
Abstract
The atomistic mechanism of chloride-induced depassivation of iron is still debated. A recent study suggests a four-stage depassivation mechanism, in general agreement with the point defect model. The proposed four-stage mechanism is based on reactive force field molecular dynamics simulations and is rather complex but here we use density functional theory to confirm the thermodynamic feasibility of the proposed mechanism. We find that the four surface species, formed in the four stages, have decreasing surface stability, which is consistent with the order of species formed in the depassivation process proposed in the reactive force field molecular dynamics study. The Fe vacancy formation energy, that is the energy needed to form a surface Fe vacancy by removing different surface species, indicates that surface species with more chlorides dissolve more easily from the surface, suggesting that chloride acts as catalyst in the iron dissolution process. The results are consistent with the suggested four-stage reaction mechanism and the point defect model.
Similar content being viewed by others

Introduction
Concrete is a highly alkaline media (pH > 13) in which a passive film forms on the steel surface, protecting it from active corrosion. The passive film has a multi-layer structure, which contains FeIII-rich oxides/oxyhydroxides in the outer layer and FeII-rich oxide/oxyhydroxides in the inner layer1,2,3,4,5,6. An intermediate layer between the inner and outer layers has also been reported1,6. However, aggressive ions such as chloride can cause depassivation and activate corrosion of iron, which is of great importance for the durability of the reinforced concrete structures that are exposed to the chloride-containing salts. Understanding the mechanism and the cause of the depassivation of this protective film is therefore a critical step in increasing the service life of current and future infrastructures.
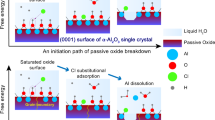
The atomistic depassivation mechanism of iron is still not well understood, but a four-stage chloride-induced depassivation mechanism was proposed recently based on reactive force field molecular dynamics (Reaxff-MD) simulations7. In this study, the depassivation process was investigated using an iron passive film created by an earlier Reaxff-MD study6 and various NaCl concentrations (2 M, 5 M and 10 M) in 0.316 M NaOH solution at a pH ≈ 13.5 with a total simulation time of 500 s at an applied external potential of 30 MeV. The proposed four-stage mechanism is illustrated in Fig. 1. Each of the four stages start with high local coverage of chloride (three chlorides per Fe atom) interacting with a surface Fe atom with lower charge (lower oxidation state), pulling the Fe atom out of the surface by approximately 0.5 Å. In OH--rich condition three and two adsorbed chlorides are replaced by hydroxide forming Fe(OH)3 and Fe(OH)2Cl respectively which are stable on the surface. Due to these first two stages, the local concentration of OH− near the surface decreases, causing OH− deficiencies and local acidification, leading to reduction of available OH− to replace the adsorbed chloride. Therefore, in the third and fourth stages, only one or none of the adsorbed chloride is replaced, forming Fe(OH)Cl2 and FeCl3 respectively. These surface species can dissolve into the solution, releasing chlorides back to the solution, while creating iron vacancies that diffuse toward the oxide-film/metal interface as suggested in previous studies7,8. This indicates that chloride can act as catalyst in the depassivation process consistent with previous density functional theory (DFT) studies8,9, while also providing support for the point defect model10,11,12,13. The four-step process for the initiation of the passive film breakdown also explains the concept of the critical chloride threshold, which requires the concentration of chlorides in the electrolyte to exceed certain threshold to initiate the depassivation process. The Reaxff-MD simulations show that a local acidification followed by iron dissolution and vacancy formation can only occur if there is a sufficient number of chloride ions in the electrolyte, in general agreement with the critical chloride threshold concept.

a Stage 1, b Stage 2, c Stage 3, d Stage 4. The solution is highly alkaline and contains high Cl concentration. The iron atoms in different colors represent the different Fe surface atoms reacting in the four stages. The reacted Fe atoms have lower charges which facilitate the Cl/OH interactions. The iron vacancy, resulting from iron complex dissolution, is represented using dashed circle. Previous studies7,8 show that these vacancies diffuse through the oxide film toward the film/metal interface to create discontinuities eventually leading to detachment/breakdown of the film, similar to the mechanism proposed by the point defect model. These stages are not shown in this figure since they were not studied herein.
Here we use DFT to investigate the thermodynamics of this four-stage mechanism, including the stability of the surface species and the dissolution of the surface species. Although it is more computationally expensive than Reaxff-MD, hence requires a more simplified analysis domain, DFT provides more accurate evaluations of relative energies and thermodynamic feasibility of the proposed depassivation mechanism. The effects of aqueous phase, external potential and pH are not included in this short communication but would surely be of interest for future studies.
Results
The structural changes caused by adsorbates
The passive film used in the Reaxff-MD study is a mixed oxide film build through simulations of the oxidation process in an alkaline media6. The mixed oxide film is too complex for periodic DFT calculations, but the critical Fe site, with lower charged Fe atom, is well represented by a step edge in a periodic crystal structure. The stepped surface used for all calculations is a stepped α-Fe2O3 (0001) surface with an oxygen vacancy near the step edge on the upper terrace (Fig. 2). More details on this model and the step structure can be found elsewhere9. Previous DFT study9 suggests that a Fe sites at low oxidation state (~FeII) near the step edge and oxygen vacancies can facilitate surface adsorption for Cl at high local coverage. These lower oxidation state sites can adsorb up to three Cl while regular Fe sites (FeIII) have weaker Cl interactions and can adsorb one Cl at most. This is consistent with the adsorption configuration observed in the Reaxff-MD study, where lower charged Fe atoms interact more readily with multiple Cl or OH adsorbates7.

The oxygen vacancy is shown using a dashed circle. The simulation box is represented using the black dotted line. Fe and O atoms are shown as larger pink and smaller red spheres, respectively. The edge Fe atom is shown in orange. This color scheme is used throughout the paper unless otherwise specified. All atoms below the blue horizontal dashed line are fixed during the calculation.
There are four types of surface species involved in the depassivation process described by the Reaxff-MD study:7 Fe(OH)3(ads), Fe(OH)2Cl(ads), Fe(OH)Cl2(ads), and FeCl3(ads). All of these surface species cause large structural changes on the step edge (Fig. 3), indicative of the strong interactions with the metal oxide. Formation of Fe(OH)3(ads) and Fe(OH)2Cl(ads) have the same effect on the surface structure, causing the edge Fe atom to move upward by 1.38 Å. This is smaller than the structural changes caused by the formation of Fe(OH)Cl2(ads) at the step edge (1.55 Å). The largest structural change is caused by the formation of FeCl3(ads), which pulls the edge Fe atom out of the surface by 1.79 Å. These structural changes are consistent with the Reaxff-MD results that the Fe(OH)3 and Fe(OH)2Cl surface species are stable on the surface while FeCl3 and Fe(OH)Cl2 dissolve into the solution.

a Fe(OH)3, b Fe(OH)2Cl, c Fe(OH)Cl2, d FeCl3. The location of the edge Fe atom prior to the adsorption is shown with an orange dashed line. White and royal blue spheres represent H and Cl respectively.
The relative stability of surface adsorbates
The four-stage mechanism suggests reactions starting from FeCl3 to form Fe(OH)nCl(3-n) (where n is an integer ≤3) by replacing surface Cl with corresponding number of OH in each of the four stages (n = 3, 2, 1 and 0 correspond to stages 1, 2, 3, and 4 respectively), as shown in reaction (1). The reaction energy (ΔE) is used to compare the stability order of the different surface species with respect to the FeCl3(ads) base case. It is defined as the total energy of the different adsorbed system, Fe(OH)nCl(3-n) (ads) + nCl(g) (final state) relative to the energy of the system where three Cl interact with the same Fe site with nOH in the gas phase, FeCl3(ads) + nOH(g) (initial state), as shown in Eq. (2).
The reaction energy of the three reactions involving change in surface species are listed in Table 1. The negative reaction energy from FeCl3 to Fe(OH)nCl(3-n) suggests that it is thermodynamically favorable to replace the Cl with OH. The final Fe(OH)3 species is lowest in energy. This is consistent with the four-stage mechanism proposed by the Reaxff-MD study where the initially formed FeCl3 reacts with OH at the film/solution interface forming initially Fe(OH)3 while sufficient OH is available.
Fe vacancy formation
The Reaxff-MD simulations suggested that Fe(OH)3 and Fe(OH)2Cl were stable on the surface while Fe(OH)Cl2 and FeCl3 would dissolve into the solution7. This is verified by comparing the energy required to make an iron vacancy, where iron vacancy is created by removing the different surface species, Fe(OH)3, Fe(OH)2Cl, Fe(OH)Cl2 and FeCl3. The Fe vacancy formation energy is calculated using Eq. (3) and results are listed in Table 2.
where \({E}_{{\mathrm{Fe}}_{\mathrm{v}}/{\mathrm{surf}}}\) is the energy of newly formed Fe vacancy surface and \({E}_{{\mathrm{Fe}}\left( {{\mathrm{OH}}} \right)_{n}{\mathrm{Cl}}_{(3 - {n})}\left( {\mathrm{g}} \right)}\) the energy of the removed surface species in the gas phase. The energy of the surface with the surface species still attached on the surface is \({E}_{{\mathrm{Fe}}\left( {{\mathrm{OH}}} \right)_{n}{\mathrm{Cl}}_{(3 - {n})}\left( {{\mathrm{ads}}} \right)}\). Here n represents the number of Cl replaced by OH in the four stages. Positive Efrom suggests endothermic Fe vacancy formation.
The Fe vacancy formed by removing FeCl3 has the lowest formation energy indicating that the dissolution of FeCl3 from the surface is easier than the other species. The second lowest formation energy is for Fe(OH)Cl2. This is consistent with the Reaxff-MD simulations which found that FeCl3 and Fe(OH)Cl2 dissolve into the solution more readily than the other two species.
Discussion
The DFT calculations of the thermodynamic feasibility of the four-stage mechanism are consistent with the Reaxff-MD simulations. We find OH to be more stable on the surface than Cl, therefore, at high pH, adsorbed Cl is thermodynamically favored to be replaced by adsorbed OH. With the decreased availability of hydroxide ions near the surface, the number of hydroxide replacing chloride decreases, forming Fe(OH)3, Fe(OH)2Cl, Fe(OH)Cl2 and FeCl3 respectively in the four stages. The Fe vacancy formation energy indicates that it is easier to form an iron vacancy by removing surface species with more chlorides, in other words, the surface species with more chlorides dissolve more readily. The dissolved chloride-iron complexes are assumed to convert to iron-hydroxides in solution releasing the chlorides back to the solution. This four-stage mechanism suggests that the role of chloride in the depassivation process of iron is to catalyze the dissolution of surface iron atoms, which is consistent with the point defect model. This thermodynamic comparison provides a high-level theory support to the Reaxff-MD simulations and also illustrates the potential for Reaxff-MD simulations as a computational tool for investigation of complex reaction mechanisms like studied herein.
Methods
The computational methods in this study are described in details elsewhere9 so only short summary is included here. Vienna Ab initio Simulation Package (VASP)14,15,16,17 is used to carry out spin-polarized DFT18,19 calculations. The generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) functional20,21 is used to describe the exchange-correlation potential. All calculations are performed using projector augmented wave potential (PAW)22 and plane wave basis set with the cutoff energy of 400 eV. The DFT+U23,24,25,26,27 method introduced by Dudarev et al.28 is applied to correct for the effects of the on-site Coulomb interactions of the Fe 3d electrons. The effective U value (Ueff = 4 eV) is determined previously29 by converging the reaction energies and the band gap for different Ueff values. A k-point grid with a 3 × 1 × 1 Monkhorst-Pack30 mesh, and a 0.02 eV/Å convergence criteria are used for all calculations.
Similar to the earlier study9, the adsorbed chloride and hydroxide from the alkaline solution are represented using neutral species (Cl, and OH), which gain negative charges upon adsorption on the α-Fe2O3 (0001) surface to be considered as Cl− and OH–8,9,29. Additionally, on the same surface, the most stable site has been suggested to be the same for adsorbates with different charges (positive, negative, or neutral)31. The interactions of adsorbates are investigated without implicit solvent. Although DFT studies using solvent models32,33 have shown that the adsorption is affected by the inclusion of solvent we do not expect that to affect the trends reported herein.
The lattice constants for the unit cell of α-Fe2O3 are optimized as a = b = 5.027 Å and c = 13.724 Å (c/a = 2.73) in this study, in good agreement with experimental values (a = b = 5.035 Å and c = 13.747 Å)34. The magnetic state for of α-Fe2O3 is applied as antiferromagnetic (+ + − −) based on our previous study29. More information regarding properties of α-Fe2O3 and surface structure can be found elsewhere8,9,29.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Gunay, H. B., Ghods, P., Isgor, O. B., Carpenter, G. J. C. & Wu, X. Characterization of atomic structure of oxide films on carbon steel in simulated concrete pore solutions using EELS. Appl. Surf. Sci. 274, 195–202 (2013).
Ghods, P., Isgor, O. B., Brown, J. R., Bensebaa, F. & Kingston, D. XPS depth profiling study on the passive oxide film of carbon steel in saturated calcium hydroxide solution and the effect of chloride on the film properties. Appl. Surf. Sci. 257, 4669–4677 (2011).
Ghods, P., Isgor, O. B., Bensebaa, F. & Kingston, D. Angle-resolved XPS study of carbon steel passivity and chloride-induced depassivation in simulated concrete pore solution. Corros. Sci. 58, 159–167 (2012).
Chen, W., Du, R.-G., Ye, C.-Q., Zhu, Y.-F. & Lin, C.-J. Study on the corrosion behavior of reinforcing steel in simulated concrete pore solutions using in situ Raman spectroscopy assisted by electrochemical techniques. Electrochim. Acta 55, 5677–5682 (2010).
Li, Y. & Cheng, Y. F. Passive film growth on carbon steel and its nanoscale features at various passivating potentials. Appl. Surf. Sci. 396, 144–153 (2017).
DorMohammadi, H., Pang, Q., Murkute, P., Árnadóttir, L. & Isgor, O. B. Investigation of iron passivity in highly alkaline media using reactive-force field molecular dynamics. Corros. Sci. 157, 31–40 (2019).
DorMohammadi, H., Pang, Q., Murkute, P., Árnadóttir, L. & Isgor, O. B. Investigation of chloride-induced depassivation of iron in alkaline media by reactive force field molecular dynamics. npj Mater. Degrad. https://doi.org/10.1038/s41529-019-0081-6 (2019).
Pang, Q., DorMohammadi, H., Isgor, O. B. & Árnadóttir, L. The effect of surface vacancies on the interactions of Cl with a α-Fe2O3 (0001) surface and the role of Cl in depassivation. Corros. Sci. 154, 61–69 (2019).
Pang, Q., DorMohammadi, H., Isgor, O. B. & Árnadóttir, L. The effect of surface defects on chloride-induced depassivation of iron – a density functional theory study. Corrosion 76, 690–697 (2020).
Macdonald, D. D. The point defect model for the passive state. J. Electrochem. Soc. 139, 3434–3449 (1992).
Nieuwoudt, M. K., Comins, J. D. & Cukrowski, I. Analysis of the composition of the passive film on iron under pitting conditions in 0.05 M NaOH/NaCl using Raman microscopy in situ with anodic polarisation and MCR-ALS. J. Raman Spectrosc. 43, 928–938 (2012).
Lin, L. F., Chao, C. Y. & Macdonald, D. D. A point defect model for anodic passive films II. Chemical breakdown and pit initiation. J. Electrochem. Soc. 128, 1194–1198 (1981).
Chao, C. Y., Lin, L. F. & Macdonald, D. D. A point defect model for anodic passive films I. Film growth kinetics. J. Electrochem. Soc. 128, 1187–1194 (1981).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)]-ERRATA. Phys. Rev. Lett. 78, 1396–1396 (1997).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943–954 (1991).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 71, 035105 (2005).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467–R5470 (1995).
Solovyev, I. V., Dederichs, P. H. & Anisimov, V. I. Corrected atomic limit in the local-density approximation and the electronic structure of d impurities in Rb. Phys. Rev. B 50, 16861–16871 (1994).
Anisimov, V. I., Aryasetiawan, F. & Lichtenstein, A. I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA+U method. J. Phys. Condens. Matter 9, 767–808 (1997).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Pang, Q., DorMohammadi, H., Isgor, O. B. & Árnadóttir, L. Density functional theory study on the effect of OH and Cl adsorption on the surface structure of α-Fe2O3. Comput. Theor. Chem. 1100, 91–101 (2017).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Pessoa, A. M., Fajín, J. L. C., Gomes, J. R. B. & Cordeiro, M. N. D. S. Ionic and radical adsorption on the Au(hkl) surfaces: A DFT study. Surf. Sci. 606, 69–77 (2012).
Zhang, R., Ling, L., Wang, B. & Huang, W. Solvent effects on adsorption of CO over CuCl(111) surface: a density functional theory study. Appl. Surf. Sci. 256, 6717–6722 (2010).
Zhang, R., Ling, L., Li, Z. & Wang, B. Solvent effects on Cu2O(111) surface properties and CO adsorption on Cu2O(111) surface: a DFT study. Appl. Catal. A Gen. 400, 142–147 (2011).
Finger, L. W. & Hazen, R. M. Crystal structure and isothermal compression of Fe2O3, Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 51, 5362–5367 (1980).
Towns, J. et al. XSEDE: accelerating scientific discovery. Comput. Sci. Eng. 16, 62–74 (2014).
Acknowledgements
This work was supported by the National Science Foundation, DMMI (Grant No. 1435417). Part of the calculations used the Extreme Science and Engineering Discovery Environment (XSEDE)35 (allocation TG-ENG170002 and TG-DMR160093) which is supported by National Science Foundation grant number ACI-1053575. The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin and the San Diego Supercomputer Center (SDSC) for providing HPC resources that have contributed to the research results reported within this paper.
Author information
Authors and Affiliations
Contributions
Q.P. performed the density functional theory calculations and analyzed the results with the help of H.D., O.B.I., and L.Á. Q.P. wrote the initial draft of the manuscript. Data interpretation was discussed among all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pang, Q., DorMohammadi, H., Isgor, O.B. et al. Thermodynamic feasibility of the four-stage chloride-induced depassivation mechanism of iron. npj Mater Degrad 4, 26 (2020). https://doi.org/10.1038/s41529-020-00131-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-00131-8
This article is cited by
-
Nitride coatings for environmental barriers: the key microscopic mechanisms and momentous applications of first-principles calculations
Surface Science and Technology (2024)
-
Substitutional adsorptions of chloride at grain boundary sites on hydroxylated alumina surfaces initialize localized corrosion
npj Materials Degradation (2021)
