
Abstract
Zirconolite ceramics present a chemically durable host matrix for waste actinides, but zirconolite dissolution rates reported in the literature often vary significantly. Here, the release of Ca and Al from a hot isostatically pressed zirconolite (Ca0.8Zr0.9Ce0.3Ti1.6Al0.4O7.0) was shown to be predominantly driven by preferential dissolution of minor perovskite and alumina phases. Both phases were undetectable by XRD, and the perovskite was difficult to detect by SEM-EDS. Whilst the zirconolite phase exhibited no signs of alteration, dissolution of the perovskite proceeded congruently without forming a hydrated altered layer or diffusion of protons into the solid that would be indicative of an ion-exchange mechanism. The weak temperature dependence of dissolution (40, 90 and 150 °C) showed that kinetics were limited by transport and a mixed transport-surface controlled reaction for Ca and Al, respectively. A significant H2O-D2O isotope effect on dissolution was observed for Ca but not for Al at all temperatures. The former was consistent with an abated rate of hydrolysis in the absence of a contribution from diffusion, whilst the latter could be attributed to differences in the activated complex for Ca and Al release through hydrolysis. These results demonstrate the role of a secondary phase perovskite in the dissolution kinetics of zirconolite even when perovskite occurs at low concentration and evades detection by bulk techniques such as XRD. This study provides a potential explanation of variations in zirconolite ceramic dissolution rates present in the literature and provides a null result to tests of an incongruent Ca release mechanism from zirconolite.
Similar content being viewed by others

Introduction
The excellent radiation resistance and actinide retention during alteration of natural zirconolite (CaZrTi2O7)1,2,3,4 makes it a promising waste actinide immobilisation host matrix5,6. Alongside Ba-hollandite and perovskite, zirconolite is a key component of Synroc-C, with zirconolite targeting smaller rare earth elements (REE) and tetravalent actinides and perovskite targeting Sr, larger REE and trivalent actinides7,8,9. Whilst the zirconolite endmember is a 2M polytype (space group C2/c)10,11, it exhibits excellent chemical flexibility. REE and actinide substitution onto the Ca and Zr sites, often facilitated by trivalent species substituting for Ti for charge compensation, leads to structural changes as a function of the substituting cation and synthesis conditions. For example, cold-pressed and sintered (CPS) Ca1-xZr1-xCe2xTi2O7+δ and spark plasma sintered (SPS) CaZr1-xCexTi2O7 zirconolites transition with x from 2M to a 4M (expanded 2M; C2/c12) to a pyrochlore structure, with perovskite impurity concentrations as a function of Ce oxidation state (Ce4+/Ce3+) and concentration13,14,15,16. Contrastingly, CPS Ca1-xCexZrTi2-xAl2xO7 (x ≤ 0.35) and hot isostatically pressed (HIP) Ca0.80Ce0.20ZrTi1.60Cr0.40O7 zirconolites retain a 2M structure17,18.
Zirconolite ceramics have been previously demonstrated to have excellent aqueous durability19,20,21. Observations of the loss of Ca and retention of sparingly soluble species (Ti, Zr and REE) in natural22,23 and synthetic samples24,25,26,27 suggest a preferential release mechanism for Ca. However, few studies have investigated zirconolite dissolution mechanisms, with acidic solutions often used to accelerate dissolution26,28,29,30. In turn, the debate on whether zirconolites undergo dissolution following a selective Ca leaching mechanism producing a relict decalcified structure24 or a congruent dissolution mechanism highly coupled to the precipitation of sparing soluble species is ongoing28.
Significant variations in zirconolite dissolution rates have been reported. For example, dissolution experiments on endmember, Ce-doped and Nd-doped zirconolites in 90 °C deionised (DI) water, using powders (PCT-B31) and monoliths (MCC-132) yielded normalised Ca release rates from 10−2 to 100 g m−2 d−1 after both 1 and 7 days, whilst normalised Ce and Nd release rates were 10−5 to 10−3 g m−2 d−1 and 10−7 to 10−4 g m−2 d−1 after 1 and 7 days, respectively15,33,34,35,36,37,38. Differences in sample preparation methods, such as the presence of adhering fine particles27,31 or differences in monolith surface finish32, between studies are known to contribute to variations in reported polycrystalline ceramic dissolution kinetics at short times. For zirconolites, differences in dissolution behaviour have also previously been correlated with perovskite impurity concentration. However, the absence of characteristic reflections for perovskite in XRD diffractograms and comparatively lower dissolution rates have been taken as indicative of its absence15,29. It is postulated here that significant variations in literature rates may in some instances be attributable to minor concentrations of undetected perovskite.
In the experiments described here, the preferential release of Ca from zirconolite as an ion-exchange process with hydronium ions was investigated through dissolution experiments on a HIP Ca0.8Zr0.9Ce0.3Ti1.6Al0.4O7.0 zirconolite. Static powder experiments varied (1) the solvent, with DI water and D2O dissolution experiments probing kinetic isotope effects associated with hydrolysis or diffusion, (2) temperature (40, 90 and 150 °C) to investigate Arrhenius temperature dependence and accelerate dissolution, and (3) time (2 to 256 h) to investigate any t−0.5 rate dependence at short timescales. Phase assemblages were first characterised (pristine sample), followed by dissolution kinetics (solution chemistry) and finally dissolution mechanisms (leached samples).
Results
Perovskite as a minor phase
Electron probe micro-analysis (EPMA) of the zirconolite (nominally Ca0.8Zr0.9Ce4+0.3Ti1.6Al0.4O7.0) monoliths yielded a stoichiometry of that targeted with an excess of Ti, attributable to the TiO2 buffer, and Hf, Sr and Ba impurities (Table 1). However, SEM-EDS showed phases with compositions consistent with alumina, ceria and a Sr-rich Zr-depleted phase were present (Fig. 1 and Table 1). The alumina phase (Fig. 1a, b) was partly attributable to contamination during precursor milling; relative area fractions yielded 3.23 ± 0.49 wt.% Al2O3, exceeding the ~0.52 wt.% milling mass loss. XPS Al concentrations (5.59 ± 1.19 at.%) agreed with EPMA concentrations alongside excess alumina (4.98 ± 1.12 at.%), but the SEM-EDS zirconolite compositions suggested incorporation of all intended precursor Al2O3. BSE images and Ce SEM-EDS maps (Fig. 1a, d) were consistent with unincorporated CeO2; Ce recoveries in the zirconolite phase were 96.0 and 80.6% for EPMA and SEM-EDS, respectively, with the latter distinguishing between zirconolite and ceria phases. Notably, alumina and ceria were observable at the pristine particles surfaces (Fig. 1h, g).

Pristine monolith BSE image labelled with phases (a) and EDS maps of Al (b), Sr and Zr (c), Ce and Zr (d), and Ca and Ti (e). Leached (256 h, 150 °C, DI water) powder BSE image (f) and Ce EDS map (g), and washed pristine powder Ca and Al (h), and Ce and Zr (i) EDS maps.
STEM-EDS of the Sr-rich Zr-depleted phase, isolated within a leached sample lamella (Fig. 2), yielded a composition consistent with a Sr-Ce-rich perovskite (Table 1). Given its depth in the lamella relative to the extent of dissolution (see later sections), this grain was considered pristine. SEM-EDS of the monoliths semi-quantitatively confirmed this perovskite contained Ba (Supplementary Table 14), whilst SAED of the perovskite grain in the lamella showed it was polycrystalline (Supplementary Fig. 18 and Supplementary Methods 1.2.3). Similarly, a phase depleted in Zr was observed on a pristine particle (Fig. 1h, i). Perovskite formation during Ce-bearing zirconolite synthesis is well known13,14,28, with Sr-perovskite solid solutions preferentially forming over zirconolite substitution39,40,41,42,43. Assuming all Sr was present in the perovskite, mass balance yielded a maximum 5.28 ± 0.32 mol.% Ca-Ce-Sr-Ba perovskite. Phase stoichiometry are summarised in Supplementary Table 14.

STEM dark field (DF) (a) and TEM bright field (BF) (b) images and STEM-EDS maps of Ca (c), Ce (d), Sr (e), Ti (f) and Zr (g) for the leached (256 h, 150 °C, DI water) lamella. The coatings applied prior to FIB are labelled in a, with labels ‘Z’ and ‘P’ on the grains corresponding to zirconolite and perovskite grains, respectively. STEM-EDS maps focus on a smaller area (centred at the meeting point of the grains) than the images. Grains deep in the lamella were considered representative of the pristine (see main text).
Surface chemistry and Ce oxidation state were investigated by XPS. Fitting Ce 3d peak models14,44,45,46,47 yielded a Ce3+ fraction, v(Ce3+), of 0.48 ± 0.05, increasing to 0.92 ± 0.05 after 80 s 200 eV sputtering (Fig. 3a). The literature confirms Ce4+ is reduced during XPS analysis48,49,50 and Ar+ sputtering51,52,53, with transition metals enhancing reduction53,54. Indeed, reanalysis of the same surface increased v(Ce3+) to 0.58 ± 0.05. Ti was only reduced (TiO1.55(41)) after 30 s 3 keV sputtering (Supplementary Figs. 8–10), and satellites in the corresponding Ce 3d spectrum (Fig. 3a) were consistent with a disordered Ce environment55. As re-oxidation from sputtered O atoms53 prevents complete reduction at short times48,50,53,55,56, the absence of a u‴ Ce4+ peak (916.7 eV) and v(Ce3+) approaching unity after 30 s 3 keV and 80 s 200 eV sputtering, respectively, are consistent with a bulk v(Ce3+) near unity. This is attributable to the HIP canister imposing reducing synthesis conditions57,58,59, consistent with significant v(Ce3+) for HIP-synthesised zirconolites, often alongside secondary-phase perovskite29,60,61. Indeed, Ce3+ is preferentially incorporated into perovskite through ionic radii considerations9,13,43.

Spectra with peak models (a) and v(Ce3+) as a function of Ar+ sputtering time and energy (b). Spectra in a are presented as measured (black), backgrounds (grey), peaks for Ce3+ (magenta) and Ce4+ (blue), and overall fits (red). Stars in the 30 s 3 keV spectrum denote shake-up satellites associated with the reduction of Ce by Ar+ sputtering and were not fitted51,55. In b, ‘narrow scans’ and ‘regions’ refer to v(Ce3+) from the fitted peak models44,45,46,55 and regions calibrated relative to a CeO2 standard62,63, respectively. Note that the zirconolite ‘regions’ surface v(Ce3+) = 0.00 arises from the CeO2 correction yielding a negative v(Ce3+). Similarly, a v(Ce3+) of 1.00 was assigned to the zirconolite ‘regions’ 30 s 30 keV sample based upon the absence of the characteristic high BE CeO2 peak, as shown in a. Uncertainties are conservatively given to ±0.05, representing the maximum uncertainty observed on all CeO2 v(Ce3+) measurements (1 S.D. of six replicates). Zirconolite and CeO2 v(Ce3+) represent single replicates and six replicate averages, respectively. As samples were analysed with multiple sputtering energies at 30 s, points are annotated with the sputtering energy used. Other points for the zirconolite and CeO2 used sputtering energies of 200 and 500 eV, respectively. Two zirconolite points are given at 0 s in b (Surface 1 and Surface 2 in a) representing analysis of the surface for Ce 3d before (Surface 1) and after analysing other species without sputtering (Surface 2). Note the sputtered sample v(Ce3+) correspond to an initial surface (0 s sputtering) which was only analysed once.
A CeO2 pellet surface v(Ce3+) of 0.57 showed the zirconolite surface v(Ce3+) reduced during analysis (Fig. 3b). Sputtering reduced CeO2 (0.57 to 0.74 ± 0.05) less than the zirconolite range (0.48 to 0.92 ± 0.05) despite using a higher sputtering energy. Following Maslakov et al. 62,63, v(Ce3+) were semi-quantitatively calibrated to CeO2 (Eq. (8), Section 4.8), yielding a reduction-corrected v(Ce3+)a0 (Fig. 3b). This yielded negative zirconolite surface v(Ce3+)a0 (set to 0.00), demonstrating bias towards lower values. However, the v(Ce3+)a0 were internally consistent, showing the increasing zirconolite v(Ce3+)a0 as a function of sputtering time was not due to analytical reduction and that the v(Ce3+)a0 = 0.00 are consistent with an oxidised surface layer. This layer likely arose from storing the monoliths in air.
Here, the approach of Zhang et al.49 modified for elastic scattering processes64,65,66,67,68 was used to approximate the depth of Ce oxidation as a function of sputtering time (further details given in Supplementary Methods 1.1.2 and Supplementary Discussion 2.1.2). Surface oxidised layer thicknesses were approximated as ≥3.34 nm (P = 95% sampling depth), reducing to near-bulk (0.06 nm) after 55 s sputtering. The non-linear calculated sputtering rates for CeO2 were consistent with an inhomogeneous Ce3+ depth distribution produced through reduction during analysis, supporting the view that the v(Ce3+)a0 are biased and the increasing zirconolite v(Ce3+) with depth was not an analytical artefact. Perovskite formation is therefore attributable to the limited solubilities of Sr and Ba in zirconolite and a predominantly Ce3+ bulk. Significant Ti reduction promoting perovskite formation69 was not observed.
XRD diffractograms were consistent with zirconolite 3T (P3121)70, with no ceria, alumina or perovskite characteristic reflections (Supplementary Figs. 35 and 37). Pawley refinements showed the structure was inconsistent with 2M (C2/c)10,11, with 3T lattice parameters (uncertainties in parentheses) of a = 7.2273(4) Å, c = 16.6496(16) Å and V = 753.60(10) Å3.
Perovskite’s role in dissolution kinetics
The dissolution behaviour of the zirconolite ceramic (nominally Ca0.8Zr0.9Ce4+0.3Ti1.6Al0.4O7.0) was qualitatively similar to other studies of Ce-bearing zirconolites with Al for charge compensation28,29. ICP-MS analysis of the aliquots taken from the DI water experiments showed Ca and Al concentrations were significant from the blanks (blank concentrations of 45.58 and 4.11 ng mL−1, respectively), with Ti and Zr concentrations below the instrument detection limit (IDL; 0.30 and 1.70 ng mL−1, respectively), and Ce concentrations insignificant from the blanks (blank concentration of 27 pg mL−1). Sr was not initially analysed, but semi-quantitative mass scans later revealed Sr and Ba concentrations an order of magnitude below Ca (Supplementary Table 19). Reanalysis of available solutions (DI water samples from all experimental times at 150 °C and only 256 hours at 90 °C) confirmed the presence of Sr and afforded Sr-Ca interference corrections (Supplementary Methods 1.3.2). By considering zirconolite mass fractions to normalise Sr releases, they were an order of magnitude higher than Ca (Fig. 4a), whilst using perovskite mass fractions to normalise the Sr releases produced excellent agreement between Ca and Sr releases from the same phase (Fig. 4b). This result was semi-quantitatively confirmed for all experiments using calculated Sr concentrations (model in Supplementary Methods 1.3.3, results in Supplementary Discussion 2.3 and Supplementary Figs. 25–28). As Ca and Sr releases were anticipated as stoichiometric from their geochemical similarity71,72 and previous Synroc-C studies42, with a generally lower solubility for Sr73, this is consistent with Ca releases being predominantly derived from preferential dissolution of the perovskite.

Releases were calculated using zirconolite (bulk sample) elemental mass fractions (a) and perovskite phase elemental mass fractions (b). Normalised releases in both cases were calculated using measured leachate concentrations with the total sample mass and bulk sample Sgeo/V ratio. Uncertainties are given to 1 S.D. propagated from those associated with the ICP-MS measurements (triplicate measurements for Ca, conservatively assigned 20% relative S.D. for Sr standard addition measurements), elemental mass fractions (30 and 20 measurements by EPMA for Ca and Sr, respectively, for zirconolite, and a single STEM-EDS map divided into regions of interest for perovskite) and Sgeo/V ratio (conservatively assigned 15% relative S.D.). The same plots using calculated Sr concentrations for all times and temperatures are presented in Supplementary Figs. 25–28.
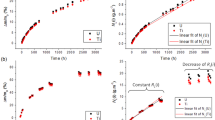
Normalised Ca and Al releases increased as a function of time up to 64 h (Fig. 5a, b), whilst pH(25 °C) remained within uncertainty of the average blank values at each temperature except 256 h at 150 °C (Fig. 5c). Ca concentrations after 256 h were 1646.9 ± 141.9, 1987.8 ± 337.3 and 5151.5 ± 1540.4 ng mL−1 at 40, 90 and 150 °C, respectively. Despite the 64 and 256 h Ca concentrations being within uncertainty at 90 and 150 °C, this was insufficient to demonstrate that a steady state was attained. Assuming that only the perovskite underwent dissolution (total sample mass), concentrations corresponded to leached fractions and altered sample thicknesses of 0.47 ± 0.07% and 2.50 ± 0.54 nm at 40 °C, 0.57 ± 0.15% and 3.02 ± 0.90 nm at 90 °C, and 1.47 ± 0.64% and 7.78 ± 3.57 nm at 150 °C, respectively. Temperature dependence was weak for Ca dissolution, with the 40 and 90 °C and the 90 and 150 °C normalised Ca releases being within uncertainty at all times, excluding 256 h 150 °C.

Normalised Ca and Al releases at 150 °C (a) and 40 and 90 °C (b), and sample leachate and blank pH(25 °C) for the DI water experiments (c). Normalised releases were calculated using measured leachate concentrations with the perovskite elemental mass fractions, the total sample mass and bulk sample Sgeo/V ratio. In a and b, uncertainties are given to 1 S.D. propagated from those associated with the triplicate ICP-MS measurements, elemental mass fractions (a single STEM-EDS map divided into regions of interest) and Sgeo/V ratio (conservatively assigned 15% relative S.D.). In c, uncertainties are given to 1 S.D. of triplicate measurements.
Given the low solubility of many ceramic constituent elements, washouts are an important component of ceramic dissolution analysis. For Ca and Al, acid washout concentrations were below or within uncertainty of the blank concentrations except for the test at 150 °C for 256 h. The washout Ti and Ce concentrations at all temperatures and Zr concentrations at 90 and 150 °C were mostly significant from the blank concentrations with no apparent trends. Additional analysis to determine Sr concentrations by standard addition yielded 26.4 ng mL−1 Sr in the 150 °C 256 h washout. Filtration tests confirmed the absence of Ca-bearing colloids >20 nm at 256 h, but contamination from the inorganic membrane filters prevented determination for Al.
SEM-EDS showed near-spherical Al-bearing precipitates after 256 h at 150 °C (Fig. 6a, b), appearing to have grown on the alumina phase (Fig. 1h), without TiO2 spherules. Small particles with compositions resembling the zirconolite surface (Supplementary Fig. 15) and regions enriched in Sr and slightly depleted in Zr (Sr0.19Zr0.61) were also observed (Fig. 6c, d). The 40 °C particles were qualitatively similar to those at 150 °C but without any apparent Sr phases, whilst a region resembling leached alumina (Al1.54O3.00)(Ca0.07Zr0.08Ce0.03Ti0.20Sr0.01) (no main phase correction) was observed at 90 °C (Fig. 6e, f). No changes were observed by XRD (Supplementary Fig. 36).

SE image (a) and EDS maps of Al and Ti (b), Ca (c) and Sr and Ca (d) for the 256 h, 150 °C, DI water leached sample. Ca and Al (e) and Sr (f) EDS maps of the 256 h, 90 °C, DI water leached sample.
XPS of the pristine and leached (256 h, 150 °C, DI water) powders showed v(Ce3+) decreased from 0.48 to 0.37 ± 0.05 following dissolution (Supplementary Table 10), indicating preferential, non-selective dissolution of Ce3+-perovskite. Note that particle geometry renders these results semi-quantitative74. Following dissolution, Sr and Ba stoichiometry increased from Sr0.05(2)Ba0.02(1) to Sr0.09(1)Ba0.09(1) (Supplementary Table 10), demonstrating dissolution occurred below the XPS sampling depth (6.051 and 3.803 nm respectively67, P = 95%) before surface precipitation; a conservative alteration depth of 7.78 ± 3.57 nm was calculated from the Ca concentrations (perovskite mass fractions, total sample mass). Contrastingly, Ca stoichiometry decreased from Ca0.84(3) to Ca0.67(1). These results qualitatively agree with aqueous solubilities for the increase in stoichiometry of the order Ca < Sr < Ba73. The absence of changes in Ti, Zr and Ce stoichiometry alongside their presence in the acid washouts demonstrates these species rapidly precipitated from solution. Alongside the surface v(Ce3+) decreasing, this suggests rapid Ce precipitation with limited bulk solution transport.
Isotopic controls on dissolution mechanisms
The D2O experiments qualitatively resembled those in DI water. Ca concentrations after 256 h corresponded to leached fractions and altered thicknesses (perovskite mass fractions) of 0.20 ± 0.05% and 1.07 ± 0.31 nm at 40 °C, 0.23 ± 0.10% and 1.24 ± 0.58 nm at 90 °C, and 0.45 ± 0.16% and 2.39 ± 0.92 nm at 150 °C, respectively. Acid washouts contained Ti and Ce concentrations significant from the blanks at all temperatures, and mostly significant Zr and Ca concentrations at 90 and 150 °C (Supplementary Tables 20 and 21).
Calculated Sr normalised releases semi-quantitatively agreed with the preferential dissolution of perovskite (Supplementary Discussion 2.3 and Supplementary Figs. 25–28), and normalised Ca releases observed a weak dependence on both time and temperature (Fig. 7a). However, normalised Ca releases were significantly lower in the D2O experiments (Fig. 7b), with an average D2O/H2O Ca release ratio of 0.43 ± 0.11 across all temperatures (within uncertainty). Contrastingly, the Al releases appeared largely unaffected at 40 and 90 °C and only showed a D2O/H2O ratio significant from unity at 150 °C (0.56 ± 0.41; Supplementary Fig. 29 and Supplementary Table 22); this was likely influenced by high uncertainties at longer times (Fig. 7b). All pH(25 °C) and pD(25 °C) remained within uncertainty across all temperatures (Fig. 7c).

Normalised Ca and Al releases for the D2O experiments (a), D2O/H2O normalised Ca release ratios calculated as the D2O experiment values divided by the corresponding DI water experiment values (b), and solution and blank pD(25 °C) for the D2O experiments (c). Normalised releases were calculated using measured leachate concentrations with the perovskite elemental mass fractions, the total sample mass and bulk sample Sgeo/V ratio. In a, uncertainties are given to 1 S.D. propagated from those associated with the triplicate ICP-MS measurements, elemental mass fractions (a single STEM-EDS map divided into regions of interest) and Sgeo/V ratio (conservatively assigned 15% relative S.D.). Uncertainties in b were propagated from those in a and are given to 1 S.D. In c, uncertainties are given to 1 S.D. of the triplicate pD(25 °C) measurements. Note the absence of 90 °C pD(25 °C) measurements at 256 h due to a technical fault.
Kinetic isotopic fractionation during diffusion is described by Eq. (1)75: the ratio of diffusivities, Di, of the isotopologues, i, HxOx-2 and DxOx-2 (x = 1–3) and H+ and D+ is a function of the ratio of their masses, mi, and an empirical exponent β. A maximum β = 0.5 for self-diffusion is achieved for an ideal gas at low pressure76,77, yielding minimum diffusivities for D+, OD− and D3O+ at 70.74, 97.17 and 92.90% of H+, OH− and H3O+, respectively. However, where diffusivities are not apparent (i.e. β is not affected by chemical or microstructural constraints), β is generally lower than 0.578,79,80,81,82,83,84. The measured D2O/H2O Ca release ratios corresponded to β = 1.26 ± 0.29, 8.23 ± 1.87 and 5.92 ± 1.34 (1 S.D.) for the D+/H+, D2O/H2O and D3O+/H3O+ mass ratios, respectively, showing these results are inconsistent with an isotope effect during the rate-limiting intrinsic (non-apparent) diffusion of reactive water species isotopologues.
The lower vibrational zero point energy, as a function of reduced mass, of the O-D bond results in a larger dissociation energy than O-H85,86. Following transition state theory, this larger dissociation activation energy is associated with kinetic isotopic fractionation as a function of reduced masses for motion along the reaction coordinate and the activated complex and dissociated product fractionation factors87,88,89,90,91. D2O/H2O Na release ratios of ~0.71 for silicate glasses have been attributed to rate-limiting water dissociation during ion-exchange92, neglecting effects on fractionation factors beyond mass. The magnitude of the D2O/H2O Ca concentration ratios measured here evidence the solvent participating in Ca release but alone cannot unequivocally elucidate the mechanism. The absence of an isotope effect at 40 and 90 °C for Al is consistent with release from a distinct phase or through a distinct mechanism to Ca, with a deviation from a D2O/H2O Al release ratio of unity at 150 °C indicating a potential change in these processes at high temperature.
Linear regressions of Ca and Al releases against t0.5 showed t−0.5 rate dependence (taken as R2 > 0.95) was not reproducible between the DI water and D2O experiments (Supplementary Table 22). In turn, these results do not evidence the diffusion of reactive species, assuming relatively negligible matrix dissolution (predominantly Ti and Zr)42,93.
An Arrhenius plot of the 2-hour normalised Ca and Al release rates demonstrates their weak temperature dependence (Supplementary Fig. 30). Analysis of the slope yielded activation energies for Ca and Al release of 6 ± 1 and 29 ± 13 kJ mol−1, respectively, (R2 > 0.980) for the DI water experiments, and 4 ± 2 and 27 ± 10 kJ mol−1, respectively, (R2 > 0.910) for the D2O experiments. These values broadly agree with those of 8 and 20 kJ mol-1 for Ca and Ti/Ce release, respectively, from Ca0.85ZrCe0.15Ti1.70Al0.30O7.00 zirconolite in 1 M HCl28, but are lower than the 15 to 30 kJ mol−1 for Ca from Ca0.8Nd0.2ZrTi1.8Al0.2O7.094, natural zirconolite (age of 550 Ma)95 and Synroc-C42,96 in DI water. Activation energies <9 kJ mol−1 are consistent with the transport of reactive water species and reacted sample species in solution to and from the reaction front, respectively, presenting a rate-limiting mechanism97,98, rather than diffusion through a hydrated solid phase28. Contrastingly, a surface-controlled reaction for minerals or bulk diffusion would exceed 40 kJ mol−197,98, showing Al releases are rate-limited by a mixed-reaction of solution transport and surface-controlled dissolution. This suggests differences in the rate-limiting mechanisms of Ca and Al release.
Limited surface alteration without Ca-H exchange
Analysis of the O 1 s XPS spectra of the leached sample (256 h, 150 °C, DI water) showed a high binding energy tail (4.85 ± 1.46% O) due to water species or organics (Fig. 8b)63,99,100,101, corresponding to 0.35 ± 0.01 mol H2O/H3O+ mol−1 leached zirconolite (Supplementary Table 10). This O was excluded from the zirconolite stoichiometry. Whilst the O 1 s defect/OH−/CO32− peak is correlated with reduction in CeO247,51,52, this effect was not observed here (simple linear regression R2 = 0.319, r = 0.56). Applying the same O 1s peak model to the pristine powders and monoliths yielded 0.00% and 0.51 ± 0.15% water/organic O, respectively. C 1 s peak models102,103 showed higher C=O and C-O concentrations for the monoliths (5.94 ± 0.64% and 15.12 ± 1.53%, respectively) than the powders (2.56 ± 0.60% and 6.82 ± 1.40%, respectively) (Fig. 8a). This confirmed the monolith O 1s water/organic peak was attributable to adsorbed atmospheric water and acetone contamination from pre-cleaning (powders were not pre-cleaned) and the leached powder peak was attributable to water species.

C 1s (a) and O 1s (b) XPS spectra for the pristine monolith, pristine powder and leached (256 h, 150 °C, DI water) powder surfaces. The original spectra and fits are given in black and red, respectively, with other colours used to denote the peak models100,102. Vertical lines denote the average position (across the three samples) of each peak for the peak models, with labels giving the source of the peak and its position (eV). Further details on the fitted peak models, their quantification and models of leached layer thicknesses are provided in Supplementary Methods 1.1 and Supplementary Discussion 2.1.
Water species in the leached powders were attributable to a hydrated altered layer or hydrated precipitates. To study the former, a leached layer thickness was estimated from the Ca 2p areas66 assuming no attenuation from precipitates and that a flat, uniform layer of complete Ca2+ ⇌ 2H3O+ exchange covered the sample (further details given in Supplementary Methods 1.1.2). This yielded a leached layer thickness of 0.4 ± 0.3 nm (75% RSD). Note that particle geometry effects render this estimation semi-quantitative74,104. Assuming this leached layer uniformly covered spherical particles yielded 7.2 ± 5.4 × 1016 H atoms per gram of sample (NH g−1). Further, assuming a perovskite density of 3.91 g cm−3 and that only the perovskite phase underwent dissolution alongside negligible zirconolite dissolution, this value yields an estimated leached layer thickness of 4.7 ± 3.5 nm localised to the perovskite phase. These values are significantly lower than the 1.5 ± 0.4 × 1019 NH g−1 estimated for ion-exchange from the Ca and Sr concentrations (256 h, 150 °C, DI water), and thicknesses of 12.6 ± 5.7 nm and 139.2 ± 63.8 nm assuming bulk sample (total mass, zirconolite mass fractions) and solely perovskite phase dissolution (maximum estimated perovskite mass, perovskite mass fractions), respectively, estimated from the Ca concentrations alone. In turn, this shows that ion-exchange and the formation of a hydrated layer through selective leaching was not responsible for dissolution.
2H MAS-NMR spectra of the 256 hour D2O samples showed no measurable signal (Supplementary Fig. 34). Assuming Ca and Sr exchange for 2D3O+ or 2D2O and condensation reactions are not significant, the estimated number of D atoms per g of sample, ND g−1, from the 90 and 150 °C Ca and Sr ICP-MS concentrations (3.3 ± 0.8 × 1018 and 7.2 ± 1.5 × 1018 ND g−1, respectively) exceeded the NMR detection limits (2.0 × 1018 and 3.6 × 1018 ND g−1, respectively), but were lower at 40 °C. These results agreed with the estimated NH g−1 from XPS in showing ion-exchange did not establish a protonated/deuterated altered layer. The low 40 °C ND g−1 and the high 1H background (1021 NH g−1) prevented investigations at 40 °C and by 1H MAS-NMR, respectively (Supplementary Table 23 and Supplementary Figs. 31–33).
TEM bright field (BF) and STEM BF and high-angle annular dark-field (HAADF) images of the Cr coating-zirconolite interface for the leached (256 h, 150 °C, DI water) sample lamella revealed no signs of alteration (Fig. 9a–c). Similarly, STEM-EDS maps averaged to provide elemental intensity profiles showed no clear compositional changes at the surface, with Ca intensities anti-correlated with Cr (Fig. 9d–g). To investigate to a first order whether Ca and Sr were released preferentially, the depths of the elemental interfaces were estimated from narrow STEM-EDS maps (0.68 nm pixel−1 resolution) as the distances at which the intensities fell below 90% of the zirconolite average (≥16.22 nm) for two consecutive pixels. Noise precluded consideration of interface topography105,106.

TEM bright-field (BF) (a), STEM high-angle annular dark-field (HAADF) (b) and STEM BF (c) images of the zirconolite-Cr coating interface, and a narrow area mapped by STEM-EDS across this interface (d) and the resulting elemental normalised intensity profiles (e–g) obtained by averaging (d) across the x-dimension and plotting along the y-dimension. Intensity profiles are plotted as steps, with each step representing the resolution of the analysis (0.46 nm2 pixel−1). The magenta box in c represents the region mapped in d.
Depths for Ca (14.2 nm) and Sr (13.5 nm) resembled Al, Ce, O, Ti and Zr (from 12.2 to 15.5 nm) with a 2.7 nm Ca-Cr interface width, showing no evidence of a Ca/Sr-depleted region. Note these profiles were significantly affected by noise, such that a depleted layer of the order 100 nm would likely be undetectable at this resolution. Further, these values are a function of the resolution, interface topography and the number of pixels normal to the interface averaged to yield the profiles106. No compositional effects were observed at zirconolite-zirconolite grain boundaries, potentially due to significant noise at high resolution (0.10 nm2 pixel-1). The broad elemental gradients at the zirconolite-perovskite interface are attributable to a topographical artefact (Supplementary Figs. 19–21).
Discussion
Characterisation of the HIP ceramic revealed zirconolite 3T, alumina, ceria and perovskite phases. The absence of alumina XRD reflections suggests this phase is poorly crystalline, whilst the absence of perovskite and ceria reflections are attributable to the highly polycrystalline nature and low concentration, respectively, of these phases. These phases arose due to the unintended reduction of Ce: Ce3+ preferentially partitioned into perovskite43, zirconolite Ce3+ preferentially partitioned onto the Ca site13,60,107, buffer Ti partitioned onto the Zr site28 and redundant charge-compensating Al was expelled. This is consistent with the observed excess Ti, lower SEM-EDS Ce recoveries than by EPMA and a higher Al concentration than estimated from milling media mass loss. The potential contribution of alumina to the zirconolite stoichiometry cannot be excluded.
Agreement in the normalised Ca and Sr releases using perovskite mass fractions and significant Ca stoichiometry and v(Ce3+) decreases at the surface showed the perovskite phase dominated the dissolution kinetics. Ti and Zr concentrations in the washouts evidenced their dissolution and transport. Further, the apparent absence of these species from surface precipitates, a negligible change in their surface stoichiometry, and a decrease in surface v(Ce3+) following dissolution suggests precipitation was highly spatially coupled to dissolution, proceeding at a locally supersaturated perovskite-solution interface. A conservative maximum leached fraction of 6.4 ± 5.5% Ca in solution arising from zirconolite dissolution was calculated using the largest Zr concentration measured in the washouts, neglecting the minor Zr content of the perovskite (Zr0.06(1))108.
XPS and 2H MAS-NMR showed no evidence of a hydrated altered layer formed through ion-exchange, in agreement with STEM-EDS of the zirconolite grains. The absence of a reproducible t-0.5 release rate dependence showed diffusion was not significant through a hydrated relict perovskite structure or a precipitated layer. Water concentrations measured by XPS may be consistent with hydrolysis or trace hydrated precipitate concentrations. Note, XPS and TEM took place only on the 150 °C samples, but the observed linear temperature dependence suggests these results may broadly extend with caution to lower temperatures.
Lower D2O Ca releases at all temperatures are attributable to an isotope effect through either (1) rate-limiting diffusion wherein apparent diffusivities are disproportionately affected by physical or chemical processes (β > > 0.5 in Eq. (1), e.g. refs. 109,110), or (2) congruent dissolution through hydrolysis in the absence of diffusion, wherein an O-H/D bond participates in the activated complex. Both mechanisms may be affected by an H2O-D2O solvent isotope effect111. Comparisons with literature data provide insight into distinguishing between these hypotheses: the absence of a measurable isotope effect for REE pyrochlores (pH/pD 2, 90 °C) was attributed to solubility controls112,113, inconsistent with the view of β << 0.5 for self-diffusion114. Whilst H2O-D2O experiments on obsidian (850 °C, 6 hours)115 and borosilicate glass (SON68, pH/pD 3.2, 50 °C, 2 h)116 yielded β of 9.55 and 11.56, respectively, 150 °C obsidian studies yielded a β of 0.006117. This suggests β during apparent diffusion is dependent upon altered layer transport properties118. Contrastingly, ~2.7 times slower rates of Ca3SiO5 hydration in D2O were attributed to an isotope effect during hydrolysis119, in agreement with that measured here for Ca (2.6 ± 1.3). The β > 0.5 and absences of measurable ion-exchange, t-0.5 rate dependence or a hydrated altered layer derived from ion-exchange were consistent with an isotope effect associated with congruent dissolution of the perovskite phase through hydrolysis.
The low activation energy for Ca release showed it was transport-limited: the small perovskite grains are postulated to spatially limit the transport of reactive water species and leached species to and from the perovskite reaction front, respectively. This local supersaturation promotes spatially coupled precipitation from a bulk solution undersaturated in Ti and Zr, potentially further limiting transport. STEM-EDS showed no evidence of such an effect along grain boundaries for zirconolite. It is emphasised this refers to the transport of species in solution and not limiting transport to diffusion through a precipitated layer. Ultimately, these results support the perovskite phase undergoing preferential dissolution through a transport-limited congruent dissolution-precipitation mechanism, without diffusion into the perovskite or through a precipitated layer.
These results are inconsistent with the formation of a Ca-depleted relict layer through selective leaching24. Rather, they support an interface-coupled dissolution-precipitation reaction, expressed for perovskite29,120,121 and zirconolite28 in Eqs. (2) and (3), respectively. Note that these reactions consider the precipitation of hydroxides, as TiO2 polymorphs are anticipated to be undersaturated in the conditions considered herein and may be formed following the kinetically limited decomposition of Ti(OH)x4-x20,21,28,120,122. These highly coupled dissolution-precipitation reactions may lead to the formation of a precipitated layer which covers the perovskite-solution interface24. At longer times, such a layer may lead to changes in the dominant dissolution mechanism.
The absence of a measurable isotope effect at 40 and 90 °C and a higher activation energy for Al dissolution are consistent with a release process distinct from Ca. This may be attributed to differences in the activated complex during hydrolysis, such as a smaller secondary isotope effect, for Al. The D2O/H2O Al release ratio significant from unity at 150 °C remains somewhat enigmatic, but this may reflect marked changes in dissolution-precipitation kinetics, the dominant phase for Al release, an artefact from large uncertainties, or a change in the hydrolysis activated complex at higher temperature; these results cannot rank these hypotheses. Preferential dissolution of an alumina phase relative to the zirconolite and Al precipitates at the zirconolite surface suggest alumina dissolution contributed to the measured Al concentrations, consistent with observations for “Al-rich oxides” in Synroc-C123,124. In turn, the absence of sparingly soluble species from alumina leads to a mixed reaction controlling kinetics. However, the relative contributions of perovskite (Al0.11(2)) and alumina dissolution to the Al releases cannot be resolved here.
These results suggest variations in reported zirconolite dissolution rates may be partially underpinned by variations in undetected minor perovskite phase concentration. Variations in crystal dissolution rates at short times are known to arise due to intrinsic heterogeneities in surface site reactivity distributions as a function of microstructure125,126. For polycrystalline ceramics, these heterogeneous distributions correspond to the presence of high-energy sites, such as intrinsic defects (e.g., point defects and dislocations) and oxygen vacancies at crystal surfaces and grain boundaries127,128,129, associated with an overall increased free energy of formation130. Such defects are known to arise from mechanical deformation during sample preparation as a function of particle size or monolith surface finish127,131. Further, changes in reactive surface area can be present arising from adhering fines27,131, surface roughness132 and sample porosity131,133, also causing variations in short-term dissolution rates. In particular, difficulties remain in meaningfully representing these effects on heterogeneous surface energy distributions using geometric or BET surface areas125.
For perovskite phases, increasing mechanical deformation, increasing surface roughness and decreasing particle size have been shown to increase Ca release rates at short times due to changes in high-energy site distributions131,132,133. Similarly, for Ce-doped zirconolites, whilst Nikoloski et al. observed Ca release rates decreased with increasing particle size27, contrastingly excellent agreement between powder and monolith Ca release rates was observed by Pöml et al.28. These results from the literature highlight the difficulties associated with identifying and predicting changes in high-energy site distributions as a function of sample preparation route. Alongside these microstructural and reactive surface area effects, variations in undetected perovskite concentration would directly cause variations in Ca release rates, with such rate variations feasibly persisting until all readily accessible perovskite is altered. However, further work is required to rank the relative contributions of these effects to observed literature dissolution rate variations.
Overall, these results demonstrate that dissolution kinetics were predominantly driven by secondary phase perovskite and alumina dissolution alongside comparatively negligible zirconolite dissolution. This perovskite was undetectable by XRD and, given its small grain size and low concentration, was only detected through the unintended presence of Sr and Ba; it would otherwise be near-undetectable by SEM-EDS. Therefore, substantial variations in literature dissolution rates may be partially explained as a function of undetected trace perovskite concentration, which is in turn a function of composition and synthesis conditions.
Methods
Sample synthesis, preparation and density
A zirconolite of nominal composition Ca0.8Zr0.9Ce0.3Ti1.6Al0.4O7.0 was synthesised as a single 577 g batch by GeoRoc International (UK). A buffer of 10% additional TiO2 above that required for zirconolite formation was included with the aim of inhibiting perovskite formation otherwise associated with rare earth substitution into zirconolite21,29,134. Target masses of reagents powders (CaTiO3 99.8%; TiO2 99.9%; ZrO2 99.9% excluding ~2% HfO2; Al2O3 99.95%; and CeO2 99.99%, Jinan Boss Chemical Industry, median particle size ≤5 µm) were wet milled using alumina media in a high alumina jar with 800 mL DI water for approximately 16 hours at 75 rotations per minute (ESSA BR06 Bottle Roller). Media mass loss of ~3 g was measured, corresponding to ~109.4% of the targeted alumina in the precursors. Milled reagents were dried for 16 hours at 120 °C before being passed through a 0.7 mm stainless steel sieve into a 304 stainless steel HIP canister (60.7 mm internal diameter, 139.2 mm internal length, 1.4 mm wall thickness), compacting the powder uniaxially in steps. Canisters were briefly dried at 120 °C and subjected to a vacuum-assisted bake-out to remove volatile species prior to canister sealing: ambient temperature to 450 °C over 8 h with a pressure drop to ~6.67 Pa over 16 h, 450–600 °C and ~46.66 Pa over 8 hours, held at 600 °C with pressure dropping to ~21.33 Pa over 16 h, and then a final heating to 650 °C and 11.33 Pa over 8 h. However, a malfunction with the thermocouple yielded a final temperature of ~800 °C during evacuation. The canisters were then cooled to ambient temperature and sealed (autogenous TIG welding and standard GeoRoc International canister sealing parameters) under vacuum at 5.47 Pa. Canisters were subjected to HIP at 1320 °C, 103 MPa for 2 h, which comprised a 4 h temperature ramp and controlled pressure ramps starting at 850 and 420 °C during heating and cooling, respectively. Mass losses of 3.5 and 0.9 g were reported during evacuation and HIP synthesis, respectively, with the former being consistent with prior loss on ignition tests of the precursors from 120 to 1000 °C. Samples were then removed by coring with a 35 mm diameter water-cooled diamond coring drill, intentionally excluding the canister-sample interaction zone to produce a 31 mm diameter, 268.54 g core.
The core was broken into smaller pieces with a hammer and checked for magnetic particles. A density of 4.63 ± 0.05 g cm−3 (1 S.D., n = 3) was measured by Archimedes’ principle using ambient temperature DI water and a calibrated (E2 class standard, OIML) analytical balance with weigh-below hook (Pioneer, Ohaus). The sample was ground using a planetary ball mill (PBM-V-2L-B, Deco) with zirconia media and jars, sieved to 38–75 µm, washed using absolute ethanol (>99.8%, anhydrous, Sigma Aldrich) without DI water following the ASTM PCT washing procedure31, and dried at 90 °C for 16 h. Secondary electron images confirmed the absence of adhering fines on the pristine particles (Supplementary Fig. 11), whilst backscattered electron images and energy-dispersive X-ray spectroscopy maps were used to confirm the presence of alumina, ceria and Zr-poor phases at the pristine particle surfaces (Supplementary Figs. 12–14 and Supplementary Tables 11–13).
Dissolution experiments
Dissolution experiments were started following the ASTM PCT-B procedure31, using 0.153 g of washed 38 to 75 µm zirconolite powder with 3.5 mL DI water (18.2 MΩ·cm, Milli-Q system, Millipore) or D2O leachant (99.9% D atoms, Sigma Aldrich) for a geometric surface area to volume, Sgeo/V, ratio of 1000 m−1. The as-received D2O (pD(25°C) > 8.5) was single distilled in-house using a PFA elbow still (Savillex) at 90 °C under Ar atmosphere ( < 5 µmol mol−1 O2, <0.1 µmol mol−1 H2O) to remove contaminants (further details in Supplementary Methods 1.3.1 and Supplementary Table 15). Experiments used 5 mL 316 stainless steel vessels with PTFE inserts and lids which were maintained at 40.0, 90.0 and 150.0 ± 0.1 °C (QBH2 dry block heater, Grant Instruments) for 2, 8, 16, 64 and 256 hours. All experiments were conducted in triplicate alongside duplicate blanks. At the end of each experiment, leachates were passed through 0.45 µm nylon syringe filters (Puradisc, Whatman) and acidified to 1% HNO3 using concentrated (~15.9 M) double distilled HNO3 (in-house, Teflon sub-boiling still). To test for colloids, unacidified aliquots of the 256 hour samples were also passed through 0.02 µm inorganic membrane syringe filters (Anotop, Whatman). Leachate pH(25 °C) or pH*(25 °C) was measured (LE438 pH electrode and FE20 FiveEasy pH metre, Mettler Toledo) using a dry block heater (Grant Instruments) to maintain samples at 25.0 ± 0.1 °C, where pH* is the measured operational pH of a D2O-matrix solution and pD was calculated as pH* + 0.40135,136. Solid samples were recovered into 0.5 mL centrifuge tubes using 2 mL fresh leachant, which were immediately sealed, transferred to an Ar atmosphere glovebox and dried at 50 °C for 48 h. Samples were kept sealed under Ar atmosphere until required. All vessels were then rinsed with DI water before 3.6 mL 1 M double distilled HNO3 was added to each vessel and heated to 90 °C for 24 h as acid washouts.
Solution analysis and data treatment
Inductively coupled plasma mass spectrometry (ICP-MS; Nexion 350D, Perkin Elmer) was used to measure the concentrations of zirconolite species (27Al, 43Ca, 44Ca, 47Ti, 90Zr and 140Ce) and 208Pb using matrix-matched calibration standards (CPI International; five standards and a blank), a Rh, In and Re internal standard (10 ng mL−1) introduced through a mixing junction, and a BCR-2 digest (USGS) and SPS-SW2 (Spectrapure Standards) as secondary standards. Available samples (256 h at 90 °C and all times at 150 °C for the DI water experiments) were later reanalysed for both zirconolite species and identified contaminants (24Mg, 86Sr, 88Sr and137Ba) through a Sr standard addition procedure (further details in Supplementary Methods 1.3, Supplementary Discussion 2.3 and Supplementary Figs. 22–28). When analysing the D2O leachant samples, the standards, acid blanks and acid washes were prepared using single-distilled D2O. Drift remained within 10% and 20% for the DI water and D2O samples, respectively, with the latter being affected by matrix effects from solutions which contained HNO3. Plasma robustness (CeO/Ce) and sensitivity (Ce) were 2.8% and 450,000 CPS ppb−1, respectively. Secondary standard recoveries were within 15% of nominal for all species except Zr for BCR-2 which was within 30%. The limits of detection and leachant blank concentrations are given in Supplementary Tables 16–18.
Notably, a significant interference of Sr on Ca was later observed: ICP-MS of all solutions first proceeded without Sr as an analyte, but semi-quantitative mass scans later confirmed the presence of notable Sr and Ba concentrations in the 256 h, 150 °C DI water sample. Subsequent tests demonstrated a significant interference of Sr on Ca associated with the low sensitivity of Ca (0.72 and 12.14 CPS ppb−1 for 43Ca and 44Ca, respectively) relative to Sr (156 and 1247 CPS ppb−1 for 86Sr and 88Sr, respectively) and the production of Sr++ (an average value of 2.7% for 86Sr on 43Ca and 88Sr on 44Ca was used in the corrections) under the conditions used. This resulted in Ca concentrations being artificially increased where Sr was not measured. As such, the available remaining solutions (256 h 90 °C and all times 150 °C, DI water) were analysed for Sr through a standard addition methodology and interferences on Ca further quantified through a calibration curve methodology. Noting a linear relationship between the 43Ca-44Ca concentration difference and the interference-corrected 44Ca concentration, simple linear regression parameters relating these variables were established using the concentrations from the standard addition ICP-MS session (where Sr was measured) and were used to correct the Ca concentrations for Sr interferences in the initial sessions (where Sr was not measured). The same approach was then used to semi-quantitatively calculate Sr concentrations from the 43Ca-44Ca concentration difference for sessions wherein Sr was not measured. Further details on the experiments used to quantify the Sr interferences, first-principles models which express the uncorrected 43Ca-44Ca concentration difference as linear functions of the corrected 44Ca concentration and Sr concentration, the corrections on the Ca concentrations by linear regression and semi-quantitative calculations of Sr concentrations by linear regression are given in Supplementary Methods 1.3.1 and 1.3.2, Supplementary Discussion 2.3 and Supplementary Figs. 22–28.
Measured sample and washout concentrations corrected for mass loss and blank concentrations, Ci (g L−1) were normalised to the Sgeo/V ratio (m2 L−1) and the elemental mass fractions, fi, for element i to yield normalised elemental releases, NLi (g m−2) (Eq. (4)).
NLi uncertainties were propagated from those of the concentration measurements (triplicate RSD, 20% for Sr standard additions), elemental mass fractions (analysis RSD, see later sections) and Sgeo/V ratio (±15%) (Eq. (5))137. Note that NLCa required an additional uncertainty term (concentration dependent) for the Sr interference corrections by linear regression (Supplementary Methods 1.3.2).
Equivalent thicknesses of altered sample, ei (µm), were then calculated from the leached mass fractions, LFi, and the mean geometric particle diameter, R0 (56.5 µm), following a Shrinking Core model approach (Eq. (6))138; where m (g) and V (mL) are the mass of sample leached and initial volume of leachant, respectively (Ci is already mass-loss corrected). CiV represents the mass of i leached, which was also used to calculate the number of atoms of i leached g-1 of sample. These LFi and ei calculations were repeated using fi from the zirconolite and perovskite phase compositions, in addition to assuming m represented the mass of sample and the mass of the perovskite phase alone (calculated from Sr at.% mass balance in the sample). NLi were solely calculated using the mass of sample and the experimental Sgeo/V ratio but with both the zirconolite (bulk sample) and perovskite phase fi, as the calculated perovskite masses (1) represented a maximum and (2) would introduce additional uncertainties into the NLi.
Temperature effects were also studied through calculating the activation energies, Ea (kJ mol−1), for the release of Ca and Al through the Arrhenius equation (Eq. (7)), which relates their 2-hour release rates, ri (g m−2 s−1), to Ea through the ideal gas constant, R (J K−1 mol−1), and temperature, T (K); Ea is then calculated as R times the gradient of a simple linear regression of ln(ri) against 103 T−1 for each element139.
XRD
The crystallinity of pristine zirconolite and one leached (256 h) sample from each triplicate at each temperature in each leachant were investigated by Cu Kα X-ray powder diffraction (XRD) (1.5406 Å; D8 Advance, Bruker) both with and without standard Si powder. Approximately 20 mg of sample was analysed on a Si low background plate from 10 to 90° (2θ) in 5475 steps of 0.015°, 0.5 s step−1 (47 min). Repeat analyses showed that similar quality diffractograms were obtained using a glass slide sample holder, 11638 steps of 0.007°, 1.0 s step−1 from 5 to 90° (2θ) (201 min). Empty sample holders with different cleaning methodologies were also analysed to assess the background. Pawley refinements used TOPAS (Bruker) software with literature data13,29. To confirm these results, the pristine sample was analysed on a Si low background sample holder by Mo Kα XRD (0.7093 Å; D8 Advance, Bruker) from 5 to 60° (2θ) in 5500 steps of 0.010°, 0.4 s step−1 (37 min).
EPMA
Solid zirconolite pieces of approximately 1 cm2 were mounted in resin (EpoFix, Struers), successively polished to 3 µm with diamond pastes and then carbon coated. The zirconolite composition was analysed by electron probe microanalysis (EPMA; Cameca SX100) using a 20 kV accelerating voltage and 40 nA beam current (1 µm beam size) for Al (corundum), Ca (diopside), Ti (rutile), Zr (zircon), Ce (CeAl2) and Hf (hafnium) in 10 spots, and a 20 kV accelerating voltage and 10 nA beam current (10 µm beam size) for Mg (olivine), Ca and Si (diopside), Al (corundum), Zr (zircon), Sr (celestine) and Ba (benitoite) in 20 spots. The significant overlap between the Ba Lα and Ti Kα X-rays introduce significant uncertainties into trace Ba concentrations when measured in the presence of wt.% Ti. Ba concentrations were estimated through using the Sr concentration measured by EPMA with the Ba/Sr ratio determined by X-ray photoelectron spectroscopy (XPS).
SEM-EDS
Scanning electron microscopy (SEM) imaging and energy dispersive spectroscopy (EDS) analysis first used a Quanta-650F (FEI) SEM with field emission gun source. Backscattered electron (BSE) images and EDS maps (20–30 min acquisition time) were acquired in triplicate for the polished and carbon-coated samples (see Section 4.5) using a centric backscatter detector (CBS) and XFlash 6|30 silicon drift detector (Bruker), respectively, under high vacuum with a 20 kV accelerating voltage, spot size of 4, working distance of 13 mm and 50 µm aperture (EDS resolution of 0.36 µm pixel−1). Pristine and leached (256 hours, DI water) powders were adhered to C tape on Al stubs and analysed uncoated. Triplicate secondary electron (SE) images were acquired using an Everhart-Thornley detector (ETD) under high vacuum, 3 kV accelerating voltage, spot size of 1, working distance of 5 mm and 30 µm aperture. Duplicate BSE images and EDS maps (15 to 30 minute acquisition time) were acquired using the same settings as the polished samples but under low vacuum to reduce charging effects.
Complementary BSE images and EDS maps were acquired for a single leached sample (256 h, 150 °C, DI water) using a TESCAN Vega SEM under high vacuum. Images were acquired using working distances of 15–5 mm, accelerating voltages of 15 or 5 kV, and beam currents of 100–300 pA. EDS maps were acquired using a 5 or 15 kV accelerating voltage at a 9 or 15 mm working distance, depending upon the element and sample particle, using a Bruker FlatQuad detector. For EDS maps acquired using a 5 kV electron beam, maps were collected in a windowless configuration to improve the signals of light elements such as Al.
EDS maps were analysed using ESPRIT 2 (Bruker). Where possible, the concentrations measured for the secondary phases were corrected for the main phase composition after selecting an element for the corrections (Ca, Ti, Zr or Ce) based upon the secondary phase stoichiometry (further details in Supplementary Methods 1.2.1, Supplementary Discussion 2.2 and Supplementary Table 13). The area fractions of each phase in the polished samples were estimated using the particle analysis tool in ImageJ (NIH) through thresholding the single element EDS maps and BSE images; note the Sr-rich phase area fraction could not be reliably resolved using this approach and instead relied on mass balance (further details in Supplementary Methods 1.2.2 and Supplementary Figs. 16 and 17).
TEM
Leached samples (256 h, 150 °C, DI water) were affixed to Al stubs using C tape and sputtered with a thin layer of Cr to protect the sample from electron beam damage. Two lamellae were prepared using a Helios NanoLab Dual Beam focussed ion beam (FIB)-SEM (FEI): protective Pt layers of approximately 1 and 2 µm were first deposited on a region of interest with the electron beam and then the Ga+ beam, respectively, before milling a trench either side of the Pt layers with the Ga+ beam, affixing the sample to an Omniprobe needle, and then affixing the lamella to a Cu transmission electron microscopy (TEM) grid. The lamellae were thinned to electron transparency (<200 nm) using a Ga+ beam from 30 to 5 kV.
Lamellae were loaded into a Tecnai Osiris TEM (FEI) and analysed using a primary beam of 200 kV with a 0.4 nA beam current. Samples were imaged by TEM (bright field (BF); UltraScan1000XP, Gatan) and scanning TEM (STEM; BF, dark field (DF) and high-angle annular dark field (HAADF); Tecnai Imaging system, FEI), and underwent STEM-EDS analysis (Super-X system, FEI) as narrow-area scans across the sample surface and grain boundaries (0.3 to 5.8 nm pixel−1) and maps of the whole lamella and specific regions of interest. Individual grains and the Pt coating were analysed by selected area electron diffraction (SAED), but a zone axis for these samples could not be successfully found. TEM images and STEM images and EDS narrow-area scans were analysed using ImageJ with Gatan Microscopy Suite and HyperSpy (Python library), respectively. STEM-EDS maps were divided into regions of interest representing individual grains using ESPRIT 2 (Bruker) and the resulting spectra analysed using HyperSpy. Given the results of the STEM-EDS narrow-area scans, analysis of the grains near the surface were considered representative of those that had undergone dissolution, whilst those deeper in the sample (approximately 1 µm) were considered pristine.
XPS
XPS analysis first took place on pristine zirconolite monoliths followed by pristine and 256 h 150 °C leached powders. Triplicate monoliths were sonicated in acetone for 5 minutes before drying at 90 °C for 16 h, whilst powders were not further treated. Samples were secured in place with Cu tape. Surface sputtering pre-cleaning did not take place. Analysis took place under ultra-high vacuum (UHV) in constant analyser energy (CAE) mode at ambient temperature using an Escalab 250Xi XPS with a monochromatic Al Kα source (1486.7 eV), with a flood gun used to minimise charging effects. Squares of 0.81 mm2 were analysed with a pass energy of 20 (powders) or 30 eV (monoliths) for narrow scans with a step size of 0.05 eV and 100 eV for survey spectra with a step size of 0.50 eV. Monolith surfaces were analysed in triplicate (C 1 s (5 scans), Ce 3d (10 scans), O 1 s (5 scans), Ti 2p (5 scans) and survey spectra), before then Ar+ sputtering at a single location at 200 eV for 5, 30, 55 and 80 s (total time) and another single location at 3 keV for 30 s (Ce 3d (25 scans), Ti 2p (5 scans) and survey spectra). Powder surfaces were analysed without Ar+ sputtering at a single random point for Al 2p (20 scans), C 1 s (10 scans), Ce 3d (15 scans), O 1 s (5 scans), Ti 2p (10 scans) and Zr 3d (10 scans) and triplicate random points for Ca 2p + Zr 3p (10 scans) and survey spectra.
As Ce4+ is readily reduced during XPS analysis44,49,54, reduction during analysis and sputtering was quantified through synthesising duplicate CeO2 pellets as controls. CeO2 powder (99.9%, Sigma Aldrich) was pressed into 5 mm diameter pellets and sintered in air at 1600 °C for 6 h, before cooling to ambient temperature in the furnace over 16 h. CeO2 pellet analysis took place on Thermo Nexsa XPS with monochromatic Al Kα source (1486.7 eV), ambient temperature under UHV in CAE mode, using a flood gun to mimimise charging. Each pellet was analysed in triplicate at random points for Ce 3d (duplicate 1 scan, single 8 scan spectra) with 20 eV pass energy and 0.05 eV step size at the pellet surface, after Ar+ sputtering for 5, 30, 55 and 80 s (sequentially) at 500 eV, and after Ar+ sputtering another location for 30 s at 5 keV. Survey spectra were acquired as single scans with a pass energy and step size of 160 and 0.50 eV, respectively. Note that the Ar+ sputtering energy was higher for the CeO2 pellets than the zirconolite samples to account for the transition metal contents of the latter potentially enhancing reduction during both analysis (beam and vacuum effects) and sputtering44,53,54, but no calculations underpinned the exact choice of higher energies for CeO2.
All spectra were analysed using CasaXPS software140. Zirconolite surface spectra were charge referenced to adventitious aliphatic carbon (C-C) at 248.80 eV, whilst Ar+ sputtered spectra were referenced to the Ce 3d u‴ peak at 916.1 eV (zirconolites, consistent with C-C) or 916.7 eV (CeO2, based on literature values44) or the u′ peak at 903.10 eV where the u‴ peak was absent. Survey spectra were analysed through integration (linear background for Ba 3d, Shirley-type for all others), and narrow scan spectra analysed using peak models established from combinations of literature data and theoretical constraints14,45,46,47,49,51,52,141,142,143. Calculated XPS stoichiometry did not consider O associated with the water or organic species O 1s peak in the 7.00 O of the zirconolite, which represented only lattice and defect O. This exclusion from the pristine monolith stoichiometry took place as this peak may also represent adventitious C contamination (acetone used in cleaning or other atmospheric contaminants) but was assigned a separate water stoichiometry for the leached powder assuming that all O was associated with H2O. The thicknesses of an oxidised Ce4+ film on a Ce3+ substrate and the thickness of a hydrated layer on a pristine substrate for the monoliths and powders, respectively, were approximated through modifications to the Hill equation49,64,65,66,67,68,144,145. Further details on the fitted peak models and the derivation and application of the overlayer film thickness equations are given in Supplementary Methods 1.1 and Supplementary Discussion 2.1, with associated results given in Supplementary Figs. 1–10 and Supplementary Tables 1–10.
Briefly for Ce 3d, v(Ce3+) was calculated as both (1) the area of the fitted Ce3+ peak model (u′, v′, u0 and v0) relative to the sum of the Ce3+ and Ce4+ peak (u‴, v‴, u″, v″, u and v) models45,51 and (2) the integrated area ratio of the u‴ peak to the v″ to v0 peak envelope, calibrated with a standard coefficient determined on CeO2 assuming v(Ce3+) = 0.00 (Eq. (8), where v(Ce3+)a0Meas is the measured v(Ce3+) calibrated to that of CeO2)62,63; where Id and I0,d are the areas of the v″ to v0 peak envelope and u‴ peak, respectively, at etch depth d in the sample and IS,d and I0,S,d are defined analogously for the CeO2 standard. Reported v(Ce3+) represented single values for the zirconolite samples and the average of six replicates for the CeO2 pellets.
NMR
1H and 2H solid-state magic-angle spinning nuclear magnetic resonance (MAS-NMR) experiments attempted to quantify the concentrations of H and D atoms, respectively, in the samples following dissolution. Samples were loaded into NMR rotors, which provide a near-hermetic seal, whilst still under Ar atmosphere. To provide a comparison, pristine 38–75 µm zirconolite powder was also dried and kept under the same conditions and analysed.
1H MAS-NMR experiments used an Infinity Plus NMR spectrometer (Varian) at 499.69 MHz (11.74 T), 30–40 mg pristine or 16, 64 and 256 h 150 °C DI water leached zirconolite powder in triplicate, and 2.5 mm zirconia rotors with PTFE inserts spun at 20 kHz with a double resonance probe (Chemagnetics). Spectra were acquired for 4800–28,800 repetitions using a Depth pulse sequence146 with a 2.75 µs pulse width \((\sim \!\frac{{{\pi }}}{2})\) and a 3 s recycle delay and were referenced to adamantane (99%, Sigma Aldrich) at +1.85 ppm147. Quantification took place through integrating the spectra (Igor Pro, WaveMetrics). 2H MAS-NMR experiments used an Infinity NMR spectrometer (Varian) at 61.47 MHz (9.39 T) with 1.3 mm zirconia rotors spun at 50 kHz using an ultra-fast spinning double resonance probe (Bruker). Triplicate samples leached for 256 h in D2O at each temperature (5–7 mg) were analysed using a spin-echo pulse sequence with a pulse width of 2.25 µs \((\sim \!\frac{{{\pi }}}{2})\) and a 10 s recycle delay for 21,846 repetitions. Spectra were referenced to D2O (99.96% D atoms, Sigma Aldrich) at +4.8 ppm148. Prior to analysing the samples, NaBD4 (98% D atoms, 90%, Sigma Aldrich; −0.55 ppm) was analysed for 2540 repetitions to optimise experimental parameters and quantify the sensitivity.
Data availability
The datasets generated and analysed during this study are available from the corresponding author on reasonable request.
References
Haifler, J., Škoda, R., Filip, J., Larsen, A. O. & Rohlíček, J. Zirconolite from Larvik Plutonic Complex, Norway, its relationship to stefanweissite and nöggerathite, and contribution to the improvement of zirconolite end-member systematics. Am. Mineral. 106, 1255–1272 (2021).
Oversby, V. M. & Ringwood, A. E. Lead isotopic studies of zirconolite and perovskite and their implications for long range SYNROC stability. Radioact. Waste Manag. 1, 289–307 (1981).
Wu, F. Y. et al. In situ U-Pb and Nd-Hf-(Sr) isotopic investigations of zirconolite and calzirtite. Chem. Geol. 277, 178–195 (2010).
Lumpkin, G. R., Hart, K. P., McGlinn, P. J. & Payne, E. Retention of Actinides in Natural Pyrochlores and Zirconolites. Radiochim. Acta 66–67, 469–474 (1994).
Yudintsev, S. V. et al. Zirconolite Matrices for the Immobilization of REE–Actinide Wastes. Ceramics 6, 1573–1622 (2023).
Strachan, D. M. et al. Radiation damage effects in candidate titanates for Pu disposition: Zirconolite. J. Nucl. Mater. 372, 16–31 (2008).
Lee, W. E., Ojovan, M. I., Stennett, M. C. & Hyatt, N. C. Immobilisation of radioactive waste in glasses, glass composite materials and ceramics. Adv. Appl. Ceram. 105, 3–12 (2006).
Ringwood, A. E. & Kelly, P. M. Immobilization of high-level waste in ceramic waste forms. Philos. Trans. R. Soc. Lond. 319, 63–82 (1986).
Gregg, D. J., Farzana, R., Dayal, P., Holmes, R. & Triani, G. Synroc technology: Perspectives and current status (Review). J. Am. Ceram. Soc. 103, 5424–5441 (2020).
Gatehouse, B. M., Grey, I. E., Hill, R. J. & Rossell, H. J. Zirconolite, CaZrxTi3-xO7; Structure Refinements for Near-End-Member Compositions with x = 0.85 and 1.30. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 37, 306–312 (1981).
Whittle, K. R. et al. Combined neutron and X-ray diffraction determination of disorder in doped zirconolite-2M. Am. Mineral. 97, 291–298 (2012).
Coelho, A. A., Cheary, R. W. & Smith, K. L. Analysis and Structural Determination of Nd-Substituted Zirconolite-4M. J. Solid State Chem. 129, 346–359 (1997).
Blackburn, L. R. et al. A systematic investigation of the phase assemblage and microstructure of the zirconolite CaZr1-xCexTi2O7 system. J. Nucl. Mater. 535, 1–11 (2020).
Li, W., Dong, F., Bian, L., Zou, H. & Song, M. Phase relations, microstructure, and valence transition studies on CaZr1–xCexTi2O7 (0.0 ≤ x ≤ 1.0) system. J. Rare Earths 36, 1184–1189 (2018).
Meng, C., Ding, X., Li, W., Zhao, J. & Yang, H. Phase structure evolution and chemical durability studies of Ce-doped zirconolite–pyrochlore synroc for radioactive waste storage. J. Mater. Sci. 51, 5207–5215 (2016).
Clark, B. M., Sundaram, S. K. & Misture, S. T. Polymorphic Transitions in Cerium-Substituted Zirconolite (CaZrTi2O7). Sci. Rep. 7, 5920 (2017).
Blackburn, L. R. et al. Synthesis and characterisation of HIP Ca0.80Ce0.20ZrTi1.60Cr0.40O7 zirconolite and observations of the ceramic–canister interface. MRS Adv. 6, 112–118 (2021).
Zhong, M. X. et al. Synthesis of Ca1-xCexZrTi2-2xAl2xO7 zirconolite ceramics for plutonium disposition. J. Nucl. Mater. 556, 153198 (2021).
Guy, C. et al. New conditionings for separated long-lived radionuclides. Comptes Rendus Phys. 3, 827–837 (2002).
Tribet, M. et al. Leaching of a zirconolite ceramic waste-form under proton and He2+ irradiation. Radiochim. Acta 96, 619–624 (2008).
Blackburn, L. R. et al. Review of zirconolite crystal chemistry and aqueous durability. Adv. Appl. Ceram. 120, 69–83 (2021).
Hart, K. P. et al. Naturally-Occurring Zirconolites - Analogues for the Long-Term Encapsulation of Actinides in Synroc. Radiochim. Acta 74, 309–312 (1996).
Lumpkin, G. R. Alpha-decay damage and aqueous durability of actinide host phases in natural systems. J. Nucl. Mater. 289, 136–166 (2001).
Leturcq, G., McGlinn, P. J., Barbe, C., Blackford, M. G. & Finnie, K. S. Aqueous alteration of nearly pure Nd-doped zirconolite (Ca0.8Nd0.2ZrTi1.8Al0.2O7), a passivating layer control. Appl. Geochem. 20, 899–906 (2005).
McGlinn, P. J., Hart, K. P., Loi, E. H. & Vance, E. R. pH Dependence of the Aqueous Dissolution Rates of Perovskite and Zirconolite at 90°C. MRS Proc. 353, 847 (1994).
Malmström, J. et al. Zirconolite corrosion in dilute acidic and basic fluids at 180-700 °C and 50 MPa. MRS Proc. 556, 165 (1999).
Nikoloski, A. N., Gilligan, R., Squire, J. & Maddrell, E. R. Chemical stability of zirconolite for proliferation resistance under conditions typically required for the leaching of highly refractory uranium minerals. Metals 9, 1–18 (2019).
Pöml, P. et al. The mechanism of the hydrothermal alteration of cerium- and plutonium-doped zirconolite. J. Nucl. Mater. 410, 10–23 (2011).
Blackburn, L. R. et al. Influence of accessory phases and surrogate type on accelerated leaching of zirconolite wasteforms. npj Mater. Degrad. 5, 1–11 (2021).
Souag, R. et al. Study of leaching of a 2M-Zirconolite (Ca0.83Ce0.17ZrTi1.66Al0.34O7) in acidic and basic media. J. Ceram. Process. Res. 16, 150–155 (2015).
ASTM International. ASTM C1285-14: Standard Test Methods for Determining Chemical Durability of Nuclear, Hazardous, and Mixed Waste Glasses and Multiphase Glass Ceramics: The Product Consistency Test (PCT) Designation (ASTM, 2014).
ASTM International. ASTM C1220-17: Standard Test Method for Static Leaching of Monolithic Waste Forms for Disposal of Radioactive Waste (ASTM, 2017).
Cai, X., Teng, Y., Wu, L., Zhang, K. & Huang, Y. The synthesis and chemical durability of Nd-doped single-phase zirconolite solid solutions. J. Nucl. Mater. 479, 455–460 (2016).
Yin, D. et al. Solid-state reaction synthesis and chemical stability studies in Nd-doped zirconolite-rich ceramics. J. Rare Earths 36, 492–498 (2018).
Zhang, K., Wen, G., Zhang, H. & Teng, Y. Self-propagating high-temperature synthesis of CeO2 incorporated zirconolite-rich waste forms and the aqueous durability. J. Eur. Ceram. Soc. 35, 3085–3093 (2015).
Zhang, K., Yin, D., Han, P. & Zhang, H. Two-step synthesis of zirconolite-rich ceramic waste matrice and its physicochemical properties. Int. J. Appl. Ceram. Technol. 15, 171–178 (2018).
Zhang, K., Luo, B. & Zhang, H. Immobilization of CeO2 using single-phase zirconolite and the chemical stability analysis. Mater. Res. Express 6, 115526 (2019).
Wen, G., Zhang, K., Yin, D. & Zhang, H. Solid-state reaction synthesis and aqueous durability of Ce-doped zirconolite-rich ceramics. J. Nucl. Mater. 466, 113–119 (2015).
Ma, J. et al. Investigating hollandite–perovskite composite ceramics as a potential waste form for immobilization of radioactive cesium and strontium. J. Mater. Sci. 56, 9644–9654 (2021).
Maddrell, E. Hot isostatically pressed wasteforms for future nuclear fuel cycles. Chem. Eng. Res. Des. 91, 735–741 (2013).
Kesson, S. E., Sinclair, W. J. & Ringwood, A. E. Solid solution limits in synroc zirconolite. Nucl. Chem. Waste Manag. 4, 259–265 (1983).
Ringwood, A. E., Kesson, S. E., Reeve, K. D., Levins, D. M. & Ramm, E. J. Synroc. in Radioactive waste forms for the future ((eds Lutze, W. & Ewing, R. C.) 233-334 (Elsevier, 1988).
Lumpkin, G. R., Smith, K. L. & Blackford, M. G. Partitioning of uranium and rare earth elements in synroc: effect of impurities, metal additive, and waste loading. J. Nucl. Mater. 224, 31–42 (1995).
Romeo, M., Bak, K., El Fallah, J., Le Normand, F. & Hilaire, L. XPS Study of the reduction of cerium dioxide. Surf. Interface Anal. 20, 508–512 (1993).
Bêche, E., Charvin, P., Perarnau, D., Abanades, S. & Flamant, G. Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf. Interface Anal. 40, 264–267 (2008).
Paparazzo, E. On the curve-fitting of XPS Ce(3d) spectra of cerium oxides. Mater. Res. Bull. 46, 323–326 (2011).
Paparazzo, E. Corrigendum: Use and mis-use of x-ray photoemission Ce3d spectra of Ce2O3 and CeO2 (2018 J. Phys.: Condens. Matter 30 343003). J. Phys. Condens. Matter 32, 099501 (2020).
Paparazzo, E., Ingo, G. M. & Zacchetti, N. X‐ray induced reduction effects at CeO2 surfaces: An x‐ray photoelectron spectroscopy study. J. Vac. Sci. Technol. A Vac. Surf. Film. 9, 1416–1420 (1991).
Zhang, F., Wang, P., Koberstein, J., Khalid, S. & Chan, S. W. Cerium oxidation state in ceria nanoparticles studied with X-ray photoelectron spectroscopy and absorption near edge spectroscopy. Surf. Sci. 563, 74–82 (2004).
Naganuma, T. & Traversa, E. Stability of the Ce3+ valence state in cerium oxide nanoparticle layers. Nanoscale 4, 4950–4953 (2012).
Pfau, A. & Schierbaum, K. D. The electronic structure of stoichiometric and reduced CeO2 surfaces: an XPS, UPS and HREELS study. Surf. Sci. 321, 71–80 (1994).
Holgado, J. P., Munuera, G., Espinós, J. P. & González-Elipe, A. R. XPS study of oxidation processes of CeOx defective layers. Appl. Surf. Sci. 158, 164–171 (2000).
Zhu, H. Y. & Hirata, T. Ce3d and Zr3d X-ray photoelectron spectroscopy spectra of ZrO2-12 mol% CeO2 after heat-treatments and Ar+ etching. J. Mater. Sci. Lett. 12, 749–751 (1993).
Dauscher, A. et al. Characterization by XPS and XAS of supported Pt/TiO2-CeO2 catalysts. Surf. Interface Anal. 16, 341–346 (1990).
Holgado, J. P., Alvarez, R. & Munuera, G. Study of CeO2 XPS spectra by factor analysis: Reduction of CeO2. Appl. Surf. Sci. 161, 301–315 (2000).
Fleming, P., Ramirez, S., Holmes, J. D. & Morris, M. A. An XPS study of the oxidation of reduced ceria-lanthana nanocrystals. Chem. Phys. Lett. 509, 51–57 (2011).
Zhang, Y. et al. Zirconolite-rich titanate ceramics for immobilisation of actinides - Waste form/HIP can interactions and chemical durability. J. Nucl. Mater. 395, 69–74 (2009).
Dixon Wilkins, M. C. J., Gausse, C., Townsend, L. T., Gardner, L. J. & Corkhill, C. L. Characterisation of a Complex CaZr0.9Ce0.1Ti2O7 Glass–Ceramic Produced by Hot Isostatic Pressing. Ceramics 5, 1035–1050 (2022).
Li, H. et al. Characterisation of stainless steel-synroc interactions under hot isostatic pressing (HIPing) conditions. J. Nucl. Mater. 355, 136–141 (2006).
Thornber, S. M., Stennett, M. C. & Hyatt, N. C. Investigation of Ce incorporation in zirconolite glass-ceramics for UK plutonium disposition. MRS Adv. 2, 699–704 (2017).
Blackburn, L. R. et al. Hot Isostatically Pressed Zirconolite Wasteforms for Actinide Immobilisation. IOP Conf. Ser. Mater. Sci. Eng. 818, 012010 (2020).
Maslakov, K. I. et al. XPS study of ion irradiated and unirradiated CeO2 bulk and thin film samples. Appl. Surf. Sci. 448, 154–162 (2018).
Teterin, Y. A. et al. X-ray photoelectron study of calcium cerium titanate ceramics. Russ. J. Inorg. Chem. 49, 87–94 (2004).
Shard, A. G. Practical guides for x-ray photoelectron spectroscopy: Quantitative XPS. J. Vac. Sci. Technol. A 38, 041201 (2020).
Powell, C. J. Practical guide for inelastic mean free paths, effective attenuation lengths, mean escape depths, and information depths in x-ray photoelectron spectroscopy. J. Vac. Sci. Technol. A Vacuum, Surfaces, Film. 38, 023209 (2020).
Jablonski, A. & Powell, C. J. Effective Attenuation Lengths for Different Quantitative Applications of X-ray Photoelectron Spectroscopy. J. Phys. Chem. Ref. Data 49, 033102 (2020).
Powell, C. J. & Jablonski, A. The NIST Electron Effective-Attenuation-Length Database. J. Surf. Anal. 9, 322–325 (2002).
Hill, J. M., Royce, D. G., Fadley, C. S., Wagner, L. F. & Grunthaner, F. J. Properties of oxidized silicon as determined by angular-dependent X-ray photoelectron spectroscopy. Chem. Phys. Lett. 44, 225–231 (1976).
Begg, B. D., Vance, E. R., Hunter, B. A. & Hanna, J. V. Zirconolite transformation under reducing conditions. J. Mater. Res. 13, 3181–3190 (1998).
Grey, I. E., Mumme, W. G., Ness, T. J., Roth, R. S. & Smith, K. L. Structural relations between weberite and zirconolite polytypes - Refinements of doped 3 T and 4 M Ca2Ta2O7 and 3 T CaZrTi2O7. J. Solid State Chem. 174, 285–295 (2003).
Hindshaw, R. S., Bourdon, B., Pogge von Strandmann, P. A. E., Vigier, N. & Burton, K. W. The stable calcium isotopic composition of rivers draining basaltic catchments in Iceland. Earth Planet. Sci. Lett. 374, 173–184 (2013).
Capo, R. C., Stewart, B. W. & Chadwick, O. A. Strontium isotopes as tracers of ecosystem processes: Theory and methods. Geoderma 82, 197–225 (1998).
Krumgalz, B. S. Temperature dependence of mineral solubility in water. Part I. Alkaline and Alkaline earth chlorides. J. Phys. Chem. Ref. Data 46, 043101 (2017).
Baer, D. R. Guide to making XPS measurements on nanoparticles. J. Vac. Sci. Technol. A 38, 031201 (2020).
Richter, F. M., Liang, Y. & Davis, A. M. Isotope fractionation by diffusion in molten oxides. Geochim. Cosmochim. Acta 63, 2853–2861 (1999).
Richter, F. M. et al. Kinetic isotopic fractionation during diffusion of ionic species in water. Geochim. Cosmochim. Acta 70, 277–289 (2006).
Richter, F. M., Davis, A. M., DePaolo, D. J. & Watson, E. B. Isotope fractionation by chemical diffusion between molten basalt and rhyolite. Geochim. Cosmochim. Acta 67, 3905–3923 (2003).
Wanner, P. & Hunkeler, D. Isotope fractionation due to aqueous phase diffusion – What do diffusion models and experiments tell? – A review. Chemosphere 219, 1032–1043 (2019).
Baur, M. E., Garl, C. W. & Stockmayer, W. H. Diffusion coefficients Of H2O-D2O mixtures. J. Am. Chem. Soc. 81, 3147–3148 (1959).
Cherniak, D. J., Watson, E. B., Meunier, V. & Kharche, N. Diffusion of helium, hydrogen and deuterium in diamond: Experiment, theory and geochemical applications. Geochim. Cosmochim. Acta 232, 206–224 (2018).
Johnson, O. W., Paek, S. H. & Deford, J. W. Diffusion of H and D in TiO2: Suppression of internal fields by isotope exchange. J. Appl. Phys. 46, 1026–1033 (1975).
Doppler, G., Bakker, R. J. & Baumgartner, M. Fluid inclusion modification by H2O and D2O diffusion: The influence of inclusion depth, size, and shape in re-equilibration experiments. Contrib. Mineral. Petrol. 165, 1259–1274 (2013).
Gonzalez, R., Chen, Y. & Tsang, K. L. Diffusion of deuterium and hydrogen in doped and undoped MgO crystals. Phys. Rev. B 26, 4637–4645 (1982).
Verney-Carron, A., Vigier, N. & Millot, R. Experimental determination of the role of diffusion on Li isotope fractionation during basaltic glass weathering. Geochim. Cosmochim. Acta 75, 3452–3468 (2011).
Chacko, T., Cole, D. R. & Horita, J. 1 Equilibrium Oxygen, Hydrogen and Carbon Isotope Fractionation Factors Applicable to Geologic Systems. Rev. Mineral. Geochem. y. 43, 1–81 (2001).
Scheiner, S. & Čuma, M. Relative stability of hydrogen and deuterium bonds. J. Am. Chem. Soc. 118, 1511–1521 (1996).
Schauble, E. A. Applying stable isotope fractionation theory to new systems. Rev. Mineral. Geochem. 55, 65–111 (2004).
Dauphas, N. & Schauble, E. A. Mass Fractionation Laws, Mass-Independent Effects, and Isotopic Anomalies. Annu. Rev. Earth Planet. Sci. 44, 709–783 (2016).
Young, E. D., Galy, A. & Nagahara, H. Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance. Geochim. Cosmochim. Acta 66, 1095–1104 (2002).
Van Hook, W. A. Isotope effects in chemistry. Nukleonika 56, 217–240 (2011).
Bigeleisen, J. The relative reaction velocities of isotopic molecules. J. Chem. Phys. 17, 675–678 (1949).
McGrail, B. P. et al. The structure of Na2O-Al2O3-SiO2 glass: Impact on sodium ion exchange in H2O and D2O. J. Non Cryst. Solids 296, 10–26 (2001).
Chave, T., Frugier, P., Ayral, A. & Gin, S. Solid state diffusion during nuclear glass residual alteration in solution. J. Nucl. Mater. 362, 466–473 (2007).
Fillet, C., Advocat, T., Bart, F., Leturcq, G. & Rabiller, H. Titanate-based ceramics for separated long-lived radionuclides. Comptes Rendus Chim. 7, 1165–1172 (2004).
Ringwood, A. E. et al. Immobilization of high-level nuclear reactor wastes in SYNROC: A current appraisal. Nucl. Chem. Waste Manag. 2, 287–305 (1981).
Woolfrey, J. L. & Levins, D. M. Effects of Water Chemistry on the Leach Resistance of Synroc C. MRS Proc. 26, 663 (1983).
Guy, C. & Schott, J. Multisite surface reaction versus transport control during the hydrolysis of a complex oxide. Chem. Geol. 78, 181–204 (1989).
Lasaga, A. C. Rate laws of chemical reactions. Rev. Mineral. Geochem. 8, 1–66 (1981).
Nelson, A. E. & Schulz, K. H. Surface chemistry and microstructural analysis of CexZr1-xO2-y model catalyst surfaces. Appl. Surf. Sci. 210, 206–221 (2003).
Beamson, G. & Briggs, D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database (Wiley, 1992).
Biesinger, M. C., Lau, L. W. M., Gerson, A. R. & Smart, R. S. C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 257, 887–898 (2010).
Biesinger, M. C. Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review. Appl. Surf. Sci. 597, 153681 (2022).
Gengenbach, T. R., Major, G. H., Linford, M. R. & Easton, C. D. Practical guides for x-ray photoelectron spectroscopy (XPS): Interpreting the carbon 1 s spectrum. J. Vac. Sci. Technol. A Vacuum, Surfaces, Film. 39, 013204 (2021).
Tougaard, S. Practical guide to the use of backgrounds in quantitative XPS. J. Vac. Sci. Technol. A 39, 011201 (2021).
Gin, S., Ryan, J. V., Schreiber, D. K., Neeway, J. & Cabié, M. Contribution of atom-probe tomography to a better understanding of glass alteration mechanisms: Application to a nuclear glass specimen altered 25 years in a granitic environment. Chem. Geol. 349–350, 99–109 (2013).
Gin, S. et al. Atom-Probe Tomography, TEM and ToF-SIMS study of borosilicate glass alteration rim: A multiscale approach to investigating rate-limiting mechanisms. Geochim. Cosmochim. Acta 202, 57–76 (2017).
Gilbert, M. & Harding, J. H. Energetics of Ce and Pu incorporation into zirconolite waste-forms. Phys. Chem. Chem. Phys. 13, 13021 (2011).
Loureiro, F. J. A., Nasani, N., Reddy, G. S., Munirathnam, N. R. & Fagg, D. P. A review on sintering technology of proton conducting BaCeO3-BaZrO3 perovskite oxide materials for Protonic Ceramic Fuel Cells. J. Power Sources 438, 226991 (2019).
Su, Y. et al. Separating water isotopologues using diffusion-regulatory porous materials. Nature 611, 289–294 (2022).
Lakatos, I. & Lakatos-Szabó, J. Diffusion of H+, H2O and D2O in polymer/silicate gels. Colloids Surf. A Physicochem. Eng. Asp. 246, 9–19 (2004).
Casey, W. H., Lasaga, A. C. & Gibbs, G. V. Mechanisms of silica dissolution as inferred from the kinetic isotope effect. Geochim. Cosmochim. Acta 54, 3369–3378 (1990).
Icenhower, J. P. et al. Dissolution of A2Ti2O7 (A = Y3+, Gd3+, or Lu3+) Pyrochlore by Experiment at pH = 2, T = 90°C: Evidence for Solubility Control Using a Linear Free Energy Model. MRS Proc. 713, JJ6.2 (2002).
Ewing, R. C., Weber, W. J. & Lian, J. Nuclear waste disposal-pyrochlore (A2B2O7): Nuclear waste form for the immobilization of plutonium and ‘minor’ actinides. J. Appl. Phys. 95, 5949–5971 (2004).
Murday, J. S. & Cotts, R. M. Self-Diffusion in Liquids: H2O, D2O, and Na. J. Chem. Phys. 53, 4724–4725 (1970).
Lapham, K. E., Holloway, J. R. & Delaney, J. R. Diffusion of H2O and D2O in obsidian at elevated temperatures and pressures. J. Non Cryst. Solids 67, 179–191 (1984).
Rébiscoul, D. et al. Water penetration mechanisms in nuclear glasses by X-ray and neutron reflectometry. J. Non Cryst. Solids 353, 2221–2230 (2007).
Anovitz, L. M., Cole, D. R. & Fayek, M. Mechanisms of rhyolitic glass hydration below the glass transition. Am. Mineral. 93, 1166–1178 (2008).
Briman, I. M. et al. Impact of pore size and pore surface composition on the dynamics of confined water in highly ordered porous silica. J. Phys. Chem. C. 116, 7021–7028 (2012).
Thomas, J. J. & Jennings, H. M. Effects of D2O and mixing on the early hydration kinetics of tricalcium silicate. Chem. Mater. 11, 1907–1914 (1999).
Nesbitt, H. W. et al. Thermodynamic stability and kinetics of perovskite dissolution. Nature 289, 358–362 (1981).
Pham, D. K., Neall, F. B., Myhra, S., Smart, R. S. & Turner, P. S. Dissolution Mechanisms of CaTiO3 - Solution Analysis, Surface Analysis and Electron Microscope Studies Implications for Synroc. MRS Proc. 127, 231 (1988).
Leturcq, G. et al. Solubility study of Ti, Zr-based ceramics designed to immobilize long-lived radionuclides. Am. Mineral. 86, 871–880 (2001).
Smith, K. L., Lumpkin, G. R., Blackford, M. G., Day, R. A. & Hart, K. P. The durability of synroc. J. Nucl. Mater. 190, 287–294 (1992).
Lumpkin, G. R. Ceramic Waste Forms for Actinides. Elements 2, 365–372 (2006).
Lüttge, A., Arvidson, R. S. & Fischer, C. A stochastic treatment of crystal dissolution kinetics. Elements 9, 183–188 (2013).
Fischer, C., Finkeldei, S., Brandt, F., Bosbach, D. & Lüttge, A. Direct Measurement of Surface Dissolution Rates in Potential Nuclear Waste Forms: The Example of Pyrochlore. ACS Appl. Mater. Interfaces 7, 17857–17865 (2015).
Corkhill, C. L. et al. Role of Microstructure and Surface Defects on the Dissolution Kinetics of CeO2, a UO2 Fuel Analogue. ACS Appl. Mater. Interfaces 8, 10562–10571 (2016).
Finkeldei, S. et al. Dissolution of ZrO2 based pyrochlores in the acid pH range: A macroscopic and electron microscopy study. Appl. Geochem. 49, 31–41 (2014).
Horlait, D. et al. Environmental SEM monitoring of Ce1-xLnxO2-x/2 mixed-oxide microstructural evolution during dissolution. J. Mater. Chem. A. 2, 5193–5203 (2014).
Wang, J. Thermodynamic equilibrium and kinetic fundamentals of oxide dissolution in aqueous solution. J. Mater. Res. 35, 898–921 (2020).
Turner, P. S. et al. Dissolution mechanisms of oxides and titanate ceramics - Electron microscope and surface analytical studies. In Surfaces Interfaces Ceramic Materials, NATO ASI Series, Vol. 173, 663–690 (Springer, 1989).
Zhang, Z., Blackford, M. G., Lumpkin, G. R., Smith, K. L. & Vance, E. R. Aqueous dissolution of perovskite (CaTiO3): Effects of surface damage and [Ca2+] in the leachant. J. Mater. Res. 20, 2462–2473 (2005).
Hsieh, Y.-H. et al. Durability of hot uniaxially pressed Synroc derivative wasteform for EURO-GANEX wastes. J. Nucl. Mater. 509, 43–53 (2018).
Rossell, H. J. Solid solution of metal oxides in the zirconolite phase CaZrTi2O7. I. Heterotype solid solutions. J. Solid State Chem. 99, 38–51 (1992).
Glasoe, P. K. & Eberson, L. Deuterium Isotope Effect in the Ionization of Substituted Succinic Acids in Water and in Deuterium Oxide. J. Physial Chem. 224, 1959–1961 (1964).
Glasoe, P. K. & Long, F. A. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64, 188–190 (1960).
Icenhower, J. P. et al. Dissolution kinetics of pyrochlore ceramics for the disposition of plutonium. Am. Mineral. 91, 39–53 (2006).
Jégou, C., Gin, S. & Larché, F. Alteration kinetics of a simplified nuclear glass in an aqueous medium: Effects of solution chemistry and of protective gel properties on diminishing the alteration rate. J. Nucl. Mater. 280, 216–229 (2000).
Pierce, E. M. et al. Experimental determination of the effect of the ratio of B/Al on glass dissolution along the nepheline (NaAlSiO4)-malinkoite (NaBSiO4) join. Geochim. Cosmochim. Acta 74, 2634–2654 (2010).
Fairley, N. et al. Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy. Appl. Surf. Sci. Adv. 5, 100112 (2021).
Thermo Fisher Scientific. Table of Elements: X-ray photoelectron spectroscopy of atomic elements. https://www.thermofisher.com/uk/en/home/materials-science/learning-center/periodic-table.html (2013).
National Institute of Standards and Technology. NIST X-ray Photoelectron Spectroscopy Database. NIST Standard Reference Database Number 20 20899, https://doi.org/10.18434/T4T88K (2000).
Zhang, Z., Vance, E. R. & Blackford, M. G. Surface alteration of Nd-bearing zirconolite following hydrothermal treatment. Mater. Res. Soc. Symp. Proc. 663, 285–292 (2001).
Tanuma, S., Powell, C. J. & Penn, D. R. Calculations of electron inelastic mean free paths. V. Data for 14 organic compounds over the 50-2000 eV range. Surf. Interface Anal. 21, 165–176 (1994).
Walton, J., Alexander, M. R., Fairley, N., Roach, P. & Shard, A. G. Film thickness measurement and contamination layer correction for quantitative XPS. Surf. Interface Anal. 48, 164–172 (2016).
Robin Bendall, M. & Gordon, R. E. Depth and refocusing pulses designed for multipulse NMR with surface coils. J. Magn. Reson. 53, 365–385 (1983).
Hayashi, S. & Hayamizu, K. Chemical Shift Standards in High-Resolution Solid-State NMR (1) 13C, 29Si, and 1H Nuclei. Bull. Chem. Soc. Jpn. 64, 685–687 (1991).
Fulmer, G. R. et al. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 29, 2176–2179 (2010).
Acknowledgements
The authors would like to thank Iris Buisman (Earth Sciences, University of Cambridge) for training and assistance throughout the EPMA analysis, Jason Day (Earth Sciences, University of Cambridge) for training and assistance throughout the ICP-MS analysis, Carmen M. Fernandez-Posada (Maxwell Centre, University of Cambridge) for training and assistance throughout the XPS analysis and for providing training in XPS data analysis, and Mark Isaacs (HarwellXPS, EPSRC National Facility for XPS) for running further analysis by XPS. The authors would also like to thank Colleen Mann (Jacobs) for discussing and proofreading the manuscript, and Paul Cook (Nuclear Decommissioning Authority) for proofreading the manuscript. T.L.G. and I.F. were funded by Nuclear Decommissioning Authority under NDA0122POH000326. A.H.M. was supported by EPSRC (grant EP/T012811/1). The funder played no role in study design, data collection, data analysis or data interpretation, or the writing of this manuscript’s technical content. The authors acknowledge use of the Cambridge XPS System (Cambridge Equipment, EPSRC grant EP/P024947/1), part of Sir Henry Royce Institute (EPSRC recurrent grant EP/R00661X/1). Further X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195.
Author information
Authors and Affiliations
Contributions
T.L.G. performed the experiments, performed the analyses (excepting STEM-EDS, TESCAN SEM-EDS and CeO2 pellet XPS analyses), interpreted the data and contributed to the writing of the manuscript. A.H.M. performed TESCAN SEM-EDS analysis, interpreted the data and contributed to the writing of the manuscript. S.M.F. performed the (S)TEM-EDS analysis, interpreted the data and contributed to the writing of the manuscript. D.P. assisted with writing the manuscript. I.F. interpreted the data, contributed to the writing of the manuscript and oversaw the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Goût, T.L., Mir, A.H., Fairclough, S.M. et al. Undetected perovskite phase interference with zirconolite dissolution measurements. npj Mater Degrad 8, 56 (2024). https://doi.org/10.1038/s41529-024-00475-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-024-00475-5
