
Abstract
Small organic molecules that modulate the degradation behavior of Mg constitute benign and useful materials to modify the service environment of light metal materials for specific applications. The vast chemical space of potentially effective compounds can be explored by machine learning-based quantitative structure-property relationship models, accelerating the discovery of potent dissolution modulators. Here, we demonstrate how unsupervised clustering of a large number of potential Mg dissolution modulators by structural similarities and sketch-maps can predict their experimental performance using a kernel ridge regression model. We compare the prediction accuracy of this approach to that of a prior artificial neural networks study. We confirm the robustness of our data-driven model by blind prediction of the dissolution modulating performance of 10 untested compounds. Finally, a workflow is presented that facilitates the automated discovery of chemicals with desired dissolution modulating properties from a commercial database. We subsequently prove this concept by blind validation of five chemicals.
Similar content being viewed by others
Introduction
As the lightest structural engineering metal, magnesium (Mg) is a promising material for advanced technologies that will ameliorate climate change through enhanced battery technologies and improved transport applications1,2. Magnesium is useful for light-weight automotive3,4,5 and aerospace components6,7, as anode material for energy storage systems8,9,10,11 and as base material for bioresorbable medical implants12,13,14,15,16,17. Due to high abundance, relatively low cost, and versatility, Mg and Mg-based alloys are being increasingly employed for these and other industrial applications. However, due to its comparably high chemical reactivity, many target implementations also require domain-specific tailoring of the degradation behavior of Mg. In transport applications, corrosion needs to be prevented to avoid material failure. In medical applications, where Mg is used in stents or bone screws, its corrosion rate needs to be controlled in an environment-specific way, as different treatments and/or patients imply different healing rates. For energy applications, for example, Mg-air primary batteries in which Mg is employed as anode material, a constant Mg dissolution rate is desired.
Clearly, benign degradation modulating strategies are needed for these applications. Several strategies, such as alloying and surface coatings, were developed to control the corrosion of Mg-based engineering materials18,19,20. However, these protective schemes need to be improved to achieve better control over the degradation properties of Mg. Small organic molecules, which form complexes with ions (e.g., iron) that accelerate the corrosion process, have shown great potential to control the dissolution properties of pure Mg materials and its alloys21. The properties of these modulators of magnesium dissolution can be tailored to specific target applications, e.g., as component of an active protective coating or as a part of the electrolyte of an Mg-air battery22,23,24,25. The massive advantage of organic dissolution modulators is their almost unlimited chemical space, providing countless potential solutions for almost all applications. The number of available organic compounds is increasing rapidly, with ∼120 million organic compounds being reported over the last decade alone26. It has been estimated that the number of organic compounds with potentially useful properties is ∼ 1063 and is thus essentially infinite27. Automation and robotics technologies are also expanding rapidly and enable modern combinatorial chemistry techniques that can synthesize larger and more diverse chemical libraries. Clearly, synergies with computer-assisted synthesis approaches will further extend this exponential rise in available organic compounds28.
Consequently, the most challenging task is to select molecules with beneficial properties for specific applications from this effectively infinite chemical space of small organic molecules. Experimental approaches alone cannot possibly explore more than a tiny fraction of the vast space of compounds with potentially useful dissolution modulating properties, despite impressive developments in high throughput techniques29,30,31,32. Fortunately, data-driven computational methods33,34,35,36,37,38,39,40 can efficiently explore larger areas of chemical space with orders of magnitude less time and effort. Hence, they offer a very efficient way to preselect a short list of promising candidates prior to experimental investigation. Additionally, computational techniques can provide deeper insight into the underlying chemical mechanisms and most important chemical functional moieties41,42,43,44,45,46. A combination of experimental and computational methods constitutes a sound foundation for a data-driven discovery of modulators. Machine learning techniques that model complex quantitative structure-property relationships can predict target properties of hitherto unsynthesized or untested compounds33,34,35,47,48. These methods require large, reliable, chemically diverse and balanced training data sets to make the most accurate predictions that can be generalizable to a broad range of materials. As data-driven models are not capable of reliably predicting the performance of molecules with poorly represented features (e.g., functional groups, elemental species) in the training data, the underlying training set has to reflect the complexity of the relevant chemical environment. Predictions made by these models are most accurate for compounds that lie within or in the neighborhood of the domain of applicability of the model (the span of values over which each molecular feature in the training set varies).
In an extensive experimental study employing hydrogen evolution experiments, Lamaka et al. measured the corrosion inhibition performance of over 150 organic compounds for nine distinct Mg-based materials21. Previously, a workflow was developed for modeling the experimentally-derived corrosion inhibition values in this database using high-throughput calculations and machine learning algorithms34. It was demonstrated in recent works33,49 that a single descriptor alone cannot adequately describe the complexity of materials degradation. In our recent study34, we employed the Smooth Overlap of Atomic Positions (SOAP) kernel50,51 and sketch-map52 to connect molecular similarities of 74 compounds to their inhibition efficiencies (IEs) for commercially pure Mg containing 220 ppm iron impurities (CPMg220). The SOAP kernel condenses the structural properties of all chemicals to pairwise similarity values, whereas sketch-map projects the resulting high-dimensional similarity matrix onto two dimensions. The resulting two-dimensional structure-property landscape elucidated the relationships between the molecular structure and corrosion inhibition performance by the formation of similarity clusters. We demonstrated that projecting untested organic compounds onto this map with out-of-sample embedding allows qualitative predictions of their corrosion inhibition performance by alignment to clusters in the similarity landscape. Importantly, the computed SOAP kernel can be employed directly as input to a kernel ridge regression (KRR)53 model that performs quantitative predictions of IEs for these unknown chemicals.
This study extends our previous modeling work and comprises three parts. Firstly, the robustness of the KRR model is benchmarked against an artificial neural network (ANN) model that was trained on a combination of atomistic and structural molecular descriptors33. To allow comparisons of the accuracy of the two approaches, both models were trained using identical training data. Secondly, the database was augmented by 74 additional compounds with unknown performance, of which 10 were subsequently used to validate the KRR model by blind prediction of their IEs. Clusters of molecular similarity in the corresponding sketch-map generated from all 152 molecules formed the basis for the selection process. Finally, a proof of concept workflow is presented that provides automated selection of untested compounds with promising properties for experimental testing by screening a large molecular database. These synergistic computational approaches should significantly improve the predictive power and model interpretation of the underlying machine learning models, thus paving the way for the discovery or rational design of bespoke Mg dissolution agents.
Results and Discussion
Comparison of model robustness
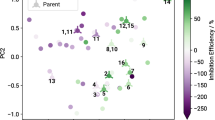
The KRR model was validated by comparing its key performance indicators to those of an ANN model generated in a recently published study33. Despite the ANN performed well in terms of prediction accuracy, its performance depends on the careful selection of molecular descriptors that can strongly influence the prediction outcome and interpretation of the models is often problematic. Hence, combining all structural features in a global similarity measure, defined by SOAP, provides an attractive approach to reduce the complexity of the model input. Employing the resulting SOAP kernel as input for KRR allows physically interpretable predictions that can be directly ascribed to the molecular structure. The KRR model was trained using the same data set and was validated using the same seven untested compounds as for the ANN model. The underlying SOAP kernel was generated for all 78 compounds, of which 71 had structural similarity values used as a training set and are represented on a sketch-map (Fig. 1). Hyperparameters for the SOAP kernel and KRR were fine-tuned in a grid search with k-fold cross validation (see Supplementary Methods and Supplementary Fig. 1 in the Supplementary Information), resulting in a hyperparameter set of rc = 3.0 Å, ξ = 0.3, ζ = 0.6 and γ = 0.3, as well as the regularization parameter σKRR = 11. All hyperparameters are defined and explained in the “Methods” section. The seven compounds used for external test set validation of the model predictions are projected onto this map and highlighted. It is clear that the four stronger inhibitors cluster together (see Fig. 1, right) while the three compounds having weak inhibition to strong dissolution properties are clustered near each other on the left-hand side of the sketch-map. The KRR model-predicted IEs (see Fig. 2) for the seven test compounds with a higher R2 of 0.79 and slightly higher root mean square error (RMSE) of 36% compared to the ANN model (R2 = 0.74, RMSE = 33%) from the earlier study (see Supplementary Fig. 2)33. However, with only seven compounds the difference between the ANN and KRR models is not highly significant. A Pearson rank correlation test was also conducted on the KRR-predicted and experimentally determined values. Similar to the ANN study there was a strong correlation (r = 0.89). The p-value of 0.007 indicates acceptable statistical significance for the model.

The sketch-map is based on molecular similarities and IEs (colored dots) of 71 compounds. Clusters with similar dissolution modulating properties indicate a structure-property relationship. Qualitative prediction of the inhibition efficiency for the seven displayed test compounds is obtained using out-of-sample embedding to project them onto the map.

Correlation of predicted test set IEs from the KRR model with experimental values from a prior study33. The blue line is a linear least square fit of the predicted and measured values. The RMSE value is in absolute percent. The orange, dashed line represents the ideal correlation. The error bars depict the standard deviation of the experimentally derived IE.
Validation of the predictive model
A blind prediction step is an excellent way to assess the predictive power of a model. Therefore, a second sketch-map was generated based on the SOAP kernel built from 74 small organic molecules with unknown experimental IE values and the 78 chemicals used to train the initial sketch-map and KRR models (Fig. 3). The untested compounds are depicted as gray dots while the already tested training set compounds are color-coded according to their IE. The untested compounds comprise chemicals with similar functional groups to those in the sketch-map training set. As these compounds sit in or near the domain of applicability of the original sketch-map model, the predictions are expected to be reliable. Only biologically benign, inexpensive, small organic molecules were included in the untested set. The molecular weight of training compounds was < 350 Da while the majority of the test set molecules (64) used for prediction had molecular weights <200 Da. Small anti-corrosion additives are required to increase the efficiency of protective coatings without impairing their structural integrity. Considering the IEs of the training structures, we identified and selected six clusters by visual inspection. Results of a k-means clustering substantiate the cluster definitions (see Supplementary Fig. 4). In Fig. 3, the mean IE values of molecules in the training set clusters decrease in the order b (71 ± 15%) > d (65 ± 6%) > c (50 ± 27%) > e (37 ± 57%) > f (3 ± 24%). Clearly, some of the untested compounds mapped onto clusters of modulators with inhibitory effects (b, c, d) while others generated new clusters (a) or were located in map regions corresponding to compounds with highly diverse properties, ranging from strong accelerators to effective inhibitors (f). The mean predicted IE values for molecules in the test set in defined clusters decrease in the order b (51 ± 9%) ≈ c (48 ± 14%) > d (39 ± 10%) ≈ a (35 ± 11%) > e (24 ± 5%) > f (−35 ± 12%), indicating a qualitatively accurate prediction.

The sketch-map is based on molecular similarities of 78 tested (colored according to IE) and 74 untested (depicted in gray) modulators. Six clusters are identified. The mean IEs of the clusters are shown in the lower left corner.
A total of 10 chemicals representative of each cluster, were randomly selected and tested experimentally under the same conditions as the compounds used for training of the sketch-map model. As the experimental performance of compounds located within cluster a is an uncharted area of the sketch-map, the number of compounds selected for the blind testing was in proportion to the size of the cluster. The general agreement between predicted and experimental values is good except for benzamide (see Table 1) that is predicted to have a moderate inhibiting effect (44%) whereas the experiment showed it was a dissolution accelerator with an IE of −43 ± 30%. The discrepancy may be due to benzamide precipitating at an inhibitor concentration of 0.01 m while the model was trained on data with a 0.05 m modulator concentration. Hence, compound 3 was excluded from correlation of experimental and predicted results (see Fig. 4). Additional information on the training and test errors of the KRR are provided in Supplementary Fig. 3. More detailed information on the experiments are provided in Supplementary Table 3. A Pearson rank correlation for the remaining nine chemicals resulted in a correlation coefficient of 0.85 and a p-value of 0.004. As Table 1 shows, most of the predicted IE values agree with the experimentally determined values within experimental error. It is noteworthy that the IE values of aliphatic compounds in the blind testing set (7, 8, 9, 10) are overestimated. The tetracarboxylic acid 7 is located in one of the tightest clusters d which might explain the small IE variation within the cluster (see Fig. 3). Chemicals in the training set that cluster in d have a mean inhibition efficiency of 65 ± 6%. Hence, untested compounds mapped close to this cluster should exhibit a similar IE. The in silico model estimates the IE value of 7 to be significantly lower than the mean value of the data points defining the cluster, a trend in agreement with the lower experimental value of 27 ± 17% IE. The complex speciation of the tetracarboxylic acid at pH between 7 and 10 may be responsible for some of the prediction error, which is nonetheless only slightly outside one standard deviation of the experimental error. The aliphatic diol 8 is located in cluster e (mean of 37 ± 57% IE). There are only two compounds allocated to this cluster. One acts as an efficient inhibitor while the other is a moderate corrosion accelerator, consistent with the very large standard error in the mean value of this cluster. However, the predicted performance of diol 8 has the correct trend with an IE value significantly lower than the mean IE of the cluster. The urea 10 and the amino acid 9 are located in cluster f that contains modulators also with highly diverse IEs ranging from weak inhibitors to potent accelerators. While the value of the piperidone compound 10 is quite accurate, the model heavily underestimates the accelerant properties of β-alanine (IEpred = −7 %) compared to the measured IE (−86 ± 17%) for CPMg220. Again, at a final pH around 10 the amino acid will be ionized, and the chemotype may not be adequately represented in the training data (a potential issue whenever the chemical diversity is large compared to the size of the data set). The accelerator properties of both compounds were confirmed by subsequent validation experiments.

Correlation between the mean of 10 experimental blind testing compounds and their predicted IE values. The error bars depict the standard deviation of the experimentally derived IE. The light blue line is the linear least squares fit of the predicted and measured values. The RMSE value is in percent. The orange, dashed line represents a perfect correlation. Benzamide was omitted because of precipitation during the experiment.
Contrary to the models’ general overestimation of IE values in the four aliphatic compounds, the predicted IE values for the five aromatic molecules 1, 2, 4, 5, 6 agree with the experimental IE values within experimental error. The isophthalate 6 is associated with the molecular cluster c and the predicted value of 55% IE is in good agreement with the mean of the compounds that were used for training of the model. The pyrazine derivative 1 and the pyridine derivative 2 are located within cluster a which represents an uncharted area of the sketch-map. Although both predictions are in good agreement with the experimentally derived values, more experimental data points are necessary to provide robustness for predictions in this region of similarity space. The neighboring cluster b includes the pyridine derivative 4 as well as the nitro-substituted modulator 5. The performance prediction of the two modulators is in good agreement with the experimental IEs. Modulators 4 and 5 have the highest predicted inhibiting effect that was confirmed by the conducted hydrogen evolution experiments. Furthermore, the similarity observed in trends for molecules in clusters a and b suggest they may in fact be members of a single larger cluster, something that could be confirmed by experimental IE measurements of chemicals lying between the two clusters. In summary, the predictions of the compounds in the blind test set is in qualitative agreement with the experimental IE values determined subsequently.
Uncharted similarity space
Of the 64 modulators whose predicted properties were not checked experimentally, 9 are located outside the defined clusters. The complete list of predicted values is provided in Supplementary Table 2 (IEKRR). One of these modulators lies close to cluster c and is likely to be a good inhibitor. Two others map to the top left area of the sketch-map close to cluster f so they will probably also be effective dissolution accelerators. The remaining six compounds map between clusters a, b and f. As a and b contain inhibiting agents while f contains weak inhibitors and accelerators, these six compounds are expected to show values of IE near zero when tested.
Chemical space – the final frontier
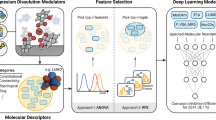
Clearly, the search for effective dissolution modulators in the vast chemical space of compounds with potentially useful properties requires very efficient tools. Manual selection of compounds for experimental screening is often biased by the individual chemical intuition, compound availability, cost, toxicity, and experience. Hence, whole regions of chemical space that may contain highly effective modulators (islands of utility) can easily be overlooked. This provided the motivation for the development of data-driven methods for unbiased identification of chemical leads, as depicted in Fig. 5. For a molecule xA with a target property yA, a molecule xB ∼ xA is likely to yield a target property yB ∼ yA, assuming a structure-property relationship. Once a cluster containing, for example, good corrosion inhibitors is identified, untested structures that map onto this cluster should have similar or superior corrosion inhibiting properties. Implementation of this approach requires a second, extensive database of potential candidates and a SOAP-REMatch kernel computed from the structures of both databases. Picking a structure of interest in the sketch-map results in the visualization of the most similar compounds of the second database, providing a basis for the automated discovery of corrosion modulators.

Picking a structure of interest in the sketch-map (based on database I) results in the visualization of the most similar compounds of the second database (database II).
The best-performing corrosion inhibitor 5 in the test set maps onto the edge of cluster b that is formed from modulators with high IE values. To check the model robustness concerning the periphery of observed clusters, this structure constitutes a promising starting point to screen a larger molecular database for similar compounds. The QM7b database54,55 contains 7211 compounds, thus potentially providing a pool of compounds not included in the training or test datasets. After computing the SOAP-REMatch kernel (rc = 3.0˚A, ξ = 0.3, ζ = 0.6, γ = 1.0) for all structures from this databases and the initial dataset (7211 + 152) the global similarity matrix (7363 × 7363 diagonally symmetric) was used to identify structurally similar compounds from the QM7b database. A sketch-map complemented with KRR-based IE predictions using this kernel is illustrated in Supplementary Fig. 5.
When searching for similar compounds in the QM7b database, a similarity submatrix (7363 × 7211) was used to eliminate hits in the 152-member training and test sets. This identified five structures with the highest similarity to 2-nitrophthalic acid (5) and predicted their IE values (Supplementary Fig. 5). However, the hit molecules found in this proof of principle example are biased because the QM7b database only contains molecules with ≤7 heavy atoms, whereas molecule 5 contains 13 heavy atoms. Clearly, the quality of hits is highly dependent on the properties of the database used. Screening of databases containing larger molecules (e.g. the GDB-13 which contains roughly 1 billion structures) will undoubtedly increase the value of the proposed workflow albeit with an increased computational cost for computation of SOAP kernels. Also, when screening large databases, it is very important to understand how the model’s domain of applicability maps onto the database, as molecules well outside the model domain will be poorly predicted. Nevertheless, the hits in this example still replicate local structural motifs in substantially smaller molecules that may play a key role in corrosion inhibition behavior.
To assess the accuracy of the workflow in the discovery of lead structures, a database provided by Thermo Fisher Scientific, containing 7094 commercially available small organic chemicals, was subsequently screened. Again, the SOAP-REMatch kernel (rc = 3.0˚A, ξ = 0.3, ζ = 0.6, γ = 1.0) was computed for all structures (7094 + 152). A sketch-map colored with KRR-based IE predictions using this kernel is depicted in Fig. 6. For the selected 2-nitrophthalic acid (5), five chemicals yielding high structural similarities are chosen from a similarity submatrix in the kernel (7246 × 7094). Albeit these chemicals exhibit high similarities in the high-dimensional space, they are not necessarily mapped in close proximity in the two-dimensional space due to the nature of the sketch-map algorithm as displayed in the inlay in the lower right corner of Fig. 6. However, all selected structures are mapped close to each other and to already tested structures. The predicted IE values are subsequently validated in hydrogen evolution experiments following the test procedure described in the preceding section. A comparison between KRR-predicted and experimental IEs is presented in Table 2. More detailed information on the experiments is summarized in Supplementary Table 4.

The dots are colored according to predicted IEs by means of KRR. The KRR model was trained on 78 experimental IEs (black-rimmed dots). By referencing the selected 2-nitrophthalic acid (5, large black-rimmed dot) to the underlying SOAP kernel, five highly similar molecules can be determined from the global similarity matrix (dashed black-rimmed dots) along with their predicted IE values. As illustrated in the inlay, high similarities in the high-dimensional space do not automatically result in close proximity in two-dimensional space.
Despite two outliers, the results show good to excellent correlation between prediction and experiment (R2 = 0.84, RMSE = 36%). All five candidates were predicted to exhibit an inhibiting effect. It is noteworthy, that the prediction for the two phthalic acid derivatives 12 and 13 as well as the tricarboxylic acid 14 are in very good agreement with the experimental investigation. However, compounds 11 and 15 only exhibit a moderate inhibiting effect on the corrosion of CPMg in contrast to the comparably high IE values that are predicted by the KRR model. An important mechanism of corrosion inhibition for Mg-based materials is the capability to form complexes with iron ions (Fe2+/Fe3+) that are released during the corrosion process. Here, substitution of the aromatic system with electron withdrawing groups also affects the interaction strength of the carboxylate with Fe which in turn influences the IE56. Although carboxylic acids 11 and 15 contain nitro groups in ortho and para position with similar electron withdrawing effects, these two compounds also lack the vicinal dicarboxylate moiety present in the other inhibitors that is an important chelating functionality for metals (as in the chelating agent EDTA)57. This explains the differences in the experimental performance. As mentioned previously, the KRR model that we employed here is based on structural similarities and does not consider electronic properties. Hence, it may only indirectly learn that there is a correlation between electronic effects and structure. It is obvious that the underlying training database does not contain a sufficient amount of structures that exhibit the substitution pattern. Thus, the performance of the two compounds is overestimated but still qualitatively correct. Contrary to this, the predicted values for the two phthalic acid derivatives 12 and 13 are highly accurate despite the presence of a nitro moiety. This can be explained by the fact that each of the compounds bears a second vicinal carboxyl group (chelating effect) that is not affected by substitution of the aromatic system with a nitro functionality. Hence, they display higher degrees of inhibition in the experiment and are in excellent agreement with the predicted values. For a larger training dataset, the cutoff radius rc of the SOAP kernel can be adapted to higher values to better capture the impact of such structural features.
In summary, machine learning models based on structural similarities and kernel ridge regression (KRR) were generated that predict the ability of small organic compounds to modulate the corrosion of commercially pure magnesium (CPMg220). The accuracies of the models were determined by test set property predictions and by comparison with an artificial neural network (ANN) model from a prior study using identical training and testing data. The ANN and the KRR-based models both make qualitatively correct predictions of the modulation properties of compounds in test sets for the corrosion of CPMg220. A total of 74 untested compounds were subsequently mapped into a sketch-map model along with 78 modulators with experimentally known IE values. The sketch-map contained six main clusters of molecular similarity. While five of the clusters mapped both tested and uninvestigated chemicals, one cluster was exclusively comprised of unknown dissolution modulating molecules. To further assess the robustness of the KRR model, 10 compounds were selected for a blind testing study, taking at least one compound from each cluster. The results support the claim that the model can predict the effect of small organic molecules on the corrosion of CPMg220, as the predicted values are in good agreement with the experimental values. This suggests that the predicted IE values for the remaining 64 compounds are likely to be good estimates. These modeling methods constitute a promising way to rapidly identify the most promising molecules for specific target applications that have reduced toxicity, environmental impact, and cost. Furthermore, the validation results of the employed model show that solubility is an additional important factor that needs to be considered in the selection process of target chemicals, where such data is available. However, further experimental validation is an important final step, especially for chemicals in unlabeled regions of the sketch-map. Expanding the size, diversity, and quality of data used to train these models is also needed to improve their robustness and domain of applicability. This could include using molecular structures closer to their “mode of action”, i.e. solvated, at correct pH or in complex formations. Additionally, clustering the data already in high-dimensional space, for example employing the probabilistic analysis of molecular motifs (PAMM) approach, could increase the accuracy of cluster definitions and thus the qualitative predictive performance of the developed model. Finally, a Python-based workflow for automated screening of large molecular databases using chemical similarities has been developed. The findings provide a proof of concept for the proposed method—a promising strategy for an unbiased identification of efficient candidates to combat the degradation of Mg-based materials. Naturally, the presented concept is not limited to Mg and can be adopted to explore the structure-property landscape of e.g. Al-, Cu- and steel-based materials in a similar fashion by employing a corresponding experimental database to train the model. Clearly, the employed machine learning-based strategies facilitate an intuitive and fast screening of large databases to identify similar compounds, simplifying the search for modulators with potentially useful properties, and dramatically decreasing the time and resources required relative to those for experimental discovery methods.
Methods
Corrosion experiments
As commercial magnesium processing includes several steps58, preventing the inclusion of metallic impurities, such as iron, is nearly impossible. These impurities generate local galvanic cells in the material that accelerate corrosion and increase hydrogen evolution and Mg dissolution. As Mg dissolution predominantly occurs in intermetallic contact areas, the process releases impurities that re-deposit on the surface, thus increasing the cathodic area and the corrosion rate59. Molecules that form stable complexes with the impurities (e.g. Fe2+/3+) constitute a promising strategy to modulate the degradation properties of Mg. They also provide starting points for building an extensive database of magnesium dissolution modulators21,60.
In corrosion experiments, hydrogen evolution is measured in the presence of modulators and referenced to the sodium chloride electrolyte in the absence of these compounds. The effect of a modulator is quantified by the inhibition efficiency (IE), which is positive for corrosion inhibitors and negative for corrosion accelerators. We used IEs for CPMg220 from an experimental database of modulator performance, collected in a prior work21, to train the machine learning model34. Only agents with a molar concentration of 0.05 m in the experiment were selected. The full chemical composition of CPMg220 from Optical Discharge Emission Spectroscopy (SPECTROLAB with Spark Analyser Vision Software) is listed in Supplementary Table 1. Additional hydrogen evolution experiments were performed to validate the predictions of the model and to extend the database using the same experimental set-up and CPMg220 alloy as reported by Feiler et al33. Eudiometers (Art. Nr. 259110-500 from Neubert-Glas, Germany) were used for these investigations. Water displaced by evolved hydrogen was automatically quantified (SKX series from OHAUS coupled with USB data logger OHAUS 30268984) and the data recorded for further processing using an in-house Python script61. A flask below the eudiometer was filled with a piece of the bulk Mg sample and 500 mL of electrolyte (0.5wt.% NaCl) without (reference) and with addition of a dissolution modulator. The reference value was determined from the normalized volume of hydrogen evolved \(\left( {V_{0,{\mathrm{H}}_2}} \right)\) after 20 h of immersion. Mg samples were also exposed to an electrolyte solution containing 0.05 m of dissolution modulator for 20 h with the initial pH being adjusted by NaOH/HCl to 6.8 ± 0.5 and the volume of evolved hydrogen quantified \(\left( {V_{{\mathrm{Inh}},{\mathrm{H}}_2}} \right)\). The testing time is considered to be sufficient as the hydrogen evolution rate is in a steady state after ∼10 h21,61. The impact of the modulator on the corrosion of magnesium is given by the inhibition efficiency according to the following equation:
Molecular similarity
Apart from the quality, quantity, and diversity of data used to train machine learning models, the largest determinant of model quality is the type of molecular descriptors or features used to represent the organic molecule modulators. SMILES strings (Simplified Molecular Input Line-Entry System, text-based representations of the structure of almost any organic molecule) were used to generate molecular structures using OpenBabel62. In contrast to the previous work where the molecular geometries were first optimized using density functional theory (DFT) and an implicit solvent model34, here the structural optimization tool of Avogadro63 is used that employs the accurate but computationally less expensive GAFF force field64 to obtain optimized geometries in vacuo. The structural and chemical similarities between the dissolution modulators were transformed to high-dimensional space using the SOAP kernel in combination with a regularized entropy match (REMatch) strategy50,51. While the SOAP kernel compares local atomic environments of the molecular compounds, the REMatch kernel condenses the local similarities between two structures into a global similarity measure. A local environment is defined in a spherical region of radius rc around an atom and is built by a superposition of Gaussian functions with width ξ. The structural information surrounding an atom directly correlates with the size of rc. The SOAP kernel contains the translationally and rotationally invariant overlap between two local environments and can be raised to a power ζ for discrimination between medium (ζ < 0.6) and large (ζ ≈ 0.9) similarities. The hyperparameter γ of the REMatch kernel controls which local similarities are combined. For small values (γ ∼ 0.01) only the best matching pairs of local environments are included while for large values (γ ∼ 10) similar weights are assigned to the local similarities for computing the global similarity (see De et al. 51 and the Supplementary Methods for more details). To facilitate the interpretation of the high-dimensional SOAP-REMatch kernel and to allow a correlation with experimental data, the similarity information was transformed into distances65 and projected into two-dimensions using a sketch-map representation52. By applying a sigmoid function to the distances (influenced by the switching distance σ, with a and b as hyperparameters), distant/close (dissimilar/similar) structures in the high-dimensional space maintain their relationships in the low-dimensional space. Due to the shape of the sigmoid function, points that are far apart in the high-dimensional space are not necessarily represented that way in the low-dimensional projection, making an interpretation of distances between basins in the two-dimensional projection unphysical. However, the relative positions of structures and the formation of clusters in the two-dimensional similarity landscape are powerful aids for intuition assessing the molecular similarity of molecules in the data set.
Supervised and unsupervised learning
Machine learning methods are trained on experimental data and molecular descriptors (features) that are mathematical representations of molecular and physicochemical properties of small organic molecules. After assigning the IEs of magnesium dissolution modulators to all structures, the SOAP-REMatch kernel and sketch-map methods can be used with KRR for qualitative and quantitative prediction of the target values, respectively. Once a two-dimensional sketch-map visualization is created and labeled with the appropriate IEs, clusters can form that predominantly contain compounds with similar molecular properties, thus indicating a structure-property relationship. Clusters can be identified by visual inspection or using a variety of different clustering algorithms in low- and high-dimensional space66,67. Untested candidates can be projected onto the sketch-map using out-of-sample embedding, a reproduction of the distances to previously defined landmark points52, to obtain qualitative predictions of the potential degradation modulating effect of these unknown materials based on their relative locations to map clusters. Clearly, an unknown compound projected into, or close to, a cluster dominated by a particular molecular property would be expected to show similar behavior. Although this approach is helpful to obtain an estimate of a compound’s effect on the corrosion rate, some applications require quantitative predictions. The sketch-map visualization can be complemented with quantitative predictions of the IE using a KRR model. The synergistic combination of both methods provides a powerful approach for the design of magnesium corrosion modulators that exploits the great efficiencies of in silico methods. Thus, a comprehensive sketch-map based on a SOAP-REMatch kernel from all structures in a training and test set can be used to virtually screen a large number of potential candidates. Concurrently, their degradation modulating performance can be predicted with KRR, either to validate the inhibition performance of a known modulator, or to predict the degradation modulating properties of unsynthesized and/or untested organic compounds.
Data availability
The authors declare that the main data supporting the findings of this study are available within the paper and its Supplementary Material. The similarity-based discovery workflow is available at www.exchem.de in form of an interactive web application called ExChem where interested readers can explore the landscape of corrosion inhibitors by themselves. Other relevant data are available from the corresponding author upon reasonable request.
Code availability
The authors declare that all written code of the similarity-based discovery workflow ExChem (www.exchem.de) is available at 10.5281/zenodo.4302405 along with the related data. The source code of ExChem is published under the GNU GPLv3 license.
References
A. Vadiraj, M. Abraham, and A. S. Bharadwaj, Trends in Automotive Light Weighting, in Light Weighting for Defense, Aerospace, and Transportation., (ed. A. Gokhale, N. Prasad, and B. Basu) 89-102 (Indian Institute of Metals Series, Singapore, 2019).
Gielen, D., Boshell, F. & Saygin, D. Climate and energy challenges for materials science. Nat. Mater. 15, 117–120 (2016).
Taub, A. I. & Luo, A. A. Advanced lightweight materials and manufacturing processes for automotive applications. MRS Bull. 40, 1045 (2015).
Joost, W. J. & Krajewski, P. E. Towards magnesium alloys for high-volume automotive applications. Scr. Mater. 128, 107 (2017).
Blawert, C., Hort, N. & Kainer, K. Automotive applications of magnesium and its alloys. Trans. Indian Inst. Met. 57, 397 (2004).
Dziubinska, A., Gontarz, A., Dziubinski, M. & Barszcz, M. The forming of magnesium alloy forgings for aircraft and automotive applications. Adv. Sci. Technol. Res. J. 10, 158 (2016).
Gupta, M. & Gupta, N. Utilizing magnesium based materials to reduce green house gas emissions in aerospace sector. Aeron. Aero. Open Access J. 1, 41–46 (2017).
Ma, Z., MacFarlane, D. R. & Kar, M. Mg cathode materials and electrolytes for rechargeable Mg batteries: a review. Batter Supercaps 2, 115 (2019).
Höche, D. et al. Performance boost for primary magnesium cells using iron complexing agents as electrolyte additives. Sci. Rep. 8, 1 (2018).
Zhang, Y. et al. Magnesium storage performance and mechanism of 2D-ultrathin nanosheet-assembled spinel MgIn2S4 cathode for high-temperature Mg batteries. Small 15, 1902236 (2019).
Yoo, H. D. et al. Intercalation of magnesium into a layered vanadium oxide with high capacity. ACS Energy Lett. 4, 1528 (2019).
R. Willumeit-Römer, N. Ahmad Agha, and B. Luthringer, Degradable magnesium implants—assessment of the current situation, in TMS Annual Meeting & Exhibition, 405–411 (Minerals, Metals & Materials Series, Springer, Cham. 2018).
Lee, J. W. et al. Long-term clinical study and multiscale analysis of in vivo biodegradation mechanism of Mg alloy. Proc. Natl Acad. Sci. USA 113, 716 (2016).
Brar, H. S., Platt, M. O., Sarntinoranont, M., Martin, P. I. & Manuel, M. V. Magnesium as a biodegradable and bioabsorbable material for medical implants. JOM 61, 31 (2009).
Witte, F. et al. Degradable biomaterials based on magnesium corrosion. Curr. Opin. Solid State Mater. Sci. 12, 63 (2008).
Luthringer, B. J., Feyerabend, F. & Willumeit-Römer, R. Magnesium-based implants: a mini-review. Magnes. Res. 27, 142 (2014).
Santos-Coquillat, A. et al. PEO coatings design for Mg-Ca alloy for cardiovascular stent and bone regeneration applications. Mater. Sci. Eng. C 105, 110026 (2019).
Blawert, C., Dietzel, W., Ghali, E. & Song, G. Anodizing treatments for magnesium alloys and their effect on corrosion resistance in various environments. Adv. Eng. Mater. 8, 511 (2006).
Jia, Z. et al. Inhibitor encapsulated, selfhealable and cytocompatible chitosan multilayer coating on biodegradable Mg alloy: a pH-responsive design. J. Mater. Chem. B 4, 2498 (2016).
Gray, J. E. & Luan, B. Protective coatings on magnesium and its alloys - a critical review. J. Alloy. Compd. 336, 88 (2002).
Lamaka, S. V. et al. Comprehensive screening of Mg corrosion inhibitors. Corros. Sci. 128, 224 (2017).
Wang, L. et al. Tailoring electrolyte additives for controlled Mg-Ca anode activity in aqueous Mg-air batteries. J. Power Sources 460, 228106 (2020).
Snihirova, D. et al. Synergistic mixture of electrolyte additives: a route to a high-efficiency Mg-air battery. J. Phys. Chem. Lett. 11, 8790–8798 (2020).
Raps, D. et al. Electrochemical study of inhibitor-containing organic–inorganic hybrid coatings on AA2024. Corros. Sci. 51, 1012–1021 (2009).
Yang, J. et al. Corrosion protection properties of inhibitor containing hybrid PEO-epoxy coating on magnesium. Corros. Sci. 140, 99–110 (2018).
Lipkus, A. H. et al. Structural diversity of organic chemistry. A scaffold analysis of the CAS Registry. J. Org. Chem. 73, 4443 (2008).
Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W. & Jhoti, H. Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15, 605–619 (2016).
Li, J. et al. Synthesis of many different types of organic small molecules using one automated process. Science 347, 1221 (2015).
García, S. J. et al. The influence of pH on corrosion inhibitor selection for 2024-T3 aluminium alloy assessed by high-throughput multielectrode and potentiodynamic testing. Electrochim. Acta 55, 2457 (2010).
White, P. A. et al. A new high throughput method for corrosion testing. Corros. Sci. 58, 327 (2012).
Muster, T. H. et al. A rapid screening multi-electrode method for the evaluation of corrosion inhibitors. Electrochim. Acta 54, 3402 (2009).
Meeusen, M. et al. A complementary electrochemical approach for time-resolved evaluation of corrosion inhibitor performance. J. Electrochem. Soc. 166, 11 (2019).
Feiler, C. et al. In silico screening of modulators of magnesium dissolution. Corros. Sci. 163, 108245 (2019).
Würger, T. et al. Data science based Mg corrosion engineering. Front. Mater. 6, 53 (2019).
Winkler, D. A. Predicting the performance of organic corrosion inhibitors. Metals 7, 553 (2017).
Fernandez, M., Breedon, M., Cole, I. S. & Barnard, A. S. Modeling corrosion inhibition efficacy of small organic molecules as non-toxic chromate alternatives using comparative molecular surface analysis (CoMSA). Chemosphere 160, 80 (2016).
Chen, F. F. et al. Correlation between molecular features and electrochemical properties using an artificial neural network. Mater. Des. 112, 410 (2016).
Winkler, D. A. et al. Towards chromate-free corrosion inhibitors: structure-property models for organic alternatives. Green. Chem. 16, 3349 (2014).
Le, T. C. & Winkler, D. A. Discovery and optimization of materials using evolutionary approaches. Chem. Rev. 116, 6107–6132 (2016).
Segler, M. H., Preuss, M. & Waller, M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604 (2018).
Yuwono, J. A., Taylor, C. D., Frankel, G. S., Birbilis, N. & Fajardo, S. Understanding the enhanced rates of hydrogen evolution on dissolving magnesium. Electrochem. Commun. 104, 106482 (2019).
Milosev, I. et al. Editors’ choice-the effect of anchor group and alkyl backbone chain on performance of organic compounds as corrosion inhibitors for aluminum investigated using an integrative experimental-modeling approach. J. Electrochem. Soc. 167, 061509 (2020).
Poberznik, M., Chiter, F., Milosev, I., Marcus, P. & Kokalj, A. DFT study of n -alkyl carboxylic acids on oxidized aluminum surfaces: from standalone molecules to self-assembled-monolayers. Appl. Surf. Sci. 525, 146156 (2020).
Feiler, C., Mei, D., Luthringer, B., Lamaka, S. V. & Zheludkevich, M. L. Rational design of effective Mg degradation modulators. Corrosion 7, 3597 (2020).
Würger, T., Feiler, C., Vonbun-Feldbauer, G. B., Zheludkevich, M. L. & Meißner, R. H. A first-principles analysis of the charge transfer in magnesium corrosion. Sci. Rep. 10, 15006 (2020).
Fockaert, L. et al. ATR-FTIR in kretschmann configuration integrated with electrochemical cell as in situ interfacial sensitive tool to study corrosion inhibitors for magnesium substrates. Electrochim. Acta 345, 136166 (2020).
Winkler, D. A. et al. Using high throughput experimental data and in silico models to discover alternatives to toxic chromate corrosion inhibitors. Corros. Sci. 106, 229 (2016).
Galvao, T. L., Novell-Leruth, G., Kuznetsova, A., Tedim, J. & Gomes, J. R. Elucidating Structure-Property Relationships in Aluminum Alloy Corrosion Inhibitors by Machine Learning. J. Phys. Chem. C. 124, 5624 (2020).
A. Kokalj et. al., Simplistic correlations between molecular electronic properties and inhibition efficiencies: Do they really exist?, Corros. Sci., https://doi.org/10.1016/j.corsci.2020.108856 (2020).
Bartók, A. P., Kondor, R. & Csanyi, G. On representing chemical environments. Phys. Rev. 87, 184115 (2013).
De, S., Bartók, A. P., Csanyi, G. & Ceriotti, M. Comparing molecules and solids across structural and alchemical space. Phys. Chem. Chem. Phys. 18, 13754 (2016).
Ceriotti, M., Tribello, G. A. & Parrinello, M. Simplifying the representation of complex free-energy landscapes using sketch-map. Proc. Natl Acad. Sci. USA 108, 13023 (2011).
V. Vovk, Kernel ridge regression, in Empirical Inference: Festschrift in Honor of Vladimir N. Vapnik 105–116 (Springer Berlin Heidelberg, 2013).
Montavon, G. et al. Machine learning of molecular electronic properties in chemical compound space. N. J. Phys. 15, 1 (2013).
Blum, L. C. & Reymond, J. L. 970 Million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 131, 8732 (2009).
Hansch, C., Leo, A. & Taft, R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195 (1991).
Byegård, J., Skarnemark, G. & Skålberg, M. The stability of some metal EDTA, DTPA and DOTA complexes: application as tracers in groundwater studies. J. Radioanal. Nucl. 241, 281–290 (1999).
M. Pekguleryuz, K. Kainer, and A. A. Kaya, Fundamentals of Magnesium Alloy Metallurgy, in Metals and Surface Engineering 1–368 (Woodhead Publishing Limited, Philadelphia, 2013).
Höche, D. et al. The effect of iron re-deposition on the corrosion of impurity-containing magnesium. Phys. Chem. Chem. Phys. 18, 1279 (2016).
Lamaka, S. V., Höche, D., Petrauskas, R. P., Blawert, C. & Zheludkevich, M. L. A new concept for corrosion inhibition of magnesium: Suppression of iron re-deposition. Electrochem. Comm. 62, 5 (2016).
Mei, D., Lamaka, S. V., Feiler, C. & Zheludkevich, M. L. The effect of small-molecule bio-relevant organic components at low concentration on the corrosion of commercially pure Mg and Mg-0.8Ca alloy: an overall perspective. Corros. Sci. 153, 258 (2019).
O’Boyle, N. M. et al. Open babel: an open chemical toolbox. J. Cheminform. 3, 33 (2011).
Hanwell, M. D. et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17 (2012).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general Amber force field. J. Comp. Chem. 25, 1157 (2004).
C. Berg, J. P. R. Christensen, and P. Ressel, Harmonic Analysis on Semigroups: Theory of Positive Definite and Related Functions (Springer, New York, 1984).
Ceriotti, M. Unsupervised machine learning in atomistic simulations, between predictions and understanding. J. Chem. Phys. 150, 150901 (2019).
Gasparotto, P., Meißner, R. H. & Ceriotti, M. Recognizing local and global structural motifs at the atomic scale. J. Chem. Theory Comput. 14, 486–498 (2018).
Acknowledgements
Funding by HZG MMDi IDEA project is gratefully acknowledged. DM thanks China Scholarship Council for the award of fellowship and funding (No. 201607040051). T.W., D.A.W., and C.F. gratefully acknowledge funding by the Deutscher Akademischer Austauschdienst (DAAD, German Academic Exchange Service) via Projektnummer 57511455. R.M. gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (D.F.G., German Research Foundation) via Projektnummer 192346071—SFB 986 and Projektnummer 390794421—GRK 2462. The authors thank Thermo Fisher Scientific for providing a chemical database that was used to validate the similarity-based discovery workflow in this study.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
T.W., D.M., B.V., D.A.W., S.V.L., M.L.Z., R.H.M., and C.F.: contributed to the conception and design of the study. D.M., B.V., and S.V.L.: provided experimental data. T.W., D.A.W., R.H.M., C.F.: provided the machine learning model. T.W. and C.F.: wrote the first draft of the manuscript. All authors contributed to the manuscript revision, read, and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Provision of the chemical database by Thermo Fisher Scientific occurred without any commercial or financial motivation.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Würger, T., Mei, D., Vaghefinazari, B. et al. Exploring structure-property relationships in magnesium dissolution modulators. npj Mater Degrad 5, 2 (2021). https://doi.org/10.1038/s41529-020-00148-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-00148-z
This article is cited by
-
Laying the experimental foundation for corrosion inhibitor discovery through machine learning
npj Materials Degradation (2024)
-
DATACORTECH: artificial intelligence platform for the virtual screen of aluminum corrosion inhibitors
npj Materials Degradation (2024)
-
Predicting corrosion inhibition efficiencies of small organic molecules using data-driven techniques
npj Materials Degradation (2023)
-
Searching the chemical space for effective magnesium dissolution modulators: a deep learning approach using sparse features
npj Materials Degradation (2023)
-
Estimating pitting descriptors of 316 L stainless steel by machine learning and statistical analysis
npj Materials Degradation (2023)
