
Abstract
High-throughput techniques have generated abundant genetic and transcriptomic data of Parkinson’s disease (PD) patients but data analysis approaches such as traditional statistical methods have not provided much in the way of insightful integrated analysis or interpretation of the data. As an advanced computational approach, machine learning, which enables people to identify complex patterns and insight from data, has consequently been harnessed to analyze and interpret large, highly complex genetic and transcriptomic data toward a better understanding of PD. In particular, machine learning models have been developed to integrate patient genotype data alone or combined with demographic, clinical, neuroimaging, and other information, for PD outcome study. They have also been used to identify biomarkers of PD based on transcriptomic data, e.g., gene expression profiles from microarrays. This study overviews the relevant literature on using machine learning models for genetic and transcriptomic data analysis in PD, points out remaining challenges, and suggests future directions accordingly. Undoubtedly, the use of machine learning is amplifying PD genetic and transcriptomic achievements for accelerating the study of PD. Existing studies have demonstrated the great potential of machine learning in discovering hidden patterns within genetic or transcriptomic information and thus revealing clues underpinning pathology and pathogenesis. Moving forward, by addressing the remaining challenges, machine learning may advance our ability to precisely diagnose, prognose, and treat PD.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is a progressively debilitating neurodegenerative disease that can lead to severe motor and nonmotor dysfunction1,2,3. Although individuals with PD share core phenotypic features, such as bradykinesia, muscle rigidity, and tremor, there is significant heterogeneity that remains incompletely understood. Over the past two decades, genetics and genomics research has revealed significant heritability of this complex disease4,5,6,7,8. Increasing number of genetic risk factors (e.g., genes and mutations) have been demonstrated to be associated to PD9,10,11,12,13,14 or PD phenotypes15,16,17. Yet, there still remain extensive gaps in our understanding of the complete heritability and pathology of PD. Today’s high-throughput techniques such as next-generation sequencing (NGS) and microarray have been continuously producing genetic and transcriptomic data of PD patients. As listed in Tables 1 and 2, a set of PD repositories are providing rich genetic and transcriptomic data of the participants10,18,19,20,21,22,23,24,25,26,27,28,29,30. This leads to a huge opportunity to investigate the disease. In this context, it’s natural to refer to the recently advanced computational technique, machine learning. Compared to the statistical methods which compute a quantitative measure of confidence to identify the correlations, machine learning has demonstrated the capacity in discovering underlying patterns and insight from rich data and hence has the potential to connect genetics and transcriptomics with clinical outcomes using more complex yet accessible approaches31. Our objective is therefore to introduce the reader to the field of machine learning and discuss its applications in genetic and transcriptomic data study in PD. Through the survey of existing studies, this review aims to discuss current achievements and remaining challenges, as well as to suggest possible future directions toward developing better machine learning algorithms with which to identify underlying patterns from genetic and transcriptomic data for advancing PD research.
Machine learning outline
The term, “machine learning,” is usually used synonymously with “artificial intelligence,” which allows computers to learn from data to uncover patterns and make decisions with minimal human intervention32. A central component of machine learning is the supervised learning and unsupervised learning (see Fig. 1a, b).

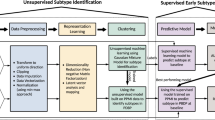
a An example of supervised learning. A supervised learning model takes input as feature vectors of the subjects and “true” labels of them, a.k.a. supervision information, and contains the following components: feature selection (optional), modeling training on training set, model evaluation on testing set, and model deployment for predicting labels of new data. b An example of unsupervised learning. An unsupervised learning model takes input as feature vectors of the subjects only, without any supervision information, and then categorizes the subjects into homogenous groups (a.k.a. clusters). c Illustration of the K-fold cross-validation. One by one, each fold is used as testing set, meanwhile one by one, each remaining K-1 folds are used as training set to train model. d Illustration of underfitting and overfitting issues. Underfitting occurs when the model doesn’t capture patterns of the data well, while overfitting occurs when the model captures details and noise of training data too well to predict new data correctly.
Figure 1a presents a canonical example of the supervised learning workflow, where we are given a set of data objects to learn from. Each object is represented as an array of measurements commonly called “features”. The array of features is then referred to as a so-called “feature vector.” In a typical supervised learning, each object is associated with a “label,” which can be a class the object belongs to such as diagnosis of PD or not, or a continuous value such as the symptom severity of a patient. The labels are then used as supervision information for training model. In this way, constructing a supervised model typically proceeds with following steps.
After some necessary data preprocessing, a model developer typically splits the data into training and testing sets, then trains the model over the training set by fitting the data using a mathematical function and evaluates the model on the testing set. Though random training-testing splitting has been a common strategy, it may result in sampling bias and fitting the model to a skewed training data. In this context, the cross-validation, especially K-fold cross-validation33, has been increasingly engaged. Typically, a K-fold cross-validation divides data into K roughly equal subsets, a.k.a. so-called folds. One by one, each fold is used as testing set, meanwhile one by one, each remaining K-1 folds are used as training set to train model (see Fig. 1c). The cross-validation strategy also helps to evaluate underfitting and overfitting issues, where the former occurs when the model doesn’t capture patterns of the data well and the latter occurs when the model captures details and noise of training data too well to predict new data correctly (see Fig. 1d). Optionally a feature selection procedure can be performed before fitting model to data to enhance the model training. The selected features can be specific ones selected from the original, large set by using statistical testing methods, as well as new, informative ones produced from the original features by using algorithms like Principal Component Analysis. Some models (such as random forest, support vector machine (SVM), and logistic regression) also allow selecting informative features during modeling fitting34,35,36. To evaluate a developed model, usually used performance measurements include accuracy, sensitivity, specificity, and area under the receiver operating characteristic curve (AUC-ROC), which estimates accuracy while comprehensively considering trade-off between true positive rate and false positive rate. Finally, the trained model with the most desirable performance is then deployed to predict unknown labels from new data. In other words, the model “trained” through supervision by labeled data is then used to predict labels of new objects.
For the purpose of accelerating understanding of the molecular biology and pathology of complex diseases, supervised learning has been used in the analysis of genetic and transcriptomic data and has achieved promising results37,38. In this context, “features” are genetic factors (e.g., single-nucleotide polymorphisms [SNPs]) or genomic variables (e.g., gene expression levels), and “labels” are disease traits, phenotypes, symptom severities. Some supervised learning models that have been successfully involved in genetics and genomics include logistic regression, Bayesian, decision tree, SVM, k-nearest neighbors (KNN), and neural network models, etc38.
In contrast to supervised learning, an unsupervised learning model takes input as feature vectors of the objects only. As shown in Fig. 1b, without any supervision information (i.e., labels), the unsupervised learning model is more straightforward, typically aiming at dividing the input data into homogenous groups (a.k.a. clusters) such that objects within a group have similar patterns somehow and that from different groups are distinct. Such nature makes the unsupervised learning important to the study of the complex diseases like PD, due to that it can, to some extent, overcome the issues with data labeling and heterogeneity of data. A usual application area of unsupervised learning is the data-driven disease subtyping39.
Rather than reviewing the taxonomy and algorithms of machine learning models in detail, we focus here on published practices of machine learning in genetic and transcriptomic data analysis in PD. We discuss the practical problems these machine learning models are trying to solve as well as the remaining challenges. For more details of machine learning on genetic and transcriptomic data analysis in general tasks, the readers may consult several previous reviews37,38,40.
Machine learning-based genetic and transcriptomic data analysis in PD
The overarching objectives of machine learning in genetic and transcriptomic data study mainly fall under two general categories: PD outcome study; and PD biomarker identification. Herein, we provide an overview of the existing studies within each category.
PD outcome study
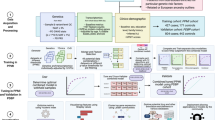
The use of noninvasive metrics for accurate diagnosis of PD in early stage and prediction of PD phenotypes are promising directions in clinical practice. With the advancement of PD genetics, machine learning models have been more and more engaged to discover heritability from these data (as shown in Table 3). Genome-Wide Association Studies (GWAS) on PD subjects has identified many genetic risk factors such as genetic locus markers, SNPs, variants and alleles7,9,10,11,12,13,14,41. Many studies have directly utilized such risk factors or genetic risk score (GRS) derived from these factors as features to build machine learning models for identification of PD. In addition, since PD has shown to be multifactorial, external information such as demographics, clinical information, and neuroimaging data, are usually combined with genetic factors for integrative analysis of PD patients (as shown in Fig. 2a). For example, Nalls et al.42 trained a logistic regression model for classifying PD case patients versus healthy controls (HCs) on the Parkinson’s Progression Markers Initiative (PPMI) population25. GRS, together with olfactory function, family history, age, and gender, were selected as predictors by using a greedy feature selection technique known as stepwise regression43. Upon the PPMI population, the model achieved an AUC-ROC of 0.92 (95% CI [0.90, 0.95]). The GRS was reported to have a higher predictive contribution than family history, age, and gender. Replication on data from 825 PD patients and 261 controls from five independent cohorts (as shown in Table 3) demonstrated the robustness of the model. Dinov et al.44 developed an end-to-end machine learning protocol from data characterization, manipulation, processing, cleaning, and analysis to validation, for PD diagnosis, which flexibly incorporated a series of machine learning models such as AdaBoost45, SVM, decision tree, etc. By combining genetic, clinical, demographic, and derived neuroimaging biomarker information from PPMI cohort, their best model achieved an average accuracy over 0.96 of fivefold cross-validation on separating PDs and HCs. Using GRS derived from 1805 variants only, Nalls et al.12 achieved an AUC-ROC of 0.69 (95% CI [0.66, 0.73]) in separating PDs and HCs.

a Applying machine learning to genetic data (usually combined with other features like demographics, clinical assessments, and neuroimaging features, etc.) for PD outcome study. b Applying machine learning to transcriptomic data (e.g., microarray data) for PD biomarker identification.
Furthermore, there are some studies focusing on predicting the phenotypes of PD. For example, in order to predict impulse control disorder (ICD) induced by dopamine replacement therapy, Kraemmer et al.46 investigated 13 candidate variants from the DRD2, DRD3, DAT1, COMT, DDC, GRIN2B, ADRA2C, SERT, TPH2, HTR2A, OPRK1, and OPRM1 genes. The results demonstrated that adding these variants as predictors significantly increased ICD predictability (AUC-ROC = 0.76, 95%CI [0.70, 0.83]) compared with the prediction results with clinical variables alone (AUC-ROC = 0.65, 95%CI [0.58, 0.73], p = 0.002). Variants of OPRK1, HTR2A, and DDC genes were found to be significant risk factors of ICD. Latourelle et al.47 designed an ensemble model to predict annual rate of change in motor signs and symptoms of PD in PPMI and LABS-PD (Longitudinal and Biomarker Study in Parkinson’s disease)21 cohorts. Along with demographic, clinical, biomarker, and dopamine transporter SPECT (DaTscan) features, a wide range of genetic data were examined including 53 known PD-related SNPs, 17,403 SNPs identified by linkage disequilibrium pruning from genome, and ten genetic principal components derived from genome48. The results showed that these genetic variations were the most predictive to motor progression comparing with other features. In another study, Liu et al.49 predicted the progression of global cognitive impairment through longitudinal analysis of the patient data from nine different cohorts. Combined with longitudinal clinical assessments (e.g., MDS-UPDRS Part I-III), mutations in the GBA (β-glucocerebrosidase) gene were fed to the proposed machine learning model, which achieved an AUC-ROCs of 0.86 (95% CI [0.82, 0.90]) and 0.85 (95% CI [0.78, 0.91]) in discovery and replication populations, respectively. Tropea et al.50 tested for predictors of progression of cognitive decline, and found that the APOE E4 allele was the best predictor.
In addition to PD diagnosis and phenotype prediction where supervised learning is largely performed, data-driven subtyping has a great potential to illuminate underlying pathologies, where an unsupervised learning is more appropriate39. One existing study51 has incorporated genetic data to identify PD subtypes using machine learning. Combining GRS derived from 28 GWAS loci and two additional risk variants GBA p.N370S and LRRK2 p.G2019S42, with demographics and clinical assessments at baseline, an unsupervised learning approach was performed. Subjects with close patterns in terms of genotype and phenotype were gathered into a group, representing a subtype of PD. Three subtypes with distinct PD progression patterns were identified. To date, there hasn’t been much work in the area of applying the unsupervised learning model to genetic and genomic data analysis in PD, probably due to that identification of the groups/clusters is usually subjective and more follow-up efforts are needed for interpreting the identified groups. Yet, it is increasingly attracting attentions as it provides a novel way to discover integrated pattern from genetic and phenotypic information and is promising to personalized medicine.
From these studies we can observe that the genetic information were usually used as predictors in constructing machine learning models for PD diagnosis, phenotype prediction, and subtyping. In many cases, the genetic information have demonstrated to be more indicative than other clinical features. Even so, it is often the combination of genetic and clinical features that can create more robust models overall, compared to the ones based on genetic or clinical features only.
PD biomarker identification
Identifying biomarkers is critical to the early diagnosis, disease prevention, as well as medication response assessment. These in turn will advance efforts to design and interpret disease-modifying clinical trials that use biomarkers for participant enrollment or as outcome measures52,53. A scenario where machine learning models can be applied is discovering the combination of multiple genes whose expression levels in a tissue of interest can discriminate PD patients from HCs, or different phenotypes of PD. Such genes together, constructing a so-called gene signature, may illuminate disease biology and if highly predictive, may provide reliable biomarkers. There have been machine learning models developed for such purpose using the transcriptomic data, e.g., microarray data38. As shown in Fig. 2b, machine learning generally transforms the problem into identifying genes as predictors that comprise the model with the best PD vs. HC predictive performance. Each microarray chip can simultaneously measure expressions of thousands of genes in a tissue of interest, e.g., brain or blood. In preparation, a first and necessary step is preprocessing the microarray data, which usually includes one or both of the following operations: removing low-intensity probes or genes; using statistical approaches such as analysis of variance (ANOVA) to originally select differentially expressed genes as candidates of biomarkers. Next, after splitting subjects into training and test sets, the machine learning classifier model is trained over the training set and evaluated for its ability to discriminate PDs from HCs over the test set. Then biomarkers may be determined by selecting predictors (i.e. genes) associated with a top classification performance, e.g., reaching a higher AUC-ROC value. Finally the identified biomarkers are validated by using an independent validation set54,55 or by using qPCR56 to detect DNA copy numbers and RT-qPCR57 to validate RNA expression levels of target genes.
Machine learning has been used to analyze transcriptomic data in PD and demonstrated its capacity to distinguish PDs from HCs58. To date, using the machine learning based framework, researchers have identified a number of biomarkers for PD from the transcriptomic data (see Table 4). For instance, Scherzer et al.54 investigated 105 individuals by scanning genome-wide expression changes in blood and trained a machine learning model to discriminate PDs from HCs, where 8 genes were identified as candidate biomarkers including VDR, HIP2, CLTB, FPRL2, CA12, CEACAM4, ACRV1, and UTX. Follow-up studies have also successfully identified multi-gene biomarkers from blood samples that are highly indicative of PD57,59,60. In addition, by using other neurodegenerative diseases that have overlapping clinical phenotypes with PD (e.g., Alzheimer’s disease [AD] and atypical parkinsonian disorders [APD] or Lewy Body Dementia) as control cohorts, there were studies demonstrated that the identified biomarkers are robust and have great potential for helping reduce misdiagnosis55,56,61.
From these studies we can observe that machine learning approaches has been used to analyze the transcriptomic data in PD and has demonstrated its capacity in advancing development of potential PD biomarkers.
Discussion: limitations and future directions
We summarize remaining limitations and challenges that the reviewed studies suffered from, and accordingly discuss potential future directions which may lead to promising machine learning approaches to address the issues (see Table 5).
Bias of sample size
In many machine learning applications, a common stumbling block to biological and medical domains is that the sample size is insufficient to achieve adequate power. For example, among all microarray data sets used for identification of biomarkers, to our knowledge the largest study in which an SVM (i.e., support vector machine) model was trained using only 205 PD subjects, 233 HCs and 48 subjects with other neurodegenerative diseases61. Genotyped subject cohorts with rich clinical data such as PPMI25 and BioFIND26 are also limited (~470 PD subjects, 80 subjects of SWEDD [i.e., scans without evidence of dopaminergic deficit] and 230 HCs for PPMI, and ~130 PD and 100 HCs for BioFIND). However, in many scenarios where machine learning has achieved clinically useful insights, we need tens or even hundreds of thousands of samples. An undesirable consequence of training on small-size data is that models can easily overfit to the data and it is then hard to generalize to new subjects. Creating a large patient cohort would be ideal but is expensive and time-consuming. Nowadays there are quite a few publicly available cohort repositories from observational studies containing both the genetic and clinical information10,18,19,21,22,23. Therefore, it would be highly valuable to develop machine learning approaches that can integrate multiple such cohort data. There are some existing studies trying to leverage multiple datasets in the learning process42,47,49, where the model is still trained on a single data set and the other data sets are mainly for replication purpose. Ideally, data from different repositories should be appropriately combined from which the machine learning model can be learned more robustly. AMP-PD (Accelerating Medicines Partnership: Parkinson’s Disease, https://amp-pd.org) established a knowledge portal which harmonized clinical, genetic, and transcriptomic data of four cohorts, PPMI, BioFIND, PDBD (Parkinson’s Disease Biomarker Program), and HBS (Harvard Biomarkers Study), hence provides the potential of applying a larger-scale machine learning-based study on PD genetic and transcriptomic data.
Handling whole spectrum genetic information
GWAS has successfully identified hundreds of genetic risk factors associated with traits of PD, however the factors identified so far only capture a small portion of the heritability and even an aggregation of these effects is often not predictive enough for clinical utility. This issue refers to “missing heritability” in which effect sizes of individual factors are too small to pass the stringent significance filters used in many studies62,63. In their current stage, machine learning models simply utilized GWAS identified genetic risk factors or GRS derived from them to make up the feature vectors. Existing studies demonstrated that such classifier is accurate enough in a cohort study. However, if we want to train the most accurate possible model that can capture “missing heritability” and can be generalized to new subjects, using only known risk factors as predictors will not suffice. There remains a need to incorporate whole-exome or even whole-genome information. In addition, when we apply analysis with the use of multiple platforms, we usually have to aggregate datasets that are generated using different sequencing technologies, which may incorporate many cleaning and calling issues that make the results unreliable and noisy. In this context, analyzing whole spectrum genetic information also helps to address such issues. Latourelle et al.47 has made an attempt to investigated a wide range of genetic information, including known risk SNPs, genome-wide SNPs, and genetic principal components derived from genome48. In addition, many optional feature engineering techniques have also been developed to reducing dimensionality of data. The state-of-the-art methods include multidimensional scaling64, linear discriminant analysis65 and autoencoder66, etc. Especially, autoencoder66, an important subcategory of deep learning, has shown impressive effectiveness and efficiency in generating low-dimensional representation from extreme high-dimensional data such as genome-wide expression data67. These approaches should be examined for their utility in future work.
Multifactorial modeling
It has been clearly demonstrated that PD is a multifactorial disease, therefore PD prediction or forecast in its early stage needs to comprehensively consider multivariate information. How to aggregate heterogeneous information, such as genetic, genomic, clinical, neuroimaging, social demographic and environmental exposure data, poses a big challenge to conventional computational approaches. The most straightforward way to handle heterogeneous data is to convert each type of data into vector format prior to processing, and orderly concatenate all vectors specific to each subject into a long vector. This has been the most common approach to current PD diagnosis and phenotype prediction. For example, in addition to genetic data, Nalls et al.42 incorporated demographics, olfactory function and self-reported family history of PD; Dinov et al.44 further used clinical and derived neuroimaging biomarker data; Latourelle et al.47 additionally utilized CSF protein biomarkers, etc. By modeling multifactorial aspects of PD, all these methods achieved high performance value (AUC-ROC over 0.80). On the other hand, the kernel approaches68 and probability models69 are alternative techniques that can fuse heterogeneous data and can be introduced to address this issue in the future.
Cohort diversity
Like other diseases, the cohort diversity issues may impact the different aspects of PD research (e.g., clinical trial design70), where machine learning is expected to play an important role. For example, one potential diversity problem with the current cohorts is ethnicity, as most existing genetic and genomic studies are highly skewed toward the European ancestry71. Genetic factors have been found to change their roles in PD risk in different ethnicities (e.g., differences in genetic risk factors have been found between the European and Asian populations72). Another is the clinical diversity. For example, PPMI recruits early-stage untreated PD patients25; while BioFIND recruits patients who are in moderate to advanced stages26. These diversity issues may hinder the generalizability of the developed machine learning models. For example, a model developed on the European cohort may not perform well on the African or Asian cohort, and a model trained from PPMI may not work for BioFIND without any adaptations. In this context, the PD community has taken great steps toward addressing the cohort diversity issues. The International Parkinson Disease Genomics Consortium71 and the Global Parkinson’s Genetics Program (GP2, https://parkinsonsroadmap.org/gp2/) have initiated efforts in analyzing data and samples worldwide. AMP-PD is also a good example that provides the platform for facilitating cross-cohort investigation. On the other hand, the model developer and end-user should be mindful of these cohorts’ diversity issues. First, a well-behaved model on a single population is not sufficient, and external validation on independent cohorts is needed. Second, training the model with data from multiple cohorts is always a good choice whenever possible. Third, new machine learning strategies like transfer learning73 could be potentially helpful here. Instead of directly duplicating the entire model, transfer learning typically fine-tunes the model parameters trained on one cohort in another cohort, which thus leverages the knowledge from both cohorts.
Model interpretation
A long-standing concern of machine learning, especially in medicine, is the model interpretation, because that not only the model’s prediction performance but also the clues for making the decision are essential. For example, in biomarker identification, a researcher would expect to see the contribution of expression level of a specific gene in discriminating PD and HCs, indicating why the gene was or was not selected as a biomarker by the model. In this context, the traditional machine learning models, including Bayesian, rule-based models (e.g., decision tree and random forest), logistic regression, SVM, etc., are instinctively capable to estimate feature contributions while training the models. This could be one reason why most of the reviewed studies rely on these approaches. Importantly, some models (e.g., SVM and logistic regression) can be extended to contain the nature of selecting informative features in two ways: (1) plus a regularization term to reduce contribution of a noninformative feature to zero34,35; or (2) being embedded in a wrapper such as greedy forward wrappers36. This results in the pipeline integrating model training, evaluation, and interpretation in an end-to-end manner.
In addition, deep learning, a new branch of machine learning, has made impressive advances in computer and data science74. A deep learning model usually appears to be a “black box” model due to its high complexity. Though preliminary studies have reported a greater computational capacity and flexibility of deep learning in genetics and genomics38,75 as well as health care76, it’s encountering a larger challenge in model interpretation. There have two potential strategies addressing this issue: to measure changes in model output while involving systematic modification of the input77; or to engage third-party tools to determine the feature contributions78. Solving the issue, PD genetic and transcriptomic data analysis may largely benefit from the prominent deep learning models.
Model evaluation
Model evaluation is essential for machine learning model development. Quantitatively, model evaluation tells us how the model performs with measures such as accuracy, sensitivity, specificity, AUC-ROC, etc. Typically, multiple random training-testing splitting or K-fold cross-validation are performed and the average performance along with standard deviation and statistical significance are reported. Yet, evaluation is limited to data in hand and it is hard to keep model performance consistent when encountering new data. To address this, researchers have tried to use isolated data sets to evaluate models trained on discovery sets42,47,49. In addition, experimental tests are also useful in model evaluation. For example, qPCR56 and RT-qPCR57 were used to validate gene expression of identified biomarkers. Such domain experts guided evaluation may enhance model confidence significantly. Alternatively, qualitative evaluation is another way for model evaluation, which often engages tools to demonstrate machine learning findings to enhance stability and interpretability of the produced model, such as visualizations of feature importance and comparisons of characteristics of identified subtypes. It helps to understand machine learning outcomes intuitively.
Interdisciplinary study
In practice, for effective application of machine learning methods to achieve good performance, good understanding on both machine learning methodology and domain-specific knowledge is necessary. Standing at the crossroads of genetics, transcriptomics, PD, and machine learning fields, it is challenging for researchers to solve this interdisciplinary problem. For neurologists in particular, it is challenging to be aware of the mathematical background of machine learning models to proficiently develop algorithms. For machine learning developers, knowing less about genetics, genomics and PD hinders designing the best model that can appropriately organize genetic and transcriptomic data as well as fully incorporate domain knowledge. In developing the most appropriate model, neurologists need to play a crucial role in the entire development life cycle, from coming up with conception practical and impactful applications, and providing domain knowledge to guide model building, to model evaluation before practice in clinic. To this end, deep collaboration between neurologists and machine learning developers is highly recommended.
Another flexible way to address the interdisciplinary issue is to incorporate prior domain knowledge to guide the model to learn. In this context, previously identified genotype-phenotype correlations can be used as domain knowledge to enhance PD predictive modeling. For instance, individuals with PD due to parkin (PARK2) gene mutations are found to be more prone to levodopa-induced dyskinesias79,80, and GBA variants have been associated with a more rapid progression of cognitive dysfunction and motor symptoms of PD patients16. Injecting such prior associations into a machine learning model may improve the model robustness in terms of both computability and interpretability. In this context, domain experts play a key role in model development by providing specialized guidance to design rule of model. In addition, gene-gene interactions are also important genomic domain knowledge which may help genetic data modeling and can be downloaded from existing public databases, such as KEGG81 and BioGRID82. To handle injection of domain knowledge, powerful techniques have been extensively developed, such as kernel68 and knowledge embedding83,84 approaches.
Conclusions
Recent years have seen a clear acceleration in our knowledge and ability to apply machine learning models to genetic and transcriptomic data in PD study. Machine learning models that combine genetic data with demographic, clinical and neuroimaging information have achieved significant refinement in PD diagnosis and disease phenotype prediction, as well PD subtype identification. In addition, many potential biomarkers in terms of gene expression levels have been identified through the use of machine learning models. Intrinsic superiority and current achievements of machine learning has demonstrated its promise in genetic and transcriptomic data analysis for advancing PD. However, remaining limitations of current studies are challenging machine learning approaches to make further breakthroughs in thoroughly understanding pathogenesis of the disease. For future research, developing appropriate machine learning models by addressing the issues may lead to great improvements in PD management.
References
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet 386, 896–912 (2015).
Chaudhuri, K. R., Healy, D. G. & Schapira, A. H. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 5, 235–245 (2006).
Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376 (2008).
Trinh, J. & Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 9, 445 (2013).
Farrer, M. J. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat. Rev. Genet. 7, 306–318 (2006).
Singleton, A. B., Farrer, M. J. & Bonifati, V. The genetics of Parkinson’s disease: progress and therapeutic implications. Mov. Disord. 28, 14–23 (2013).
Singleton, A. & Hardy, J. The evolution of genetics: Alzheimer’s and Parkinson’s diseases. Neuron 90, 1154–1163 (2016).
Lin, M. K. & Farrer, M. J. Genetics and genomics of Parkinson’s disease. Genome Med. 6, 48 (2014).
Bandrés-Ciga, S. et al. Genome-wide assessment of Parkinson’s disease in a Southern Spanish population. Neurobiol. Aging 45, 213. e213–213. e219 (2016).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989 (2014).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511 (2017).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Abeliovich, A. & Gitler, A. D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207 (2016).
Blauwendraat, C. et al. Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol. 75, 1416–1422 (2018).
Mata, I. F. et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 71, 1405–1412 (2014).
Davis, M. Y. et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 73, 1217–1224 (2016).
Paul, K. C. et al. APOE, MAPT, and COMT and Parkinson’s disease susceptibility and cognitive symptom progression. J. Parkinsons Dis. 6, 349–359 (2016).
Locascio, J. J. et al. Association between α-synuclein blood transcripts and early, neuroimaging-supported Parkinson’s disease. Brain 138, 2659–2671 (2015).
Williams-Gray, C. H. et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 84, 1258–1264 (2013).
Zhu, K., van Hilten, J. J. & Marinus, J. Predictors of dementia in Parkinson’s disease; findings from a 5-year prospective study using the SCOPA-COG. Parkinsonism Relat. Disord. 20, 980–985 (2014).
Ravina, B. et al. A longitudinal program for biomarker development in Parkinson’s disease: a feasibility study. Mov. Disord. 24, 2081–2090 (2009).
Breen, D. P., Evans, J. R., Farrell, K., Brayne, C. & Barker, R. A. Determinants of delayed diagnosis in Parkinson’s disease. J. Neurol. 260, 1978–1981 (2013).
Jankovic, J. et al. Variable expression of Parkinson’s disease A base‐line analysis of the DAT ATOP cohort. Neurology 40, 1529–1529 (1990).
Rosenthal, L. S. et al. The NINDS Parkinson’s disease biomarkers program. Mov. Disord. 31, 915–923 (2016).
Marek, K. et al. The parkinson progression marker initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Kang, U. J. et al. The BioFIND study: Characteristics of a clinically typical Parkinson’s disease biomarker cohort. Mov. Disord. 31, 924–932 (2016).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Brazma, A. et al. ArrayExpress—a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 31, 68–71 (2003).
Taccioli, C. et al. ParkDB: a Parkinson’s disease gene expression database. Database 2011 (2011).
International Parkinson’s Disease Genomics, C. & Wellcome Trust Case Control A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 7, e1002142 (2011).
Bzdok, D., Altman, N. & Krzywinski, M. Statistics versus machine learning. Nat. Methods 15, 233–234 (2018).
Deo, R. C. Machine learning in medicine. Circulation 132, 1920–1930 (2015).
Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Stat. Methodol. 36, 111–133 (1974).
Roth, V. The generalized LASSO. IEEE T. Neural. Networ. 15, 16–28 (2004).
Shevade, S. K. & Keerthi, S. S. A simple and efficient algorithm for gene selection using sparse logistic regression. Bioinformatics 19, 2246–2253 (2003).
Pudil, P., Novovičová, J. & Kittler, J. Floating search methods in feature selection. Pattern Recognit. Lett. 15, 1119–1125 (1994).
Okser, S., Pahikkala, T. & Aittokallio, T. Genetic variants and their interactions in disease risk prediction–machine learning and network perspectives. BioData Min. 6, 5 (2013).
Libbrecht, M. W. & Noble, W. S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 16, 321 (2015).
Fereshtehnejad, S. M. & Postuma, R. B. Subtypes of Parkinson’s disease: what do they tell us about disease progression? Curr. Neurol. Neurosci. Rep. 17, 34 (2017).
Swan, A. L., Mobasheri, A., Allaway, D., Liddell, S. & Bacardit, J. Application of machine learning to proteomics data: classification and biomarker identification in postgenomics biology. OMICS 17, 595–610 (2013).
Billingsley, K. J., Bandres-Ciga, S., Saez-Atienzar, S. & Singleton, A. B. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 373, 9–20 (2018).
Nalls, M. A. et al. Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: a population-based modelling study. Lancet Neurol. 14, 1002–1009 (2015).
Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Contr. 19, 716–723 (1974).
Dinov, I. D. et al. Predictive big data analytics: a study of Parkinson’s disease using large, complex, heterogeneous, incongruent, multi-source and incomplete observations. PLoS ONE 11, 1–28 (2016).
Rätsch, G., Onoda, T. & Müller, K. R. Soft margins for AdaBoost. Mach. Learn. 42, 287–320 (2001).
Kraemmer, J. et al. Clinical-genetic model predicts incident impulse control disorders in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 87, 1106–1111 (2016).
Latourelle, J. C. et al. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: a longitudinal cohort study and validation. Lancet Neurol. 16, 908–916 (2017).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Liu, G. et al. Prediction of cognition in Parkinson’s disease with a clinical–genetic score: a longitudinal analysis of nine cohorts. Lancet Neurol. 16, 620–629 (2017).
Tropea, T. F. et al. APOE, thought disorder, and SPARE-AD predict cognitive decline in established Parkinson’s disease. Mov. Disord. 33, 289–297 (2018).
Fereshtehnejad, S. M., Zeighami, Y., Dagher, A. & Postuma, R. B. Clinical criteria for subtyping Parkinson’s disease: biomarkers and longitudinal progression. Brain 140, 1959–1976 (2017).
Gomperts, S. N. et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol. 73, 1334–1341 (2016).
Irwin, D. J. et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 16, 55–65 (2017).
Scherzer, C. R. et al. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc. Natl Acad. Sci. USA 104, 955–960 (2007).
Molochnikov, L. et al. A molecular signature in blood identifies early Parkinson’s disease. Mol. Neurodegener. 7, 26 (2012).
Potashkin, J. A., Santiago, J. A., Ravina, B. M., Watts, A. & Leontovich, A. A. Biosignatures for Parkinson’s disease and atypical parkinsonian disorders patients. PLoS ONE 7, 1–13 (2012).
Calligaris, R. et al. Blood transcriptomics of drug-naive sporadic Parkinson’s disease patients. BMC Genom. 16, 876 (2015).
Babu, G. S. & Suresh, S. Parkinson’s disease prediction using gene expression–A projection based learning meta-cognitive neural classifier approach. Expert Syst. Appl. 40, 1519–1529 (2013).
Santiago, J. A., Scherzer, C. R., Study, H. B. & Potashkin, J. A. Specific splice variants are associated with Parkinson’s disease. Mov. Disord. 28, 1724–1727 (2013).
Karlsson, M. K. et al. Found in transcription: accurate Parkinson’s disease classification in peripheral blood. J. Parkinsons Dis. 3, 19–29 (2013).
Shamir, R. et al. Analysis of blood-based gene expression in idiopathic Parkinson disease. Neurology 89, 1676–1683 (2017).
Jakobsdottir, J., Gorin, M. B., Conley, Y. P., Ferrell, R. E. & Weeks, D. E. Interpretation of genetic association studies: markers with replicated highly significant odds ratios may be poor classifiers. PLoS Genet. 5, 1–8 (2009).
Wei, Z. et al. From disease association to risk assessment: an optimistic view from genome-wide association studies on type 1 diabetes. PLoS Genet. 5, e1000678 (2009).
Saeed, N., Nam, H., Haq, M. I. U., Saqib, M. & Bhatti, D. A survey on multidimensional scaling. ACM Comput. Surv. 51, 1–25 (2018).
Balakrishnama, S. & Ganapathiraju, A. Linear discriminant analysis-a brief tutorial. Inst. Signal Inf. Process. 18, 1–8 (1998).
Kramer, M. A. Nonlinear principal component analysis using autoassociative neural networks. AIChE J. 37, 233–243 (1991).
Tan, J., Ung, M., Cheng, C. & Greene, C. S. Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders. In Biocomputing 2015, pp 132–143 (World Scientific, 2015).
Hofmann, T., Schölkopf, B. & Smola, A. J. Kernel methods in machine learning. Ann. Stat. 36, 1171–1220 (2008).
Nasrabadi, N. M. Pattern recognition and machine learning. J. Electron. Imaging 16, 049901 (2007).
Leonard, H. et al. Genetic variability and potential effects on clinical trial outcomes: perspectives in Parkinson’s disease. J. Med. Genet. 57, 331–338 (2020).
The International Parkinson Disease Genomics, C. Ten years of the international Parkinson disease genomics consortium: progress and next steps. J. Parkinsons Dis. 10, 19–30 (2020).
Foo, J. N. et al. Identification of risk loci for Parkinson disease in asians and comparison of risk between Asians and Europeans: a genome-wide association study. JAMA Neurol. 77, 746–754 (2020).
Pan, S. J. & Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 22, 1345–1359 (2010).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436 (2015).
Zou, J. et al. A primer on deep learning in genomics. Nat. Genet. 51, 12–18 (2019).
Su, C., Xu, Z., Pathak, J. & Wang, F. Deep learning in mental health outcome research: a scoping review. Transl. Psychiatry 10, 116 (2020).
Zhang, Q. & Zhu, S. Visual interpretability for deep learning: a survey. Front. Inf. Technol. Electron. Eng. 19, 27–39 (2018).
Ribeiro, M. T., Singh, S. & Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp 1135–1144 (San Francisco, California, USA, 2016).
Khan, N. L. et al. Parkin disease: a phenotypic study of a large case series. Brain 126, 1279–1292 (2003).
Nishioka, K. et al. A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 81, 391–395 (2010).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Stark, C. et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539 (2006).
Cho, H., Berger, B. & Peng, J. Diffusion component analysis: unraveling functional topology in biological networks. Proc. International Conference on Research in Computational Molecular Biology, pp 62–64 (2015).
Su, C., Tong, J., Zhu, Y., Cui, P. & Wang, F. Network embedding in biomedical data science. Brief. Bioinform. 21, 182–197 (2018).
Acknowledgements
We would like to thank the valuable comments and suggestions from Olivier Elemento, Margaret Elizabeth Ross and Claire Henchcliffe. The work is supported by Michael J. Fox Foundation funding number 14858, 14858.01, and 15914.
Author information
Authors and Affiliations
Contributions
Study concept and design: C.S., F.W. Collection, analysis, and interpretation of data: C.S., F.W. Drafting of the manuscript: C.S., F.W. Critical revision of the manuscript: C.S., J.T., F.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, C., Tong, J. & Wang, F. Mining genetic and transcriptomic data using machine learning approaches in Parkinson’s disease. npj Parkinsons Dis. 6, 24 (2020). https://doi.org/10.1038/s41531-020-00127-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-020-00127-w
This article is cited by
-
Preclinical translational platform of neuroinflammatory disease biology relevant to neurodegenerative disease
Journal of Neuroinflammation (2024)
-
Transcriptomic analysis reveals associations of blood-based A-to-I editing with Parkinson’s disease
Journal of Neurology (2024)
-
Bioinformatics analysis and machine learning approach applied to the identification of novel key genes involved in non-alcoholic fatty liver disease
Scientific Reports (2023)
-
Machine learning-based prediction of cognitive outcomes in de novo Parkinson’s disease
npj Parkinson's Disease (2022)
-
Exploring photoacoustic spectroscopy-based machine learning together with metabolomics to assess breast tumor progression in a xenograft model ex vivo
Laboratory Investigation (2021)
