
Abstract
Widespread application of magnesium (Mg) has been prevented by its low strength and poor corrosion resistance. Core of this limitation is Mg’s low electrochemical potential and low solubility for most elements, favoring secondary phase precipitation acting as effective micro-galvanic elements. Mg-based metal–metal composites, while benefiting strength, are similarly active galvanic couples. We show that related detrimental corrosion susceptibility is overcome by nanoscale composite architecture design. Nanoscale phase spacings enable high-strength Mg–Fe composites with degradation rates as low as ultra-high purity Mg. Our concept thus fundamentally changes today’s understanding of Mg’s corrosion and significantly widens the property space of Mg-based materials.
Similar content being viewed by others

Introduction
Magnesium (Mg) and iron (Fe) are largely different materials. On the one hand, Fe’s strength and allotropic phase transformations along with the possibility to form corrosion resistant alloys, i.e., stainless steels, makes it the most important structural material. On the other hand, Mg has the lowest density of all structural metals, making it a prosperous aspirant for energy-efficient lightweight structures in the mobility sector. Additionally, Mg’s excellent biocompatibility combined with low corrosion resistance enabling biodegradability can be utilized for temporary medical implants, such as bone-fracture fixations in orthopedics and trauma surgery1,2,3. However, a widespread use of Mg in these fields has been prevented so far by the limited strength and high corrosion susceptibility in aqueous environments for technically pure Mg. Fe, a natural impurity element in Mg, presents a main source for Mg’s low degradation resistance. The Fe content, in fact, directly relates to the degradation rate, with an increase in Fe resulting in faster degradation4,5,6,7.
The detrimental role of Fe in Mg is multifold: Because of the large standard potential difference between Mg and Fe (ΔE = 1.9 V), and Fe’s high exchange current density for the hydrogen reduction reaction (Fe: i0,H2/H+ = 1.0 × 10−6 A cm−2, for comparison Zn has a i0,H2/H+ = 3.2 × 10−11 A cm−2)8, the contact of Mg with Fe entails a massive driving force for dissolution of the less noble Mg phase (with −2.7 V against SHE) through galvanic-coupling induced corrosion. Moreover, Fe-rich phases acting as effective cathodic sites can easily form, either through precipitation owing to its extremely low solubility in Mg (<10 ppm for wrought Mg5,9) or through electrochemical redeposition and accumulation on the corroding Mg surface10. In either case, Fe provokes a localized corrosion attack around Fe-rich areas through micro-galvanic corrosion and induces accelerated dissolution. This also holds true for other more noble trace elements in Mg, such as nickel, copper or cobalt, which likewise form cathodic sites in the Mg-matrix, though their effectiveness in facilitating the cathodic reaction is less severe than for Fe6,11,12. The importance of impurity elements and their distribution on the corrosion rate of Mg has been recognized almost 80 years ago12. In fact, Fe has been identified as one of the most detrimental elements to the corrosion resistance of Mg5,9.
While the development of vacuum-distilled ultra-high purity (UHP) Mg could successfully address this impurity-related shortcoming and offers extremely low degradation rates13,14, the strength of UHP Mg is too low to be used in structural applications. Alloying or strengthening with lattice defects such as grain boundaries or stacking faults are hence necessary to strengthen Mg15,16,17. However, achieving simultaneously high strength and high corrosion resistance remains a challenge – particularly in the biomedical application field, where the choice of alloying elements is severely limited to ensure biological safety. Two aspects contribute to the problem of designing slowly degrading Mg-alloys: Passivation through alloying, comparable to the effect that chromium has on Fe to form stainless steels, is challenging for Mg2,18,19 because (i) Mg typically forms non-dense, readily dissolvable oxides and (ii) Mg has a low solubility for most elements at typical forming temperatures (~523–623 K). The associated ease in precipitation of secondary phases possibly favors strength but, in most cases, accelerates (local) corrosive attack. New strategies to significantly expand the property space of Mg-based materials are thus required to combine simultaneously high strength and high corrosion resistance.
An attractive answer are composites, which hold the potential to combine the benefits of the individual material components they are made of. Traditional metal‒metal composites, however, are generally not considered because of the aforementioned effective galvanic couple that Mg forms with most other metals2. Here, we scrutinize a deliberate manipulation of the composite architecture (e.g., phase fraction, distributions, phase size and spacings), hypothesizing that phase architecture at the nanoscale may provide the necessary lever to subdue this detrimental effect. Our assumptions are built on recent studies on Mg-alloys that indicate a significant impact of the size and distribution of second phase particles (i.e., micro-galvanic elements) on the corrosion rates20,21,22. We expect similar size effects for Mg-based metal-metal composites, if the phase spacing is sufficiently refined, possibly facilitated through a confinement of the active corrosion mechanisms at the nanoscale. If proven successful, nanostructured Mg-composites would finally unlock the highly demanded property combination in lightweight and biodegradable Mg-alloys by exhibiting exceptional strength and corrosion resistance.
Nanostructured Mg–Fe composites with largely improved corrosion resistance
As a proof of concept for the raised hypothesis, we focus in the following on Mg–Fe composites. Fe was chosen as composite constituent, because of its large difference in the electrochemical potential, as stated above. Mg–Fe composites are hence ideal to answer if targeted nanoscale design of the composite architecture can indeed be utilized to lower the detrimental effects of galvanic coupling. To this end, Mg–Fe composites with different phase spacings were fabricated and the effects of composite architecture on the degradation rate investigated. High pressure torsion (HPT) processing, a method allowing to apply severe shear strains to a material, was chosen for bulk composite synthesis, because (i) it overcomes the limitations of conventional metallurgical methods in synthesizing such composites (namely Mg boils way before Fe melts23) through the use of metallic powder blends of arbitrary composition, and (ii) it allows for control of the phase spacings over up to four orders of magnitude through adjustment of the applied strain24.
Two different architectures were produced for this proof of concept using monotonic and cyclic high pressure torsion (HPT and CHPT) to fabricate a nanostructured Mg–Fe composite and a composite with micrometer-sized phase spacing, respectively. For both composites, a phase fraction of 50 vol.% was chosen. Representative micrographs of the composites’ architecture are displayed in Fig. 1. Severe cyclic strains (CHPT) consolidate the powder blend into a bulk piece while preserving its original shape and phase dimensions of ~20 µm. The slight elongation in tangential direction results from the initial compression step preceding the cyclic deformation. Accumulation of cyclic strain resulted in the formation of grains and subgrains within the phases, evident from the backscattered electron (BSE) images (Fig. 1a), in which an Fe particle is outlined with a white dashed line and some subgrains within the particle are highlighted by yellow dotted lines. This composite is referred to as the coarse Mg–Fe composite hereinafter. In contrast, severe monotonic strains (HPT) significantly refine the composite structure (Fig. 1b). In fact, the phase spacing was substantially reduced to <300 nm for most lamellae (Fig. 1b), yielding a nanostructured Mg–Fe composite. Despite the substantially different mechanical properties of Mg and Fe, a fairly homogeneous composite architecture was obtained, without obvious signs of severe strain localization (e.g., in shear bands which would misalign the lamellae with respect to the principle shear direction). Overall, Mg–Fe composites of identical phase fraction but structural scales differing by two orders of magnitude could thus be successfully produced. Moreover, as the applied strain in HPT and CHPT increases linearly with the radius, the Mg–Fe composites possess an inherent structural gradient. However, as the applied cyclic strains do not manipulate the phase spacing considerably, this gradient is most prominent for the composites processed by monotonic HPT, which consist of nanostructured phase spacings (Fig. 1b) at the outermost radii, while coarser spacings prevail towards the center of the disk. As such, these two types of composites are ideally suited to study length scale effects on corrosion mechanisms and rates.

SEM images in BSE contrast acquired in radial direction of the disk-shaped samples. a Coarse Mg–Fe processed by CHPT. The dashed white line outlines an Fe particle with subgrains developed within (marked by dotted yellow lines); b Nanostructured Mg–Fe processed by monotonic HPT.
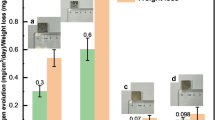
Our corrosion behavior investigation of the composites is summarized in Fig. 2a. The hydrogen gas (H2) evolution is taken as a measure for the Mg-degradation rate25. Given the known dependence of Mg’s corrosion rate on the electrolyte, we analyzed the composites upon immersion in either (i) phosphate buffered saline (PBS, pH value of 7.4), (ii) unbuffered 3.5% NaCl solution or (iii) Hanks balanced salt solution (HBSS) without glucose26. Detailed compositions of the three solutions can be found in the Methods section (Table 1). Degradation rates of UHP Mg were measured in comparison. For the Mg–Fe composites, the degradation rates were observed to be largely independent of the electrolyte with slightly slower degradation rates in HBSS. For the UHP Mg, the differences are larger and, as expected, the slowest rates are measured for the unbuffered NaCl solution, which can be rationalized by the pronounced increase of the pH value establishing in surface-near regions.

a Time-resolved degradation, measured by H2 evolution, of the coarse and nanostructured Mg–Fe composites compared to UHP Mg reference in three different electrolytes. For decreasing composite phase spacing, the degradation rates slow down significantly. b Cross-sectional SEM images of the nanostructured Mg–Fe composite after nine days of immersion in PBS. Triangles denote the interface with solution contact, arrows show exemplary ingress of solution (dark contrast, filled with embedding resin) causing lamellae exfoliation and structural bulging (stars). Pentagon marks the intact composite.
As expected, the coarse composite corroded rapidly upon immersion. Within only 40–60 min, the measured amount of evolved H2 saturated and the composite completely disintegrated, indicative of complete Mg dissolution. In contrast, the composite processed by monotonic HPT, consisting of a gradient from coarse (center) to nanostructured (rim) phase spacings, degraded considerably slower. In fact, the measured H2 volume suggests presence of the Mg phase up to ~24 hours, a reduction in degradation rate by a factor of ~24 compared to the coarse composite. Notably, microscopic inspection of this composite sample following immersion revealed larger corrosion attack towards the center of the HPT disk, corresponding to lower strain and hence larger phase spacings. This observation provides strong indication that the phase spacing plays a crucial role in governing the degradation rate. For confirmation, the nanostructured composite was tested without the low-strained center part, removed by slow-advance drilling of a 2.5 mm hole. The resulting sample consisted of fine and largely homogeneous phase spacings only (Fig. 1b). Indeed, this drastically reduced degradation rates judged from small H2 evolution (Fig. 2a), thus confirming that the nanoscale architecture induces only minor degradation rates. In fact, the H2 data suggests, even after nine days only ~10% of the Mg phase has degraded, corresponding to a >2000-fold reduction of the degradation rate compared to a coarse microstructure of the same composite composition. Importantly, dissolution even slowed down further with times. Our observations suggest that nanoscale engineering of the Mg–Fe composite architecture modifies the active corrosion mechanisms, which origin in corrosion-hindering structures and result in H2-evolution rates close to those of UHP Mg. Potential mechanisms enabling this superior performance are discussed in the following.
Interplay of processes underlying the stronlgy enhanced corrosion resistance of nanostructured Mg–Fe
To make use of the composite’s enhanced corrosion resistance in functional material solutions for engineering or biomedical applications, a profound understanding of the underlying reaction kinetics and mechanistic principles must be established. Aiming at stimulating their in-depth study, we here outline possible solid-state and interface phenomena acting on the nanostructured Mg‒Fe composites, Fig. 3a. The processes described are not limited to those leading to reduced degradation rates, but those that lead to changes in the underlying degradation mechanisms when composites with nanoscale instead of coarse phase spacing are considered:
-
Mechanical intermixing. The severe strains applied in monotonic HPT do not only induce a substantial refinement of the phase spacing (Fig. 1b), but for sufficiently large strains probably cause a breakdown of the lamellar structure or at least an incorporation of fragments into the other phase. This can result in mechanical alloying even in systems having zero solubility in thermodynamic equilibrium (e.g., refs. 27,28,29), such as the Mg–Fe system. Mechanical alloying has already been reported for the Mg–Fe system, with alloying occurring potentially in both phases depending on the concentration and treatment applied30,31,32. Depending on the applied strain and degree of intermixing, two scenarios may occur. First, if Fe-particles from lamella fragmentation are incorporated into the Mg matrix and are sub-critical sized (i.e., ‘Fe-nanosheets’), the effective catalytical activity for the hydrogen-molecule formation on Mg is virtually ‘deactivated’ by the fast hydrogen diffusion into the Fe nanoscale phase33. Second, if Fe is not only dispersed but mechanically forced into solid solution within the Mg phase, Mg's dissolution rate would decrease due to elevated electrochemical potential of the Mg phase and due to Fe surface enrichment, reducing the potential difference to the cathodically active Fe phase, and ultimately decreasing the driving force for galvanic coupling-assisted dissolution.
-
Fe redeposition. Fe constituting the electrochemically more noble element in these composites can redeposit through electrochemical reduction on the dissolving Mg surface10,34, similar to the well-known redeposition phenomenon of Cu during aluminum-alloy corrosion35. This phenomenon is particularly likely when Fe presents a solute in the Mg phase, such as favored through mechanical intermixing. While microscopic noble-element redeposits on Mg act as efficient cathodes for the hydrogen reduction reaction (micro-galvanic-coupling assisted corrosion) and as such are detrimental to its corrosion resistance10,34,36,37,38,39, the effect can reverse when noble-element contents are sufficiently high as to allow coalescence of redeposits40. The high Fe content in the Mg–Fe composites and its potential mechanical mixing in the Mg phase might enable, through dissolution and redeposition processes, the formation of a continuous – presumably dense – Fe layer. A continuous Fe layer would act as a quasi-protective film and thus ultimately slow down further Mg dissolution. It should be noted, that even defects in this Fe layer would not result in dramatic localized Mg dissolution, because of the alkaline pH created locally, see description below.
-
Near-surface solution alkalization. Mg dissolution with its low level of hydrolysis and the concomitant hydrogen reduction on the surface generate an increased pH of the surface-near liquid. Alkalization stabilizes hydroxides formed in aqueous solutions41 and favors precipitation of insoluble carbonates or phosphates in biological fluids42,43. The presence of effective reduction on Fe cathodes will even amplify this phenomenon. Common corrosion products forming on Mg surfaces in aqueous solutions are filamentous or porous44, and present a quasi-protective film that can slow down the dissolution rates.
-
Confined phase volumes. The nanoscale phase spacings may further retard the corrosion attack through solution confining geometric factors. Near-surface Mg dissolves preferentially in an aqueous solution, leading to a progressively increasing exposed Fe phase at the surface. Fe – with an intrinsically lower reactivity compared to Mg – will then dictate the further dissolution rates. Moreover, the preferential dissolution of submicron-sized Mg lamellae will create narrow channels enclosed by catalytically active Fe, which limits fluid flow and element diffusion, and in turn hinders equilibration with the bulk solution. Consequently, local solution alkalization and corrosion-product stabilization are expected to be particularly pronounced for confined Mg volumes, which slow down or annihilate their continuous corrosion attack45.
-
Material exfoliation. The confined lamellar geometry and concomitant eased formation of voluminous corrosion products can give raise to separation of the Fe lamellae, referred to as exfoliation corrosion. Exfoliation occurs for anisotropic microstructures and elongated grain structures, characteristic for wrought processes, such as extrusion, rolling or HPT46,47. In all cases, the separation occurs parallel to the elongated grain or phase structure, and was also observed for the slowly degrading Mg–Fe composites in this study (Fig. 2b). While classically defined as a type of intergranular attack, exfoliation can occur in lamellar nanostructured Mg‒Fe composites due to the confined phase geometry. Exfoliation is particularly favored in environments that stabilize corrosion products of larger volume compared to metallic Mg, provoking exfoliation through volume expansion46,48. While the Fe phase in the composite is (partially or fully) cathodically protected, its dissolution once electrically decoupled through exfoliation will be independent from Mg. Thus, Fe will establish its own free corrosion potential, whose dissolution rate is a function of the prevalent environment. The large surface-to-volume ratio and the high defect densities within the exposed or disintegrated nanometric Fe phase are assumed to foster dissolution rates larger than those of bulk Fe49,50. The nanoscale architecture is hence not only beneficial to retard Mg dissolution, but also to accelerate the one of Fe. This is particularly relevant for biomedical applications, for which Fe alloys show overly low degradation rate in vivo51. By nanoscale composite architectures, dissolution in physiological environments seems finally possible.
-
Hydrogen trapping. Atomic hydrogen formed on the corroding surface upon its contact to humid air or aqueous solution (H2O + e− → OH− + H) may diffuse into the metal to form hydrides. This originates from Mg’s large affinity to form hydrides and Fe’s high diffusivity for hydrogen52. The assembly of hydride-forming and hydrogen-diffusing solid has been recognized as potential hydrogen storage units33,53. Although MgH2 is not stable in contact with water and reacts to form gaseous hydrogen54, it can be stabilized in a multi-layered assembly of Mg with Fe33. Additionally, as microstructural defects are efficient trapping sites for hydrogen55, their high density in the composites brought in through the HPT process, makes the nanostructured Mg‒Fe composites potentially effective for hydrogen traps. As such, they may reduce the effective amount of evolving hydrogen. The potential formation of brittle hydrides or occurring hydrogen embrittlement of the Fe-phase may – if sufficiently slow – indeed be beneficial for applications aiming at complete biodegradability. Presence of trapped hydrogen could support complete dissolution via mechanical-assisted disintegration of the Fe lamellae, enabling it to occur under the absence of hydrogen-gas formation. Contrary, if the process of hydrogen embrittlement occurs too fast, the bcc Fe phase could be easily replaced by an fcc Fe phase (e.g., an Fe–Mn alloy) that is characterized by a significantly reduced hydrogen diffusivity56.

a Schematic of discussed corrosion processes active for the nanostructured Mg‒Fe composite. b Left: Property space of Mg and Fe combinations at different length scales (from macroscopic at the top to nanostructured at the bottom) and, right: their technological use. Top: Mg’s low corrosion resistance is used as sacrificial anodes in shipbuilding or offshore protection. Center: microscopic Fe particles in Mg as impurities do not have any technological use but are detrimental to Mg’s corrosion resistance. Technical pure Mg corrodes significantly faster than high purity Mg (photographs after 2 h in 0.15 M NaCl). Bottom: nanostructured Mg–Fe composites have low corrosion rate and high mechanical strength. Photograph is a screw fabricated from the nanostructured Mg–Fe composite, intended to be used in orthopedics and trauma.
Further in-depth analysis is required to answer which role these individual processes play in the overall Mg‒Fe composite performance. However, it seems safe to assume that multiple solid-state and solid-liquid interface phenomena contribute simultaneously to the unexpected composite corrosion resistance. Any of the processes proposed above are facilitated by an increased structural confinement, in line with our observation of reduced H2 evolution for decreasing phase spacings. We further expect that their effectiveness in controlling the composites dissolution is additionally affected by the composite composition, i.e., Mg:Fe ratio, stimulating further investigations to optimize it. Notably, we confidently exclude the role of insulating oxide layers along the phase boundaries decoupling the galvanic element, because the native oxide layer on the powder-particles surfaces was found to fracture or even to dissolve during the HPT process57,58.
Benefitting from simultaneously reduced degradation and higher strength
Besides the strongly efficient lever to tailor degradation rates, the composite architecture further allows to tailor mechanical properties, which are effectively controlled by the phase fraction, geometry and spacing. As such, the Mg–Fe composites allow to access a much wider property space than what can be achieved with traditional Mg-alloys. For illustration, we measured the microhardness of the Mg–Fe composites as a function of the applied (accumulated cyclic) strain in HPT (Fig. 4a). Data of the UHP Mg and a well-annealed, i.e. defect-scarce, coarse Mg–Fe composite are shown for comparison. A reduction of the phase spacing to the nanoscale gives rise to exceptional strength levels (based on a conversion of hardness to strength >450 MPa can be expected59), as the free path for dislocation gliding is drastically shortened. For both composite types, hardness increases with the applied (accumulated) strain. This behavior is less pronounced for the coarse composite, rationalized by the accumulation and rearrangement of dislocations into grains and subgrains as the main strength contributor. Because the grain or subgrain size that evolves upon cyclic straining mainly depends on the applied strain amplitude rather than the number of cycles60, the effect of accumulated strain is apparent as an initial sharp increase but vanishes after a few cycles. With about 1.1 GPa hardness (~300 MPa strength), even the coarse Mg–Fe composite is substantially stronger than UHP Mg (~0.3 GPa) and many biocompatible Mg alloys61, compare Fig. 4b.

a Hardness of the two composite types as a function of the applied (accumulated) strain. Data points of the monotonically deformed Mg–Fe composite for strains larger than ~140 correspond to the nanostructured spacing in Fig. 2. Data of UHP Mg and an annealed coarse composite are shown for comparison. The Young’s modulus of the nanostructured composite (best-performing condition, circled in blue) is given in its pristine state and after 9 days immersion in PBS. b Yield strength and degradation rate of various biodegradable alloys prepared by conventional methods and severe plastic deformation (cyclic extrusion and compression) compared to the new Mg–Fe composites3,14,70,71,72. Note that for this comparison, yield strength includes data measured in tension, compression as well as from a conversion of hardness. Slight deviations for the same test method may thus occur. Please note the inverted scaling in second (upper) x-axis in (a) and second (right) y-axis in (b).
Similarly, the monotonically deformed composites, showed a significant strengthening at low strains (ε < 20, Fig. 4a), which continuously increased with applied strain. With 1.4 GPa hardness obtained for the largest strains applied here, strength levels of up to 450 MPa can be expected for the nanostructured composite according to the Tabor relation59. This conversion is known to give reasonable predictions for severely deformed metals or nanocomposites, and even underestimates the maximum strength in the latter case62. With that, the nanostructured composite significantly outperforms the to-date strongest rare-earth element free (REE) Mg alloys17,61, see Fig. 4b. Notably, further optimization of either HPT strains and/or Fe to Mg ratios may yield even higher strength levels.
Prospects and potential avenues enabled through nanostructured Mg–Fe composites
The wider property space of the Mg–Fe composites compared to Mg-based alloys (Fig. 4b) leverages the use of low-density and biodegradable Mg in so far inaccessible applications. Mg–Fe composites are an ideal aspirant for load-bearing degradable biomedical applications and thus a serious alternative to the monopoly of REE-based Mg-alloys to achieve the highest strength levels without compromising degradation rates63,64. This replacement is important as REE alloys are considered risky. In fact, REE were found to accumulate in organs, with thus far unknown long-term effects65. Moreover, the stepwise degradation of the nanostructured Mg–Fe composites with initial dissolution of Mg followed by that of Fe, would allow for close-to-ideal material properties for load-bearing bone-fracture fixation, Fig. 5: (i) Its strength exceeds the requirements for the initial healing phase (400 MPa17). (ii) Preferential Mg dissolution results in a less rigid Fe-rich skeleton (compare nanoindentation data in Fig. 4a). The Young’s modulus reduced significantly between the pristine (94.7 ± 6 GPa) and degraded samples (59.8 ± 7 GPa after 9 days immersion). Different from state-of-the-art titanium implants, this progressive reduction of strength and modulus enables a continuous load-transfer back to the bone as it heals – a pre-requisite for healthy bone regrowth and remodeling. (iii) The release of Mg2+ ions stimulates new bone formation66. (iv) The limitation of insufficient in vivo degradation rates for bulk Fe-alloys51 may be overcome by the nanoscale phase geometry, facilitating the desired complete dissolution within a reasonable time (<2 years). The extended property space, tailorable by the composite architecture (Fig. 4b), may even enable more customized solutions (‘personalized medicine’).

Immediately following the material’s implantation, the surficial Mg starts to dissolve, leading to Mg2+ release and near-surface solution alkalization, as well as formation of voluminous corrosion products, which lead to bulging and exfoliation. With ongoing degradation and formation of larger pores, bone ingrowth is facilitated. New bone replaces the site of the implant within a targeted time of two years. The grayscale image on top represents the Kikuchi pattern contrast from electron backscatter diffraction, showing the remaining Fe skeleton of a partially degraded composite structure.
Certainly, prior to any product development, the composite’s properties and their scaling with architecture need to be established. This involves not only in-depth studies of the corrosion processes at play but also understanding of deformation, fracture, strength, and efficient synthesis of the Mg–Fe composites. The promising finding that the detrimental effects of galvanic corrosion can be mitigated by deliberate manipulation of the composite architecture is thus expected to stimulate a wealth of interdisciplinary research efforts. Finally, one should note, that apart from the investigated Mg–Fe composites with the suggested use in biomedical applications, new avenues for other high-performance composites might arise, as the proposed concept is expected to be generally applicable to any combinations of noble-active metal matrix composites.
Methods
Materials
Metallic powders of Fe (MaTecK, 99.9%, 70 µm particle size) and Mg (Alfa Aesar, 99.8%, 40 µm particle size) were blended in a 50:50 volume ratio and served as a precursor for the synthesis of the Mg–Fe composites. Distilled ultra-high purity (UHP, 99.999%) Mg13 with a total impurity content of only 19 wt. ppm was used as a reference for the lowest accessible Mg degradation rate14. The UHP Mg was tested in an undeformed condition to avoid potential accelerating effects of lattice defects on corrosion. To provide a reference for the hardness of a coarse, defect scarce Mg–Fe composite (Fig. 4), an as-prepared coarse Mg–Fe composite sample was additionally annealed at 773 K for 30 min to widely remove the defects induced during the cyclic HPT process.
Sample synthesis
The Mg–Fe powder blends were pressed into disks (8 mm diameter, 0.8 mm thickness) at 7.8 GPa nominal pressure and subsequently deformed using high pressure torsion (HPT). Two different HPT protocols were used to synthesize Mg–Fe composites with different average phase spacing. Coarse Mg–Fe composites were fabricated using cyclic HPT for 20 cycles at a twist angle of 10 degrees at room temperature, while the nanostructured Mg–Fe composites were prepared by conventional monotonic HPT at 573 K (15 rotations at a speed of 0.6 rot min−1) using a device described in detail in ref. 67. Elevated temperatures in case of monotonic HPT were chosen to ensure better co-deformation of the two different phases.
The applied equivalent von Mises strain in Fig. 4a, ε, (corresponding to the accumulated strain, εacc, in case of CHPT) can be calculated according to Eqs. (1) and (2)68 and yield maximum values of ε = 272 and εacc = 40, both calculated at the disk edge (i.e., disk radius of 4 mm). These strains are sufficient to achieve seamless bonding of the powder particles. In Eqs. (1) and (2), r denotes the disk radius, t the disk thickness, N the number of rotations applied in case of monotonic HPT or the number of cycles in case of CHPT and θ the twist angle.
Microstructural characterization
Composite architectures were examined in radial direction of the HPT disks (at disk radius ~3.5 mm) using a scanning electron microscope (SEM, Zeiss Leo 1525; JEOL 7200 F). The samples were mechanically ground and polished (water free) followed by a gentle mechanochemical polishing step using colloidal silica. Images were obtained at 20 kV and using a backscattered-electron (BSE) detector. The pattern contrast map of the immersed sample was obtained at 20 kV and a sample pre-tilt of 70° using the electron backscatter diffraction detector Nordlys Nano from Oxford Instruments. The step size was set to 40 nm and Fe (bcc) and Mg (hcp) were loaded as options for orientation solutions. The areas of the remaining skeleton were to 98% indexed as Fe with a high certainty of the solutions (average mean angular deviation 0.58°, average band contrast 125––both from the indexed areas).
Mechanical characterization
Vickers microhardness (0.3 gf load, 15 s dwell time) was determined along the disk radius to provide an estimate for the composite strength and its evolution with strain.
Hardness and Young’s moduli of an as-prepared and partially degraded (9 days in PBS) nanostructured composite sample were measured using an instrumented ultra-micro indentation system (UMIS, Fischer-Cripps) equipped with a diamond Berkovich tip. 15 individual quasi-static indents were taken on each sample (50 mN load), with the standard deviation reported as the error bar. The resulting indent depth and width (~1 µm and 5 µm, respectively) are considered sufficient to probe a representative volume of the selected nanocomposite structure. The obtained data was evaluated according to Oliver and Pharr69. Sample preparation followed the same routine as for the microstructural analysis and the sample was indented in tangential direction.
Evaluation of corrosion rates
The Mg-phase degradation was assessed with the hydrogen evolution method25, which relies on the formation of hydrogen gas (H2) according to Mg + 2H2O→Mg2+ + 2OH− + H2. Quarters of the HPT disks were prepared such that they had a comparable surface area exposed to the corrosive environment, however, the absolute sample mass varied by about 20%. The burr was removed, and the disks finally ultrasonically cleaned using isopropanol. Each sample was immersed separately at 298 K in either (i) phosphate buffered saline (PBS, pH value of 7.4, Roth, Germany), (ii) unbuffered 3.5% NaCl solution or (iii) Hanks balanced salt solution without Glucose26, and the time-resolved H2 evolution was monitored. Compositions of the three solutions are summarized in Table 1. Additionally, the H2 volume evolving from UHP Mg was measured as a slowly degrading reference14.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Pollock, T. M. Weight loss with magnesium alloys. Science 328, 986–987 (2010).
Virtanen, S. Biodegradable Mg and Mg alloys: corrosion and biocompatibility. Mater. Sci. Eng. B 176, 1600–1608 (2011).
Zheng, Y. F., Gu, X. N. & Witte, F. Biodegradable Metals. Mater. Sci. Eng. R: Rep. 77, 1–34 (2014).
Hofstetter, J. et al. Influence of trace impurities on the in vitro and in vivo degradation of biodegradable Mg-5Zn-0.3Ca alloys. Acta Biomater. 23, 347–353 (2015).
Liu, M., Uggowitzer, P. J., Schmutz, P. & Atrens, A. Calculated phase diagrams, iron tolerance limits, and corrosion of Mg-Al alloys. JOM 60, 39–44 (2008).
Hillis, J. E. The effects of heavy metal contamination on magnesium corrosion performance. SAE Tech. Pap. https://doi.org/10.4271/830523 (1983).
Yang, L. et al. Effect of iron content on the corrosion of pure magnesium: critical factor for iron tolerance limit. Corros. Sci. 139, 421–429 (2018).
McCafferty, E. Kinetics of corrosion, in: Introduction to corrosion science. 1st edn, (Springer New York, 2010). https://doi.org/10.1007/978-1-4419-0455-3.
Liu, M. et al. Calculated phase diagrams and the corrosion of die-cast Mg-Al alloys. Corros. Sci. 51, 602–619 (2009).
Höche, D. et al. The effect of iron re-deposition on the corrosion of impurity-containing magnesium. Phys. Chem. Chem. Phys. 18, 1279–1291 (2015).
Song, G. & Atrens, A. Understanding magnesium corrosion. A framework for improved alloy performance. Adv. Eng. Mater. 5, 837–858 (2003).
Hanawalt, J. O., Nelson, C. E. & Peloubet, J. A. Corrosion studies of magnesium and its alloys. AIME Trans. 147, 273–299 (1942).
Löffler, J. F., Uggowitzer, P. J., Wegmann, C., Becker, M. & Feichtinger, H. Process and apparatus for vacuum distlllation of high-purity magnesium, European Patent EP 2804964. European Patent EP 2804964 1–25 (2012).
Hofstetter, J. et al. Assessing the degradation performance of ultrahigh-purity magnesium in vitro and in vivo. Corros. Sci. 91, 29–36 (2015).
Homma, T., Kunito, N. & Kamado, S. Fabrication of extraordinary high-strength magnesium alloy by hot extrusion. Scr. Mater. 61, 644–647 (2009).
Jian, W. W. et al. Ultrastrong Mg alloy via nano-spaced stacking faults. Mater. Res. Lett. 1, 61–66 (2013).
Ikeo, N., Nishioka, M. & Mukai, T. Fabrication of biodegradable materials with high strength by grain refinement of Mg–0.3at.% Ca alloys. Mater. Lett. 223, 65–68 (2018).
Ghali, E. Corrosion Resistance of Aluminum and Magnesium Alloys: Understanding, Performance, and Testing. (John Wiley & Sons, Inc., 2010). https://doi.org/10.1002/9780470531778.
Deng, M. et al. Approaching “stainless magnesium” by Ca micro-alloying. Mater. Horiz. 8, 589–596 (2021).
Gao, J. H. et al. Homogeneous corrosion of high pressure torsion treated Mg-Zn-Ca alloy in simulated body fluid. Mater. Lett. 65, 691–693 (2011).
Seong, J. W. & Kim, W. J. Development of biodegradable Mg-Ca alloy sheets with enhanced strength and corrosion properties through the refinement and uniform dispersion of the Mg2Ca phase by high-ratio differential speed rolling. Acta Biomater. 11, 531–542 (2015).
Cheng, W. Li, Ma, S. Chao, Bai, Y., Cui, Z. Qin & Wang, H. Xia. Corrosion behavior of Mg-6Bi-2Sn alloy in the simulated body fluid solution: the influence of microstructural characteristics. J. Alloys Compd. 731, 945–954 (2018).
Nayeb-Hashemi, A. A., Clark, J. B. & Swartzendruber, L. J. The Fe-Mg (Iron-Magnesium) system. Bull. Alloy Phase Diagr. 6, 235–238 (1985).
Kormout, K. S., Ghosh, P., Bachmaier, A., Hohenwarter, A. & Pippan, R. Effect of processing temperature on the microstructural characteristics of Cu-Ag nanocomposites: From supersaturation to complete phase decomposition. Acta Mater. 154, 33–44 (2018).
Song, G., Atrens, A. & St John, D. An hydrogen evolution method for the estimation of the corrosion rate of magnesium alloys. in TMS Annual Meeting 255–262 (2001). https://doi.org/10.1002/9781118805497.ch44.
Quest CalculateTM HBSS (Hank’s Balanced Salt Solution) Solution Preparation and Recipe. Available at: https://www.aatbio.com/resources/buffer-preparations-and-recipes/hbss-hanks-balanced-salt-solution. (Accessed: 17th March 2022).
Sauvage, X., Jessner, P., Vurpillot, F. & Pippan, R. Nanostructure and properties of a Cu-Cr composite processed by severe plastic deformation. Scr. Mater. 58, 1125–1128 (2008).
Bachmaier, A., Kerber, M., Setman, D. & Pippan, R. The formation of supersaturated solid solutions in Fe-Cu alloys deformed by high-pressure torsion. Acta Mater. 60, 860–871 (2012).
Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 46, 1–184 (2001).
Hightower, A., Fultz, B. & Bowman, R. C. Mechanical alloying of Fe and Mg. J. Alloy. Compd. 252, 238–244 (1997).
Konstanchuk, I. G. et al. The hydriding properties of a mechanical alloy with composition Mg-25%Fe. J. Less-Common Met 131, 181–189 (1987).
Ivanov, E., Konstanchuk, I., Stepanov, A. & Boldyrev, V. Magnesium mechanical alloys for hydrogen storage. J. Less-Common Met 131, 25–29 (1987).
Mooij, L. et al. The effect of microstructure on the hydrogenation of Mg/Fe thin film multilayers. Int. J. Hydrog. Energy 39, 17092–17103 (2014).
Taheri, M. et al. Towards a physical description for the origin of enhanced catalytic activity of corroding magnesium surfaces. Electrochim. Acta 116, 396–403 (2014).
Buchheit, R. G., Grant, R. P., Hlava, P. F., Mckenzie, B. & Zender, G. L. Local dissolution phenomena associated with S Phase (Al2CuMg) particles in aluminum alloy 2024‐T3. J. Electrochem. Soc. 144, 2621–2628 (1997).
Birbilis, N., King, A. D., Thomas, S., Frankel, G. S. & Scully, J. R. Evidence for enhanced catalytic activity of magnesium arising from anodic dissolution. Electrochim. Acta 132, 277–283 (2014).
Lysne, D., Thomas, S., Hurley, M. F. & Birbilis, N. On the Fe enrichment during anodic polarization of Mg and its impact on hydrogen evolution. J. Electrochem. Soc. 162, C396–C402 (2015).
Michailidou, E., McMurray, H. N. & Williams, G. Quantifying the role of transition metal plating in the cathodic activation of corroding magnesium. ECS Trans. 75, 141–148 (2017).
Cihova, M. et al. The role of zinc in the biocorrosion behavior of resorbable Mg‒Zn‒Ca alloys. Acta Biomater. 100, 398–414 (2019).
Danaie, M., Asmussen, R. M., Jakupi, P., Shoesmith, D. W. & Botton, G. A. The role of aluminum distribution on the local corrosion resistance of the microstructure in a sand-cast AM50 alloy. Corros. Sci. 77, 151–163 (2013).
Pourbaix, M. Atlas of electrochemical equilibria in aqueous solutions. 2nd edn, (National Association of Corrosion Engineers, 1974).
Lamaka, S. V. et al. Local pH and its evolution near Mg alloy surfaces exposed to simulated body fluids. Adv. Mater. Interfaces 5, 1800169 (2018).
Esmaily, M. et al. Fundamentals and advances in magnesium alloy corrosion. Prog. Mater. Sci. 89, 92–193 (2017).
Cihova, M., Schmutz, P., Schäublin, R. & Löffler, J. F. Biocorrosion zoomed in: evidence for dealloying of nanometric intermetallic particles in magnesium alloys. Adv. Mater. 31, 1903080 (2019).
Ng, W. F., Chiu, K. Y. & Cheng, F. T. Effect of pH on the in vitro corrosion rate of magnesium degradable implant material. Mater. Sci. Eng. C. 30, 898–903 (2010).
Ding, Z. Y. et al. Exfoliation corrosion of extruded Mg-Li-Ca alloy. J. Mater. Sci. Technol. 34, 1550–1557 (2018).
Morishige, T., Doi, H., Goto, T., Nakamura, E. & Takenaka, T. Exfoliation corrosion behavior of cold-rolled Mg-14 mass% Li-1 mass% Al alloy in NaCl solution. Mater. Trans. 54, 1863–1866 (2013).
Robinson, M. J. The role of wedging stresses in the exfoliation corrosion of high strength aluminium alloys. Corros. Sci. 23, 887–899 (1983).
Zhou, J. et al. Accelerated degradation behavior and cytocompatibility of pure iron treated with sandblasting. ACS Appl. Mater. Interfaces 8, 26482–26492 (2016).
Bagherifard, S. et al. Accelerated biodegradation and improved mechanical performance of pure iron through surface grain refinement. Acta Biomater. 98, 88–102 (2019).
Kraus, T. et al. Biodegradable Fe-based alloys for use in osteosynthesis: outcome of an in vivo study after 52 weeks. Acta Biomater. 10, 3346–3353 (2014).
Hagi, H. Diffusion coefficient of hydrogen in iron without trapping by dislocations and impurities. Mater. Trans. JIM 35, 112–117 (1994).
Zheng, S., Wang, K., Oleshko, V. P. & Bendersky, L. A. Mg-Fe thin films: a phase-separated structure with fast kinetics of hydrogenation. J. Phys. Chem. C. 116, 21277–21284 (2012).
Song, G. L. in Corrosion of Magnesium Alloys, 1st edn, (Woodhead Publishing, 2011). https://doi.org/10.1533/9780857091413.1.3.
Koyama, M. et al. Recent progress in microstructural hydrogen mapping in steels: quantification, kinetic analysis, and multi-scale characterisation. Mater. Sci. Technol. 33, 1481–1496 (2017).
Nagumo, M. Fundamentals of hydrogen embrittlement, 1st edn, (Springer, Singapore, 2016). https://doi.org/10.1007/978-981-10-0161-1.
Guo, J. et al. Oxygen-mediated deformation and grain refinement in Cu-Fe nanocrystalline alloys. Acta Mater. 166, 281–293 (2019).
Guo, J. et al. Combined Fe and O effects on microstructural evolution and strengthening in CuFe nanocrystalline alloys. Mater. Sci. Eng. A 772, 138800 (2020).
Tabor, D. The Hardness and strength of metals. J. Inst. Met. 79, 1–18 (1951).
Suresh, S. Fatigue of Materials. 2nd edn, (Cambridge University Press, 1998). https://doi.org/10.1017/CBO9780511806575.
Li, N. & Zheng, Y. Novel magnesium alloys developed for biomedical application: a review. J. Mater. Sci. Technol. 29, 489–502 (2013).
Kapp, M. W., Hohenwarter, A., Wurster, S., Yang, B. & Pippan, R. Anisotropic deformation characteristics of an ultrafine- and nanolamellar pearlitic steel. Acta Mater. 106, 239–248 (2016).
Jin, W. et al. Improvement of corrosion resistance and biocompatibility of rare-earth WE43 magnesium alloy by neodymium self-ion implantation. Corros. Sci. 94, 142–155 (2015).
Martynenko, N. S. et al. Increasing strength and ductility of magnesium alloy WE43 by equal-channel angular pressing. Mater. Sci. Eng. A 712, 625–629 (2018).
Myrissa, A. et al. Gadolinium accumulation in organs of Sprague–Dawley® rats after implantation of a biodegradable magnesium-gadolinium alloy. Acta Biomater. 48, 521–529 (2017).
Zhang, Y. et al. Implant-derived magnesium induces local neuronal production of CGRP to improve bone-fracture healing in rats. Nat. Med. 22, 1160–1169 (2016).
Kapp, M. W. et al. Cyclically induced grain growth within shear bands investigated in UFG Ni by cyclic high pressure torsion. J. Mater. Res. 32, 4317–4326 (2017).
Zhilyaev, A. P. & Langdon, T. G. Using high-pressure torsion for metal processing: Fundamentals and applications. Prog. Mater. Sci. 53, 893–979 (2008).
Oliver, W. & Pharr, G. An improved technique for determining hardness and elastic modulus using load and displacement-sensing indentation systems. J. Mater. Res. 7, 1564–1583 (1992).
Hufenbach, J., Wendrock, H., Kochta, F., Kühn, U. & Gebert, A. Novel biodegradable Fe-Mn-C-S alloy with superior mechanical and corrosion properties. Mater. Lett. 186, 330–333 (2017).
Hofstetter, J. et al. High-strength low-alloy (HSLA) Mg-Zn-Ca alloys with excellent biodegradation performance. JOM 66, 566–572 (2014).
Zhang, E., He, W., Du, H. & Yang, K. Microstructure, mechanical properties and corrosion properties of Mg-Zn-Y alloys with low Zn content. Mater. Sci. Eng. A 488, 102–111 (2008).
Acknowledgements
This work was partly funded by the Austrian Academy of Sciences under via Innovation Fund project IF 2019–37 and by the Christian Doppler Research Association within the framework of the Christian Doppler Laboratory for Advanced Aluminum Alloys. Financial support from the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development and the Christian Doppler Research Association is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
O.R.: Conceptualization, Investigation, Writing – original draft. Irmgard Weißensteiner: Conceptualization, Investigation, Writing - original draft. M.C.: Data analysis and concept/experiment discussion, Writing –original draft. Eva Maria Steyskal: Conceptualization, Formal analysis, Writing – review & editing. N.S.: Conceptualization, Writing – review & editing. M.T.: Investigation and data analysis, Writing – review & editing. S.P.: Conceptualization, Methodology, Supervision, Writing - review & editing. P.S.: Data analysis and concept/experiment discussion, Writing – review & editing. J.E.: Conceptualization, Supervision, Writing – review & editing. P.J.U.: Conceptualization, Methodology, Supervision, Writing – review & editing. Reinhard Pippan: Conceptualization, Supervision, Writing – review & editing. A.M.W.: Conceptualization, Supervision, Writing – original draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Renk, O., Weißensteiner, I., Cihova, M. et al. Mitigating the detrimental effects of galvanic corrosion by nanoscale composite architecture design. npj Mater Degrad 6, 47 (2022). https://doi.org/10.1038/s41529-022-00256-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-022-00256-y
