. G. The synucleinopathies: twenty years on. J. Parkinsons Dis. 7, S51–S69 (2017).
Ibanez, P. et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171 (2004).
Chartier-Harlin, M. C. et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Pihlstrom, L. et al. A comprehensive analysis of SNCA-related genetic risk in sporadic parkinson disease. Ann. Neurol. 84, 117–129 (2018).
Blauwendraat, C. et al. Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov. Disord. 34, 866–875 (2019).
Chung, S. J. et al. Common variants in PARK loci and related genes and Parkinson’s disease. Mov. Disord. 26, 280–288 (2011).
Kay, D. M. et al. Genetic association between alpha-synuclein and idiopathic Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1222–1230 (2008).
Ross, O. A. et al. Familial genes in sporadic disease: common variants of alpha-synuclein gene associate with Parkinson’s disease. Mech. Ageing Dev. 128, 378–382 (2007).
Winkler, S. et al. alpha-Synuclein and Parkinson disease susceptibility. Neurology 69, 1745–1750 (2007).
Maraganore, D. M. et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296, 661–670 (2006).
Tan, E. K. et al. Polymorphism of NACP-Rep1 in Parkinson’s disease: an etiologic link with essential tremor? Neurology 54, 1195–1198 (2000).
Tan, E. K. et al. Alpha synuclein promoter and risk of Parkinson’s disease: microsatellite and allelic size variability. Neurosci. Lett. 336, 70–72 (2003).
Corrado, L. et al. The length of SNCA Rep1 microsatellite may influence cognitive evolution in Parkinson’s disease. Front. Neurol. 9, 213 (2018).
Huang, Y. et al. SNCA gene, but not MAPT, influences onset age of Parkinson’s disease in Chinese and Australians. Biomed. Res. Int. 2015, 135674 (2015).
Hadjigeorgiou, G. M. et al. Association of alpha-synuclein Rep1 polymorphism and Parkinson’s disease: influence of Rep1 on age at onset. Mov. Disord. 21, 534–539 (2006).
Chung, S. J. et al. Alpha-synuclein repeat variants and survival in Parkinson’s disease. Mov. Disord. 29, 1053–1057 (2014).
Davis, A. A. et al. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol. Aging 37, 209.e1–209.e7 (2016).
Brockmann, K. et al. SNCA: major genetic modifier of age at onset of Parkinson’s disease. Mov. Disord. 28, 1217–1221 (2013).
Fernandez-Santiago, R. et al. SNCA and mTOR pathway single nucleotide polymorphisms interact to modulate the age at onset of Parkinson’s disease. Mov. Disord. 34, 1333–1344 (2019).
Li, N. N. et al. SNCA rs356219 variant increases risk of sporadic Parkinson’s disease in ethnic Chinese. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 452–456 (2013).
Kim, H. J. et al. Motor complications in Parkinson’s disease: 13-year follow-up of the CamPaIGN cohort. Mov. Disord. 35, 185–190 (2020).
Si, Q. Q. et al. SNCA rs11931074 polymorphism correlates with spontaneous brain activity and motor symptoms in Chinese patients with Parkinson’s disease. J. Neural Transm. 126, 1037–1045 (2019).
Shu, L. et al. Genetic impact on clinical features in Parkinson’s disease: a study on SCNA-rs11931074. Parkinsons Dis. 2018, 2754541 (2018).
Cooper, C. A. et al. Common variant rs356182 near SNCA defines a Parkinson’s disease endophenotype. Ann. Clin. Transl. Neurol. 4, 15–25 (2017).
Zheng, J. et al. Festination correlates with SNCA polymorphism in Chinese patients with Parkinson’s disease. Parkinsons Dis. 2017, 3176805 (2017).
Wang, G. et al. Variants in the SNCA gene associate with motor progression while variants in the MAPT gene associate with the severity of Parkinson’s disease. Parkinsonism Relat. Disord. 24, 89–94 (2016).
Ritz, B., Rhodes, S. L., Bordelon, Y. & Bronstein, J. alpha-Synuclein genetic variants predict faster motor symptom progression in idiopathic Parkinson disease. PLoS ONE 7, e36199 (2012).
Huang, Y., Rowe, D. B. & Halliday, G. M. Interaction between alpha-synuclein and tau genotypes and the progression of Parkinson’s disease. J. Parkinsons Dis. 1, 271–276 (2011).
Ng, A. S. L. et al. SNCA Rep1 promoter variability influences cognition in Parkinson’s disease. Mov. Disord. 34, 1232–1236 (2019).
Campelo, C. L. C. et al. Variants in SNCA gene are associated with Parkinson’s disease risk and cognitive symptoms in a Brazilian sample. Front. Aging Neurosci. 9, 198 (2017).
Shi, C. et al. Exploring the effects of genetic variants on clinical profiles of Parkinson’s disease assessed by the Unified Parkinson’s Disease Rating Scale and the Hoehn-Yahr Stage. PLoS ONE 11, e0155758 (2016).
Luo, N. et al. Variants in the SNCA locus are associated with the progression of Parkinson’s disease. Front. Aging Neurosci. 11, 110 (2019).
Markopoulou, K. et al. Does alpha-synuclein have a dual and opposing effect in preclinical vs. clinical Parkinson’s disease? Parkinsonism Relat. Disord. 20, 584–589 (2014).
Zheng, J. et al. Alpha-synuclein gene polymorphism affects risk of dementia in Han Chinese with Parkinson’s disease. Neurosci. Lett. 706, 146–150 (2019).
Mata, I. F. et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 71, 1405–1412 (2014).
Williams-Gray, C. H. et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 84, 1258–1264 (2013).
Goris, A. et al. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann. Neurol. 62, 145–153 (2007).
Huertas, I. et al. Genetic factors influencing frontostriatal dysfunction and the development of dementia in Parkinson’s disease. PLoS ONE 12, e0175560 (2017).
Dan, X. et al. Association between common genetic risk variants and depression in Parkinson’s disease: a dPD study in Chinese. Parkinsonism Relat. Disord. 33, 122–126 (2016).
Factor, S. A. et al. Disease-related and genetic correlates of psychotic symptoms in Parkinson’s disease. Mov. Disord. 26, 2190–2195 (2011).
Gjerstad, M. D., Boeve, B., Wentzel-Larsen, T., Aarsland, D. & Larsen, J. P. Occurrence and clinical correlates of REM sleep behaviour disorder in patients with Parkinson’s disease over time. J. Neurol. Neurosurg. Psychiatry 79, 387–391 (2008).
Bjørnarå, K. A., Pihlstrom, L., Dietrichs, E. & Toft, M. Risk variants of the alpha-synuclein locus and REM sleep behavior disorder in Parkinson’s disease: a genetic association study. BMC Neurol. 18, 20 (2018).
Krohn, L. et al. Fine-mapping of SNCA in rapid eye movement sleep behavior disorder and overt synucleinopathies. Ann. Neurol. 87, 584–598 (2020).
Li, Y. et al. Hyposmia is associated with RBD for PD patients with variants of SNCA. Front. Aging Neurosci. 9, 303 (2017).
Toffoli, M. et al. SNCA 3’UTR genetic variants in patients with Parkinson’s disease and REM sleep behavior disorder. Neurol. Sci. 38, 1233–1240 (2017).
American Academy of Sleep Medicine Board of Directors. Diagnostic and Coding Manual (American Academy of Sleep Medicine, 2005).
Noyce, A. J., Lees, A. J. & Schrag, A. E. The prediagnostic phase of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 87, 871–878 (2016).
Chen, W. et al. Hyposmia correlates with SNCA variant and non-motor symptoms in Chinese patients with Parkinson’s disease. Parkinsonism Relat. Disord. 21, 610–614 (2015).
Verbaan, D. et al. Is olfactory impairment in Parkinson disease related to phenotypic or genotypic characteristics? Neurology 71, 1877–1882 (2008).
Farrer, M. et al. alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum. Mol. Genet. 10, 1847–1851 (2001).
Mueller, J. C. et al. Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann. Neurol. 57, 535–541 (2005).
Mata, I. F. et al. SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch. Neurol. 67, 1350–1356 (2010).
Chiba-Falek, O. & Nussbaum, R. L. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 10, 3101–3109 (2001).
Fuchs, J. et al. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 22, 1327–1334 (2008).
Linnertz, C. et al. Genetic regulation of alpha-synuclein mRNA expression in various human brain tissues. PLoS ONE 4, e7480 (2009).
Iwaki, H. et al. Genetic risk of Parkinson disease and progression: an analysis of 13 longitudinal cohorts. Neurol. Genet. 5, e348 (2019).
Tan, M. M. X. et al. Genome-wide association studies of cognitive and motor progression in Parkinson’s disease. Mov. Disord. https://doi.org/10.1002/mds.28342 (2020).
Macleod, A. D. et al. Age-related selection bias in Parkinson’s disease research: are we recruiting the right participants? Parkinsonism Relat. Disord. 55, 128–133 (2018).
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G. & Group, P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J. Clin. Epidemiol. 62, 1006–1012 (2009).
Stoker, T. B. et al. A common polymorphism in SNCA is associated with accelerated motor decline in GBA-Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 91, 673–674 (2020).
Zhang, K. et al. The effects of SNCA rs894278 on resting-state brain activity in Parkinson’s disease. Front. Neurosci. 13, 47 (2019).
Sampedro, F., Marin-Lahoz, J., Martinez-Horta, S., Pagonabarraga, J. & Kulisevsky, J. Cortical thinning associated with age and CSF biomarkers in early Parkinson’s disease is modified by the SNCA rs356181 polymorphism. Neurodegener. Dis. 18, 233–238 (2018).
Caspell-Garcia, C. et al. Multiple modality biomarker prediction of cognitive impairment in prospectively followed de novo Parkinson disease. PLoS ONE 12, e0175674 (2017).
Zheng, J. et al. Association between gene polymorphism and depression in Parkinson’s disease: a case-control study. J. Neurol. Sci. 375, 231–234 (2017).
Cheng, L. et al. SNCA rs356182 variant increases risk of sporadic Parkinson’s disease in ethnic Chinese. J. Neurol. Sci. 368, 231–234 (2016).
Chen, Y. et al. Genetic variants of SNCA are associated with susceptibility to Parkinson’s disease but not amyotrophic lateral sclerosis or multiple system atrophy in a Chinese population. PLoS ONE 10, e0133776 (2015).
Guo, X. Y. et al. SNCA variants rs2736990 and rs356220 as risk factors for Parkinson’s disease but not for amyotrophic lateral sclerosis and multiple system atrophy in a Chinese population. Neurobiol. Aging 35, 2882.e1–2882.e6 (2014).
Yarnall, A. J. et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology 82, 308–316 (2014).
Cardo, L. F. et al. A search for SNCA 3’ UTR variants identified SNP rs356165 as a determinant of disease risk and onset age in Parkinson’s disease. J. Mol. Neurosci. 47, 425–430 (2012).
Ding, H. et al. Association of SNCA with Parkinson: replication in the Harvard NeuroDiscovery Center Biomarker Study. Mov. Disord. 26, 2283–2286 (2011).
Elbaz, A. et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann. Neurol. 69, 778–792 (2011).
Factor, S. A. et al. Postural instability/gait disturbance in Parkinson’s disease has distinct subtypes: an exploratory analysis. J. Neurol. Neurosurg. Psychiatry 82, 564–568 (2011).
Kim, H. J., Kim, J. M., Lee, J. Y., Park, S. S. & Jeon, B. S. alpha-Synuclein polymorphism and Parkinson’s disease in a tau homogeneous population. Neurol. Asia 15, 61–63 (2010).
Yu, L. et al. SNP rs7684318 of the alpha-synuclein gene is associated with Parkinson’s disease in the Han Chinese population. Brain Res. 1346, 262–265 (2010).
De Marco, E. V. et al. Alpha-synuclein promoter haplotypes and dementia in Parkinson’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147, 403–407 (2008).
Pals, P. et al. Case-control study of environmental risk factors for Parkinson’s disease in Belgium. Eur. J. Epidemiol. 18, 1133–1142 (2003).
Parkinson’s Progression Markers Initiative. The Parkinson’s Progression Markers Initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Acknowledgements
C.C.P. is supported by HELSEVEST (29604). M.G.G.F. is supported by The National Association for Public Health (16152). J.M.-G. and G.A. are supported by the Research Council of Norway (287842).
Author information
Authors and Affiliations
Contributions
C.C.P. and J.M.-G. were responsible for the conception of the review, the literature research, and writing the manuscript. J.L. contributed to interpretation of data and writing the manuscript. M.G.G.F., A.D.M. and G.A. each contributed to the critical revision of the manuscript. All authors revised, read, and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pedersen, C.C., Lange, J., Førland, M.G.G. et al. A systematic review of associations between common SNCA variants and clinical heterogeneity in Parkinson’s disease. npj Parkinsons Dis. 7, 54 (2021). https://doi.org/10.1038/s41531-021-00196-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-021-00196-5
This article is cited by
-
Interplay between α-synuclein and parkin genes: Insights of Parkinson’s disease
Molecular Biology Reports (2024)
-
Epigenetic Orchestration of Neurodegenerative Disorders: A Possible Target for Curcumin as a Therapeutic
Neurochemical Research (2024)
-
Unraveling Dysregulated Cell Signaling Pathways, Genetic and Epigenetic Mysteries of Parkinson’s Disease
Molecular Neurobiology (2024)
