
Abstract
Exome sequencing (ES) has become one of the important diagnostic tools in clinical genetics with a reported diagnostic rate of 25–58%. Many studies have illustrated the diagnostic and immediate clinical impact of ES. However, up to 75% of individuals remain undiagnosed and there is scarce evidence supporting clinical utility beyond a follow-up period of >1 year. This is a 3-year follow-up analysis to our previous publication by Mak et al. (NPJ Genom. Med. 3:19, 2018), to evaluate the long-term clinical utility of ES and the diagnostic potential of exome reanalysis. The diagnostic yield of the initial study was 41% (43/104). Exome reanalysis in 46 undiagnosed individuals has achieved 12 new diagnoses. The additional yield compared with the initial analysis was at least 12% (increased from 41% to at least 53%). After a median follow-up period of 3.4 years, change in clinical management was observed in 72.2% of the individuals (26/36), leading to positive change in clinical outcome in four individuals (11%). There was a minimum healthcare cost saving of HKD$152,078 (USD$19,497; €17,282) annually for these four individuals. There were a total of six pregnancies from five families within the period. Prenatal diagnosis was performed in four pregnancies; one fetus was affected and resulted in termination. None of the parents underwent preimplantation genetic diagnosis. This 3-year follow-up study demonstrated the long-term clinical utility of ES at individual, familial and health system level, and the promising diagnostic potential of subsequent reanalysis. This highlights the benefits of implementing ES and regular reanalysis in the clinical setting.
Similar content being viewed by others

Introduction
Since the advancement of next-generation sequencing (NGS) technology, exome sequencing (ES), and genome sequencing (GS) have received increasing attention and application in clinical diagnostics, owing to its comprehensive and timely diagnostic capacity. The diagnostic and clinical utility of ES/GS have been well-studied and proven to be greater than chromosomal microarray or other conventional molecular testing in children with suspected genetic disorders1,2,3,4,5. However, in practice, ES is more widely used than GS because ES has a lower cost, while covering all known exons with majority of the pathogenic variants. Although there are relatively few studies on the diagnostic utility of GS, a meta-analysis of these studies showed that clinically there is a minimal difference between using GS and ES methods5.
The mean diagnostic rate of ES is estimated to be 36%, ranging from 25 to 58%, depending on the patient selection criteria and the disease type3,5,6,7,8. ES can be a powerful diagnostic tool compared to conventional methods, but a large proportion of individuals remain undiagnosed. One reason for this may be owing to the clinical and genetic heterogeneity of rare diseases. On the other hand, causal variants in the exome may be unrecognized due to limitations in the analytical methods or inadequate knowledge in the literature regarding the disease genetics. These unsolved cases fall into the “experimental maze”, where the attempt to make a diagnosis becomes a trial of novel approaches. Frésard and Montgomery discussed multiple strategies to address this issue, including exome reanalysis, GS, long-read sequencing, and other omics studies9. Among these methods, reanalysis of the exome is the most accessible and inexpensive. Multiple studies with predominantly Caucasian individuals have shown that the diagnostic rate of reanalysis ranges from 6 to 47%10,11,12,13,14,15,16,17,18,19,20,21,22,23. Stark et al. has estimated that 18-months is the most cost-effective time point to perform reanalysis19. In comparison, other approaches such as GS as a second-tier test for negative ES are less well-studied.
There is no doubt that a positive genetic diagnosis could provide further information to aid clinical decisions and patient management. The change in medical management is thus commonly used as an indicator of clinical utility, where a measure of outcome can be captured at three instances: (i) the return of results, (ii) documented changes to clinical management, and (iii) measures of long-term clinical outcome24. While many studies focused on describing the changes in acute management brought about by a positive genetic diagnosis, very few have assessed the long-term impact by longitudinal follow-up of >1 year3,19,25,26,27,28,29. Furthermore, the impact on reproductive decisions for both the affected individuals and their families is often limited. Lastly, there is also scarcity of health economic evidence on long-term follow-up after ES, which is important to guide the healthcare policy development30.
Based on our previously published cohort of predominant Chinese pediatric patients who underwent ES in 2013–2017, the current study aims to address two main questions: (i) the potential of ES reanalysis in making further diagnoses; and (ii) the long-term outcome of initial ES-positive individuals, measured at the individual level (change in clinical management and/or outcome); the family level (effect on reproductive decision making of the parents); and health system level (healthcare utilization and associated costs).
Results
Improved diagnostic yield through exome reanalysis
All undiagnosed individuals in the initial analysis (n = 61) were invited to participate in the reanalysis. Of the 61 undiagnosed individuals, 46 of them consented for reanalysis (13 were unable to be contacted and two refused to participate). Thus, for this study, our reanalysis is limited to these 46 individuals summarized in Supplementary Table 1. ES was performed on stored DNA of 34 individuals, and reanalysis on existing raw data was performed in 12 individuals. Twenty-six were analyzed as singletons, one as duo (proband and affected sibling), and 19 as trios. The average exome read coverage was 76.2×, while the percent of targeted bases over 20× coverage was 87.1%.
An additional 12 diagnoses were made through ES reanalysis (26% of the reanalyzed cohort), boosting the overall diagnostic rate from 41% (43/104) to at least 53% (55/104). Nine are autosomal dominant conditions, with heterozygous pathogenic/likely pathogenic variants identified in ATP1A3, COL11A1, GNB1, MN1, MFN2, PACS1, PTPN11, and SPTAN1 (two probands); three are autosomal recessive conditions (COQ7, PRF1, and SKIV2L). The summary of the findings is summarized in Table 1 and Supplementary Table 2.
Among the 12 new positive diagnoses, the majority (6/12; 50%) of individuals harbored variants in genes that had weak/no gene–disease association during the time of initial ES. The pathogenicity was established in reanalysis with increasing evidence from the latest literature.
For individual U018, a novel pathogenic variant was identified in ATP1A3. This gene has been known and thought to be the cause of three distinct diseases since 2004: rapid-onset dystonia Parkinsonism (RDP), alternating hemiplegia of childhood (AHC), and CAPOS (cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss) syndrome31,32,33,34. It was not until 2014 that it was suggested that they belong to the same spectrum of disease35. An updated review of the growing evidence of patients presenting with intermediate/mixed/atypical phenotypes was published in 201835,36. This clinical spectrum of disease is now summarized as ATP1A3-related neurological disorder, and our patient’s phenotype belongs to mixed phenotype of RDP and AHC.
ES reanalysis diagnosed individual U022 with Schuss-Hoeijmakers syndrome caused by a pathogenic variant in the gene PACS1. PACS1 was first discovered in 2012 in which two individuals who shared similar phenotypes were found to harbor the same variant37. The sample size remained limited until a case series of 19 individuals were reported in 2016, while the initial ES of U022 was performed in 201438.
U077 presented with global developmental delay, hypotonia, and MRI finding of mega cisterna magna. Exome reanalysis revealed a de novo missense mutation in SPTAN1 that explained his phenotype. This gene was first reported to be associated with a distinctive form of West syndrome with hypomyelination in 2008, later classified as early infantile epileptic encephalopathy39,40. Syrbe et al. reported six individuals with milder phenotypes of less severe intellectual disability with or without epilepsy; in particular, the variant identified in U077 was also reported41.
An MFN2 variant has been identified in individual U066. This variant was detected in the initial analysis performed in 2016, but after extensive review was concluded as unsolved, as he did not have any symptoms of Charcot-Marie-Tooth (CMT) disease. Rather, he presented with intellectual disability, behavioral problems, and seizure. After independent reanalysis, the same variant has been identified and classified as likely pathogenic, and now, there is stronger variant–disease association. This variant has been reported as pathogenic/likely pathogenic in multiple individuals with CMT disease in ClinVar, in which multiple submitters providing assertion criteria provided the same interpretation (two stars evidence). Upon discussion with neurologist and further literature review, there are individuals carrying pathogenic variant in MFN2 reported to present with developmental delay and other central nervous system involvement in addition to neuropathies42. Variable age of onset (up to 50 years old), expressivity and incomplete penetrance have been observed43,44,45. Therefore, although individual U066 did not present with the typical phenotype of neuropathy of CMT disease, it may due to late disease-onset (U066 is currently 14 years old), reduced expressivity or non-penetrance. This likely pathogenic variant is reported as it may have an implication to the further care and management.
During the initial ES in 2016, the association of COQ7 and coenzyme Q10 (CoQ10) deficiency was limited as there was only one article reporting an individual with homozygous variant in 201546. After reanalysis, individual U094 became the third case reported with this diagnosis, caused by compound heterozygous variants in COQ747. He has clinical phenotype compatible to CoQ10 deficiency and further skin fibroblast testing showed a reduction in CoQ10 level and decreased combined complex II + III activity, supporting that the biallelic variants may contribute to U094’s phenotype47.
Lastly, GNB1 was a new gene discovered near the time of the initial ES of individual U086. The first paper on GNB1 was published in May 2016, while initial ES was requested in March 2016 (report issued on in June 2016)48. This variant has been reported as a de novo pathogenic variant in multiple patients with similar phenotype, and considered as a “mutation hotspot” within the gene48,49. Therefore, the genetic diagnosis is established in U086.
New diagnoses were made in four patients because of an update on phenotype that became more apparent with age or recognition of nonclassical/atypical symptoms that masked the core features when the patient was first assessed (33%).
For example, as discussed above, individual U018 was presented with mixed phenotype of RDP and AHC, which is atypical to the early reports on the association between ATP1A3 and diseases.
Reanalysis yielded the diagnosis of Noonan syndrome in individual U043, who first presented as a new born with congenital arthrogryposis multiplex rather than typical features of Noonan syndrome. In retrospect, U043 has mild developmental delay and the facial feature resembling Noonan syndrome became more apparent upon clinical evaluation at 4 years of age. Interestingly, multiple joint contractures only occur in 4% of patients with Noonan syndrome50.
Individual U057 was born from a consanguineous family, presented with developmental delay and epilepsy, and later passed away due to acute deterioration after high swinging fever with multi-organ failure. Initial ES at that time was unrevealing. Reanalysis found a homozygous variant in the PRF1 gene, which is associated with hemophagocytic lymphohistiocytosis (HLH) type 2. His predominant neurological manifestation at onset was nonclassical for HLH, but in retrospect, the clinical course is compatible and the diagnosis was supported by the additional phenotype of T-cell lymphoma and intracranial lesion from postmortem examination.
Lastly, for individual U071, the diagnosis was Stickler syndrome/Marshall syndrome (COL11A1). In addition to his connective tissue problem explainable by the diagnosis, he had hypotonia during infancy, gross motor delay, Marfanoid habitus, scoliosis, prominent aortic sinus, and limited intelligence that may have prompted clinicians to consider other differential diagnosis, such as Marfan syndrome, Sprinzten Golderg syndrome, or neuromuscular diseases. The diagnosis of COL11A1 could only partially explain his phenotype of connective tissue disorder. We have evaluated other genes in relation to these additional phenotypes, but so far no other candidate genetic variant has been identified. It is uncertain if there is other genetic factor contributing to his clinical presentation.
Additional sequencing for trios (individuals U018 and U077) and other similarly affected family member (U057) aided analysis in three patients (25%). The variants identified in U018 and U077 were found de novo, which trio analyses allowed immediate delineation of the inheritance pattern and support the pathogenicity. For U057, reanalysis was initiated when a younger sibling presented with similar phenotype of development regression and status epilepticus. The reanalysis with the affected sibling found homozygous variants in PRF1 in both siblings.
Improved sequencing technology also contributed to the increased yield (17%), where two variants had increased sequence coverage with re-sequencing: NM_002834.5(PTPN11):c.5 C > T (U043) has increased coverage from 8× (singleton) to >20× (in trios sample) and NM_006929.5(SKIV2L):c.1404-2 A > G (U045) has increased coverage from 5× (singleton) to >30× (in trios sample). Due to the low coverage in these regions in the initial exome, the variants were likely filtered in the initial analysis and thus we were unable to reach a diagnosis. Also, improved bioinformatics by new copy number variants (CNVs) caller has allowed the detection of a heterozygous multi-exon deletion in individual U075 (in-frame deletion of SPTAN1 exons 10–12), who presented with developmental delay and hypotonia. Individuals with large in-frame deletions located outside of the heterodimer domain have been reported to have milder phenotypes (varying from developmental delay with or without epilepsy to epileptic encephalopathy)41.
Lastly, the diagnosis of individual U036 was confirmed through international collaboration for new syndrome discovery: MN1 C-terminal truncation syndrome, where 23 individuals who harbor truncating variants at C-terminal of MN1 gene were found to share strikingly similar neurodevelopmental and craniofacial features51. It is postulated that these C-terminal truncating variants have escaped nonsense-mediated mRNA decay, thus creating a gain-of-function effect that increased protein stability, diminished cell proliferation, and enhanced MN1 aggregation51,52.
Diagnostic ES resulted in change in clinical management, outcome, and healthcare costs
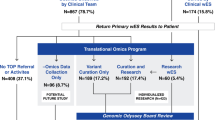
We clinically followed up 36 individuals with diagnoses made in the initial study. The median length of follow-up was 1255 days (3.4 years; mean = 1223.6 days; range: 72–2250 days). Families who only came for second opinion/diagnostic purpose (no other consultation record in Hong Kong public hospitals; n = 5; U001, U039, U055, U081, and U100), proband who passed away before the diagnosis was made (n = 1; U049), and those who refused to receive the genetic result (n = 1; U017) were excluded. The survival rate is 97% at the time of this publication as one individual (U023) passed away during this period (follow-up length of 72 days).
A change in clinical management was observed in 72.2% of the families (26/36) that are comparable to the 84% (36/43) prediction by Mak et al. at the time of molecular diagnosis27. Table 2 showed the actual change in clinical management on the six major types of interventions in accordance to Riggs et al.53. The change in management led to a positive impact in the clinical outcome in four individuals (11%). The costs associated with these changes in clinical management and outcomes are presented in Table 3. The diagnosis of Fanconi anemia for infant U005 has facilitated timely bone marrow transplantation, using the cord blood from his unaffected, HLA-matched sibling. This transplantation avoided life-long blood transfusions, and led to surveillance for non-hemic malignancy. In individual U090, the diagnosis of Costello syndrome in a child with unexplained failure-to-thrive allowed early detection of hypertrophic cardiomyopathy, which is a possibly fatal complication of the syndrome. In contrast to other myasthenic syndrome patients where acetylcholinesterase inhibitors were usually used as the treatment, in individual U092, the genetic diagnosis of myasthenic syndrome type 8 allowed genotype-directed therapy. Acetylcholinesterase inhibitors such as Mestinon (pyridostigamine) were found to have negative response in this specific subtype54. Instead, Ventolin (salbutamol), a common drug for treating asthma, has better treatment response and prevented individual U092 from recurrent respiratory infection and ICU admissions. Lastly, individual U102 has undergone pallidal deep brain stimulation, a surgical procedure commonly used for treating Parkinsonism, to stabilize his progressive dystonia that was shown to be an effective treatment for patients with the genetic defect in KMT2B55. The patient had good response to the treatment and showed improvements in speech, head extension, and increased voluntary movement in lower limbs.
Diagnostic ES influenced parents’ reproductive decision
A questionnaire on the reproductive planning and decision was distributed to each parent of initial ES-positive individuals. A total of 36 questionnaires (19 fathers and 17 mothers from 22 families) were completed and returned with a response rate of 59%. The marital status was indicated as ‘married’ in 19 families, ‘remarried’ in two, and ‘divorced’ in one family. Of the 36 respondents, 61% (22/36) agreed that the genetic diagnosis affected his/her decision to have more biological children.
Regarding the use of prenatal diagnosis (PND) and preimplantation genetic diagnosis (PGD), of all the respondents, 78% (28/36) thought PND should be offered to families with the same genetic diagnosis and 75% (27/36) would consider using PND for themselves. Interestingly, 56% (19/36) of the respondents agreed PGD should be offered to families with the same genetic diagnosis, but only 36% (13/36) of them would consider PGD at their personal level. In fact, most of the respondents (47%; 17/36) would not consider PGD for themselves.
The reproductive outcome of the 37 families with diagnostic ES were collected (including the family with proband passed away before genetic diagnosis), using the electronic patient record. Five couples had sought advice from reproductive services (Fig. 1). There were a total of six pregnancies over this follow-up period from these five families. Family U023 had two pregnancies. Two couples (parents of U062 and U080) did not receive any reproductive intervention after consultation as the recurrence risk is low (de novo variant). Three couples (four pregnancies) underwent PND for autosomal recessive conditions. Of note, mother of U005 was pregnant with a pair of twins at the time of diagnosis, therefore, only PND could be offered. One of the four pregnancies was affected and resulted in termination of pregnancy. None of the family underwent PGD in this cohort.

PND prenatal diagnosis, TOP termination of pregnancy; *: ongoing twin pregnancy at the time of genetic diagnosis.
Discussion
This study demonstrated the diagnostic capacity of ES reanalysis for patients suspected of genetic diseases. Twelve additional diagnoses were made by ES reanalysis in 46 individuals at a time frame of 3.2 years after the initial analysis. The diagnostic rate within the reanalysis group is 26% (12/46), while the additional yield compared with the initial analysis is at least 12% (increased yield from 41% to at least 53%). The diagnostic yield in this study showed comparable results to other published studies with reanalysis diagnostic yield of 5–36%; and additional diagnostic yield of 5–22% at 6–36 months after initial analysis (Fig. 2)10,11,12,13,14,15,16,17,18,19,20,21,22,23.

We searched PubMed with the search terms “exome” and “reanalysis” to identify relevant studies. There were no language and date restrictions. Only studies that provide both the initial analysis and reanalysis data were included. *Liu et al. cohort 1 and Liu et al. cohort 2 are two separate patient cohorts from the same reanalysis study with different approach and time frame (cohort 1: manual reanalysis over a 5-year period; cohort 2: semiautomated reanalysis over a 4-year period)17.
A higher additional diagnostic yield would be expected if all families participated in the reanalysis (13 could not be contacted and two refused to participate). There was difficulty in recontacting all families as some of the patients are from mainland China and Taiwan. One of the families refused to participate because the child has been improving so they do not think there is a need for reanalysis.
Among the 46 negative cases reviewed, three individuals (U022, U077, and U104) received a genetic diagnosis through genetic evaluations in other centers during this period, either via the clinical or research route. No diagnosis was made by routine nongenetic clinical evaluation. One individual participated in another ES research, in which the diagnosis made was consistent with our finding (U077; NM_001130438.3(SPTAN1):c.4828 C > T). Two individuals had the diagnoses through standard of care genetic testing available in the Hong Kong health system. U022 received the same genetic diagnosis of PACS1 variant through NGS panel testing. U104 had a clinical suspicion of myotonic dystrophy on reassessment and therefore targeted testing was performed. It was found that she has expanded repeats (>667) in DMPK, confirming the diagnosis. The diagnosis of U104 was not detectable by ES reanalysis as the repeat expansion is located in 3′ untranslated region of DMPK, which would not be captured by a typical exome. Furthermore, short read sequencing from ES/GS has technical limitations in detecting these expanded repeats that specialized tools have modest success in detecting expansion outliers56,57. By comparing the additional yield, ES reanalysis remains as a strong candidate to tackle the “experimental maze” after negative exome result. However, clinicians should be aware of its limitation and consider other possible approaches.
Currently, GS is viewed to be the ultimate genetic diagnostic test to detect variations in the genome. Therefore, after negative exome, clinicians often consider GS to be one of the possible solutions. In fact, ES reanalysis is recommended before GS due to GS’s relatively high cost yet low diagnostic rate21. To date, there is a lack of back-to-back comparison between the diagnostic capacity of ES reanalysis and GS. Alfare et al. proceeded to GS following negative ES, they postulated that 30% of the new findings should be detectable by ES reanalysis, and therefore GS could only achieve 7% higher detection rate than ES reanalysis, further supporting the role of ES reanalysis in clinical setting for patients with suspected genetic disease58.
This study presented a variety of reasons for the improved diagnostic yield: strengthened gene–disease association and literature updates (50%), additional information on patient phenotypes (33%), additional sequencing for trios/other affected family members (25%), improvement in sequencing data by re-sequencing (17%), improvement in bioinformatics tools (8%), as well as research collaborations through international case sharing platforms (8%; Table 1). This reinforces the need for reanalysis, as with time, new gene– and variant–disease associations may be discovered, patients may develop new phenotypes, in combination with advancing sequencing and bioinformatic technology, could contribute to an increased diagnostic yield of ES. Although there is no marked difference in the detection rate by reanalysis of raw data (4/11, 36.3%) and re-sequencing (8/35, 22.8%), the result has illustrated that re-sequencing to generate new exome data could be critical for additional diagnosis. Two diagnoses were achieved due to the increase in coverage in region containing the variant (U043 and U045), it could be missed if only data reanalysis has been performed. Therefore, re-sequencing may have an advantage of better capturing the variant as compared to data reanalysis. However, since the initial ES of this study was performed in commercial laboratories, we do not have access to all data for parallel comparison of the performance in reanalysis and re-sequencing. More systematic study should be conducted to evaluate and compare the diagnostic capacity, as well as the cost-effectiveness of keeping data for reanalysis versus keeping DNA for re-sequencing.
In this cohort of 104 pediatric patients, two patients have the same diagnosis of Schuss-Hoeijmakers syndrome; U006 was diagnosed in the initial ES in 2013 (trios), while U022 was diagnosed in the reanalysis phase (singleton). Reviewing the clinical history of the two patients in our cohort, it was surprising to find that U022’s initial negative ES was performed in 2014, later than the diagnosis of U006. The initial ES for these two cases were performed in different laboratories with different sequencing and analytical methodologies. Although from a diagnostic point of view, missing a diagnosis is not ideal, this reflected the difficult reality. Apart from the sequencing and variant calling capacity, laboratories have different analytical pipelines, and therefore may have different interpretation toward the new and limited evidence, especially before the publication of ACMG guidelines on variant interpretation in 201559. During this rapidly evolving era of genomics where new genes and its disease association are discovered, genetic diagnosis and variant classification are dynamic and changeable over time, stressing the importance of data reanalysis. Also, this example illustrated the benefits of having trio samples. It allowed the possible detection of de novo variants, which was the key to reaching the diagnosis in U006. The use of the “de novo” paradigm is known to be important in the analysis of certain diseases and variant types, such as in neurodevelopmental disorders. A meta-analysis has shown that the odds of reaching diagnosis with a trio design is double that of singleton5. Within this reanalysis cohort, samples from 20 individuals were reanalyzed with additional family members (19 trios and 1 duo with affected sibling), while 26 were singletons. The diagnostic yield from trio samples (6/19, 31.5%) was higher than that of singleton samples (5/26, 19.2%). Therefore, we recommend trio analysis for maximizing the diagnostic utility of ES. However, if there is a resource constraint, a stepped approach should be adopted, starting with singleton ES and if negative, proceed to trio sequencing during reanalysis.
To date, this is the third longitudinal follow-up study, with the longest study duration, on investigating the long-term benefit of ES diagnosis on patient care and outcome. Over the 3-year follow-up period, 26 out of 36 diagnosed individuals (72%) have implemented changes in their management as a result of ES diagnosis, of which four (11%) had a long-term change in clinical outcome, consequently saving costs from the healthcare system perspective in the long term. This is comparable to a similar study by Stark et al., which reported at over a year of follow-up, 16 out of 48 diagnosed infants had a change in management, and among these, four had a change in clinical outcome or hospital service use19.
At the initial report of this cohort, changes in management were recommended by clinical geneticists based on disease-specific management guidelines, case reports, or known function of genes of the diagnosed patient. From the result of the longitudinal follow-up, the change in clinical management is generally in line with the prediction. However, there are slight discrepancies, especially on the aspects of specialist referrals (predicted = 44%, actual = 25%), lifestyle change (predicted = 14%, actual = 6%), and medication (predicted = 28%, actual = 17%). This discrepancy might be attributed to case-specific applicability of the advised changes (e.g., some patients have already developed complications prior to genetic diagnosis and therefore already seeing specialists or taking appropriate medications due to the presenting symptoms), ineffective communication (genetic diagnosis was not communicated to other responsible specialists in some cases), and clinicians’ perceived importance of the ES results (some clinicians did not make management changes based on the genetic diagnosis provided by geneticists). Furthermore, lifestyle changes would be difficult to measure in a clinical setting and may not be recorded in the medical consultations. For example, sun protection is recommended for individual U076 with keratitis-ichthyosis-deafness syndrome. The inability to measure whether certain lifestyle change has been implemented would be a limitation to this study. The discrepancy also demonstrated the importance of long-term follow-up data, as acute changes in management might not necessarily be followed through over the clinical course of patients. Further exploring the factors affecting the delivery and actionability of ES results in the clinical setting could be important to maximize the value of diagnostic ES.
Consistent with the initial prediction, ES mostly assisted the clinical management in the aspects of surveillance for potential complications (47%, 17/36), further diagnostic testing (36%, 13/36), and specialist referrals (25%, 9/36) within the individual health domain. This is also in line with Niguidula et al., which demonstrated majority of patients benefitted from a positive ES diagnosis in the areas of prognosis and enhanced surveillance29. Indeed, it is also the clinicians’ perception that impact on prognostication and disease surveillance are some of the most important outcomes of a positive exome result60. In contrast to the individuals reported by Stark et al.19, where changes in health outcome were all resulted from changes in medication or procedural interventions, one individual (U090) in our cohort had a significant change in health outcome due to implementation of surveillance. With the diagnosis of Costello syndrome, cardiac surveillance by echocardiogram was initiated as cardiomyopathy is a known complication of the syndrome. In fact, early asymmetrical hypertrophic cardiomyopathy was detected 2 months after the disclosure of genetic result. It could be otherwise unnoticed and likely fatal until symptoms present. The genetic diagnosis has led to the prevention insidious life-threatening event of cardiomyopathy.
The genetic diagnosis did not change the clinical management in ten individuals. Nine of them have intellectual disability syndrome, in which the developmental support and management plan do not fall into one of the six categories. For individual U087 with the diagnosis of Auriculocondylar syndrome, no new treatment was initiated, but we connected the family to the surgical expert in doing surgery for this syndrome. U087 is now in joint care by local hospital and expert surgeons from Taiwan.
A diagnostic genetic test does not only benefit the clinical management and outcome, it also provides information that aids reproduction and family planning. By knowing the mode of inheritance and carrier status, the reproductive risk of parents can be determined and couple can make use of the information to decide on the reproductive plan. From our survey result, the confidence in the use of PND is considerably higher than that of PGD. Over 70% of the respondents agree that PND should be offered to families with the same genetic diagnosis, and they would consider PND if they are planning for future pregnancies. Only 56% of them agree PGD should be offered and even less (36%) would consider it for themselves. Looking into the actual pregnancy outcomes, in fact, none of the families chose PGD as the alternative assisted reproduction option. Here, we outline several reasons for such. Firstly, among the surveyed families with inheritance determined (n = 21), the majority (76%, 16/21) have a recurrence risk of <1% as the variant identified in the proband is a de novo or mosaic variant. Usually, PGD would be more attractive to couples with higher recurrence risks. In these 21 families, only 24% (5/21) of them has a 25% recurrence risk (parents being a carrier of an autosomal or X-linked recessive disease) and none of them has an inherited autosomal dominant disorder. Secondly, the proactiveness of having more babies. PGD is a more active intervention that the couple needs to decide and seek assistance before being pregnant, while PND may be accepted as a test or a solution after getting pregnant. Having an ill child may have created struggle in the family that would hinder the wanting of another child despite PGD could significantly reduce the risk, as 61% of the respondents reflected that the genetic diagnosis affected their decision to have more children. Similarly, van Rij et al. observed a negative association between the presence of a living, affected child and PGD intention (OR 0.5, 95% CI 0.3–1.1), and use (OR 0.3, 95% CI 0.1–0.6) in 246 couples with high reproductive risk of a range of chromosomal or Mendelian disorders61. On the other hand, a study from Hong Kong in 2002 showed parents with children affected with homozygous beta thalassemia showed more positive attitude toward the use of PGD than PND as an alternative62. This may be due to the potential advantage of selecting HLA-matched embryos for the affected child. Thirdly, couples may take into consideration that the PGD-IVF ongoing pregnancy rate in Hong Kong is only 33.1%63. Also, there is a maternal age requirement, PGD request will not be accepted if the maternal age is over 40 years old in Hong Kong. Given people are getting married and giving birth at a later age in modern society like Hong Kong, the applicability and feasibility of PGD are limited even if the couples want to have another child. The median age of mothers filling in the questionnaire was 41 (mean = 41.8; range = 33–52) years old. Lastly, couples have to bear the cost of PGD-IVF themselves as it is not subsidized in the Hong Kong healthcare system. The high cost of at least HKD$40,000 (United State dollars (USD)$5128; €4545) per cycle would be another factor that prevented couples from choosing PGD. Two couples who had sought advice from assisted reproductive service expressed their concern on the financial cost of PGD, and eventually decided not to proceed with PGD.
From a healthcare system standpoint, unnecessary hospital admissions, management procedures, and treatment medications could be avoided as a result of the genetic diagnosis. Four individuals (11%) had a long-term change in clinical outcome; of which changes in routine management, such as avoidance of routine procedures (i.e., blood transfusions), avoidance of frequent hospital admissions, and initiation of routine clinical follow-ups have led to a minimum healthcare cost saving of HKD$152,078 (USD$19,497; €17,282) annually for these four individuals. Further cost savings would be seen in a longer term, and levelled off in 6.5 years for these four individuals, as total cost of certain operations, such as bone marrow transplantation and deep brain stimulation would have been incurred as a one-off cost, with minimal follow-up costs in the following years. In Stark et al.’s cohort, it was found to save $1578 Australian dollars (HKD$8456) per quality-adjusted life year (QALY) gained at 1-year-follow-up19. Although a comprehensive cost-utility analysis is not performed in our study as QALY was not collected, it could be anticipated that the positive outcome of additional life span, quality of life gained, lifetime value of unaffected children through assisted reproductive methods, etc. would lead to clear dominance (better outcome at a lower cost) of ES from the individual, family, healthcare system, and societal perspective.
To conclude, this 3-year follow-up study demonstrated the diagnostic and clinical utility of ES and reanalysis in a cohort of 104 pediatric individuals. ES reanalysis is a useful tool that provides promising additional diagnoses compared to routine clinical evaluation. ES and subsequent periodic reanalysis are recommended in undiagnosed individuals as genetic knowledge and literature is regularly updated. This study also provided quantification to the long-term clinical utility of genetic diagnoses, which has a positive impact on individual’s clinical management, parents’ reproductive planning, and healthcare cost savings in a long run. Our study highlights the benefits of implementing ES and regular reanalysis in the clinical setting.
Methods
Cohort characteristics
This is a follow-up analysis to our previous study, Mak et al., where 104 individuals with suspected genetic disorder were recruited for ES in 2013–201727. ES was performed in one of the two clinical commercial laboratories, 95 by Genome Diagnostic Nijmegen and nine by Ambry Genetics. A total of 78% (81/104) of the individuals were analyzed as singletons, followed by targeted Sanger sequencing of parental samples in HKU if candidate variant was identified. The remaining 23 individuals with ES were analyzed as trios. Detailed sequencing methodology and variant interpretation approach of the initial ES were described in the original paper by Mak et al.27. The paper concluded a diagnostic yield of 41% (43/104 received a genetic diagnosis) and predicted that 84% of them would result in a change in clinical management.
Exome reanalysis: patient recruitment and data processing
Individuals without a diagnosis in the initial ES study (n = 61) were recruited for reanalysis, in collaboration with Yale University. The median time frame from the initial analysis to reanalysis was 1178.5 days (3.2 years; mean: 1150 days; range: 301–1039 days). Families that were unable to be contacted or refused consent for reanalysis were excluded. Depending on the availability of raw data, either raw sequencing data were reanalyzed, or stored DNA were re-sequenced and analyzed. For re-sequencing, the IDT xGen Exome Panel exome capture kit was used and 100 bp paired-end reads sequencing was performed on the Illumina NovaSeq 6000 platform. Reads were aligned to hg19 reference genome using bwa-mem and processed following GATK best practice guidelines, including variant calling with HaplotypeCaller64,65,66,67. CNVs were called using gcnv (part of GATK4). The exact data processing and variant calling pipeline can be accessed at https://github.com/leklab/cromwell_wdl/tree/master/gatk4_multisample. The variants were annotated using Variant Effect Predictor through Hail (https://github.com/hail-is/hail) and then uploaded to seqr (https://github.com/macarthur-lab/seqr) for analysis68. For CNV analysis, variant calls from gcnv were annotated using AnnotSV69.
Exome reanalysis: data interpretation
Using seqr interface, only rare coding and splice-site variants (allele frequency <1% in gnomAD genomes, gnomAD exomes, ExAC, 1000 genomes, and TOPMED bravo databases) were included in downstream analysis. We filtered variants with a genotype quality < 20, alternate allele balance <25%, and non-PASSing variants sites determined by variant quality score recalibration. Missense, in-frame, frameshift, nonsense, and essential splice site variants are included in downstream analysis; synonymous and intronic variants were included only when there was a strong suspicion in a candidate gene.
For trio exomes, de novo and biallelic rare variants were prioritized. For singletons, genes with heterozygous variants absent from gnomAD, rare homozygous variants or two rare heterozygous variants were selected to match with individual’s clinical phenotype. A diagnosis is reached when the selected variant could be classified as pathogenic/likely pathogenic by the ACMG guidelines59. Sanger sequencing was performed to confirm the finding and segregation studies were undertaken if possible.
If a CNV overlapped with coding regions of a gene that had been associated with a phenotype matching with the individual’s clinical features, it was considered for disease causality. CNVs were validated by Seqplexing (Valencia, Spain) using EOSAL-CNV (Easy One-Step Amplification and Labeling procedure for CNV detection), which utilized a tailed primer with fluorescent probe for amplification of samples and control70. Amplified products were then loaded onto a capillary DNA sequencer for sizing and quantification. Analysis was done by comparing results of the samples and controls.
Long-term follow-up: clinical outcome
For individuals with an earlier diagnosis from initial ES (n = 43), we measured the long-term clinical utility by clinical follow-up and review of their medical records. The length of follow-up was measured from the date of result disclosure to the latest follow-up date in the electronic patient health record. We evaluated whether the genetic diagnosis has led to a change in individuals’ clinical management and/or outcome in accordance to the six aspects suggested by Riggs et al., in which we have adopted this classification in our initial study: referral, diagnostic testing, procedure, surveillance, lifestyle changes, and medication27,53.
Long-term follow-up: cost analysis
For individuals with changes in management and clinical outcomes as a result of the diagnosis, we have estimated the costs of investigations, treatment that were initiated and/or avoided, and healthcare service utilization. Costs were obtained from the Hong Kong Hospital Authority. For procedural costs that were not publicly available in the Hong Kong Hospital Authority setting, costs were obtained using the United Kingdom National Health Service National schedule of reference costs71. All costs in this study are reported in Hong Kong dollars, with an exchange rate of ~7.8 per USD and 8.8 per European dollar (Euro) at the time of study.
Long-term follow-up: reproductive outcome
Information related to parents’ use and views on assisted reproductive service and their pregnancy outcome were collected. There are two major options in assisting families with genetic conditions: PGD and PND. A questionnaire on the opinion toward these options was set out: whether PGD and PND should be offered to families with the same genetic diagnosis, and whether they would consider undergoing PGD and PND themselves. The questionnaire was distributed to each parent during their child’s medical follow-up or mailed to them for completion. Electronic patient record was reviewed to collect information on actual service use on these reproductive methods.
Ethics approval
Ethics approval was granted by the Institutional Review Board, the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW 12–211). Written informed consents have been obtained from the parents of the participants.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequencing and phenotype data that support the findings of this study have been deposited in dbGaP and AnVIL with the accession code phs000744.
Code availability
Bioinformatics codes used to process the data in this study are deposited in GitHub with no access restriction, details are provided in the “Methods” section.
References
Dixon-Salazar, T. J. et al. Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 4, 138ra178 (2012).
Katsanis, S. H. & Katsanis, N. Molecular genetic testing and the future of clinical genomics. Nat. Rev. Genet. 14, 415–426 (2013).
Stark, Z. et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med. 18, 1090–1096 (2016).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511 (2013).
Clark, M. M. et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 3, 16 (2018).
Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879 (2014).
Farwell, K. D. et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586 (2015).
Wright, C. F. et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314 (2015).
Fresard, L. & Montgomery, S. B. Diagnosing rare diseases after the exome. Cold Spring Harb. Mol. Case Stud. 4, a003392 (2018).
Wenger, A. M., Guturu, H., Bernstein, J. A. & Bejerano, G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet. Med. 19, 209–214 (2017).
Eldomery, M. K. et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 9, 26 (2017).
Al-Nabhani, M. et al. Reanalysis of exome sequencing data of intellectual disability samples: yields and benefits. Clin. Genet. 94, 495–501 (2018).
Ewans, L. J. et al. Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet. Med. 20, 1564–1574 (2018).
Nambot, S. et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet. Med. 20, 645–654 (2018).
Wright, C. F. et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet. Med. 20, 1216–1223 (2018).
Jalkh, N. et al. The added value of WES reanalysis in the field of genetic diagnosis: lessons learned from 200 exomes in the Lebanese population. BMC Med Genomics 12, 11 (2019).
Liu, P. et al. Reanalysis of clinical exome sequencing data. N. Engl. J. Med. 380, 2478–2480 (2019).
Salmon, L. B. et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet. Med. 21, 1443–1451 (2019).
Stark, Z. et al. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Genet. Med. 21, 173–180 (2019).
Shashi, V. et al. A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet. Med. 21, 161–172 (2019).
Schmitz-Abe, K. et al. Unique bioinformatic approach and comprehensive reanalysis improve diagnostic yield of clinical exomes. Eur. J. Hum. Genet. 27, 1398–1405 (2019).
Li, J. et al. Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene 700, 168–175 (2019).
The Epilepsy Genetics Initiative. Systematic reanalysis of diagnostic exomes increases yield. Epilepsia 60, 797–806 (2019).
Peterson, J. F. et al. Building evidence and measuring clinical outcomes for genomic medicine. Lancet 394, 604–610 (2019).
Hayeems, R. Z. et al. Care and cost consequences of pediatric whole genome sequencing compared to chromosome microarray. Eur. J. Hum. Genet. 25, 1303–1312 (2017).
Meng, L. et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171, e173438 (2017).
Mak, C. C. et al. Exome sequencing for paediatric-onset diseases: impact of the extensive involvement of medical geneticists in the diagnostic odyssey. NPJ Genom. Med. 3, 19 (2018).
Willig, L. K. et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 3, 377–387 (2015).
Niguidula, N. et al. Clinical whole-exome sequencing results impact medical management. Mol. Genet. Genom. Med. 6, 1068–1078 (2018).
Schwarze, K., Buchanan, J., Taylor, J. C. & Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 20, 1122–1130 (2018).
de Carvalho Aguiar, P. et al. Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175 (2004).
Demos, M. K. et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J. Rare Dis. 9, 15 (2014).
Heinzen, E. L. et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 44, 1030–1034 (2012).
Rosewich, H. et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol. 11, 764–773 (2012).
Rosewich, H. et al. The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology 82, 945–955 (2014).
Carecchio, M., Zorzi, G., Ragona, F., Zibordi, F. & Nardocci, N. ATP1A3-related disorders: an update. Eur. J. Paediatr. Neurol. 22, 257–263 (2018).
Schuurs-Hoeijmakers, J. H. et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet. 91, 1122–1127 (2012).
Schuurs-Hoeijmakers, J. H. et al. Clinical delineation of the PACS1-related syndrome-Report on 19 patients. Am. J. Med. Genet. A 170, 670–675 (2016).
Tohyama, J. et al. Early onset West syndrome with cerebral hypomyelination and reduced cerebral white matter. Brain Dev. 30, 349–355 (2008).
Saitsu, H. et al. Dominant-negative mutations in alpha-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am. J. Hum. Genet. 86, 881–891 (2010).
Syrbe, S. et al. Delineating SPTAN1 associated phenotypes: from isolated epilepsy to encephalopathy with progressive brain atrophy. Brain 140, 2322–2336 (2017).
Del, Bo,R. et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 71, 1959–1966 (2008).
Kostera-Pruszczyk, A. et al. Exome sequencing reveals mutations in MFN2 and GDAP1 in severe Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 19, 242–245 (2014).
Lawson, V. H., Graham, B. V. & Flanigan, K. M. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 65, 197–204 (2005).
Muglia, M. et al. A novel founder mutation in the MFN2 gene associated with variable Charcot-Marie-Tooth type 2 phenotype in two families from Southern Italy. J. Neurol. Neurosurg. Psychiatry 78, 1286–1287 (2007).
Freyer, C. et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 52, 779–783 (2015).
Kwong, A. K. et al. A fatal case of COQ7-associated primary coenzyme Q10 deficiency. JIMD Rep. 47, 23–29 (2019).
Petrovski, S. et al. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am. J. Hum. Genet. 98, 1001–1010 (2016).
Hemati, P. et al. Refining the phenotype associated with GNB1 mutations: clinical data on 18 newly identified patients and review of the literature. Am. J. Med. Genet. A 176, 2259–2275 (2018).
Romano, A. A. et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126, 746–759 (2010).
Mak, C. C. Y. et al. MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain 143, 55–68 (2019).
Miyake, N. et al. Gain-of-function MN1 truncation variants cause a recognizable syndrome with craniofacial and brain abnormalities. Am. J. Hum. Genet. 106, 13–25 (2020).
Riggs, E. R. et al. Chromosomal microarray impacts clinical management. Clin. Genet. 85, 147–153 (2014).
Nicole, S. et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain 137, 2429–2443 (2014).
Meyer, E. et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet. 49, 223–237 (2017).
Dashnow, H. et al. STRetch: detecting and discovering pathogenic short tandem repeat expansions. Genome Biol. 19, 121 (2018).
Dolzhenko, E. et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019).
Alfares, A. et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet. Med. 20, 1328–1333 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Stevens Smith, H., Russell, H. V., Lee, B. H. & Morain, S. R. Using the Delphi method to identify clinicians' perceived importance of pediatric exome sequencing results. Genet. Med. 22, 69–76 (2019).
van Rij, M. C. et al. Profiles and motives for PGD: a prospective cohort study of couples referred for PGD in the Netherlands. Hum. Reprod. 26, 1826–1835 (2011).
Hui, P. W. et al. Attitude of at-risk subjects towards preimplantation genetic diagnosis of alpha- and beta-thalassaemias in Hong Kong. Prenat. Diagn. 22, 508–511 (2002).
Chow, J. F. et al. Experience of more than 100 preimplantation genetic diagnosis cycles for monogenetic diseases using whole genome amplification and linkage analysis in a single centre. Hong Kong Med. J. 21, 299–303 (2015).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at https://www.biorxiv.org/content/10.1101/201178v3 (2017).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11 10 11–11 10 33 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Geoffroy, V. et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574 (2018).
Blesa, S. et al. Easy one-step amplification and labeling procedure for copy number variation detection. Clin. Chem. 66, 463–473 (2020).
National Health Service. National schedule of reference costs. https://improvement.nhs.uk/resources/reference-costs/ (2018).
Acknowledgements
We thank the patients and their families for participating in the study. This study was supported by the Society of the Relief of Disabled Children and the Edward and Yolanda Wong Fund. The Yale Center for Mendelian Genomics (UM1HG006504) is funded by the National Human Genome Research Institute. The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives, and provided logistical and general study coordination. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. If applicable: funds were also provided by the National Heart, Lung, and Blood Institute/National Eye Institute. S.H. was supported by the Chinese Scholarship Council (CSC; File No. 201808320197). S.P. received support from Estonian Research Council grant PUTJD827.
Author information
Authors and Affiliations
Contributions
J.L.F.F., M.H.C.Y., S.H., and C.C.Y.C. drafted the manuscript. S.H. and J.L.F.F. performed exome reanalysis with technical support and insight from S.P. and M.H.C.Y. B.H.Y.C., J.L.F.F., C.C.Y.C., and M.C.Y.C. collected and analyzed the longitudinal follow-up data. Cost analysis was performed by C.C.Y.C. Literature review was performed by J.L.F.F., S.H., and V.C.C.H. C.C.Y.M., M.H.Y.T., and K.S.Y. gave advice and provided intellectual input to the manuscript. B.H.Y.C. and M.L. conceptualized, oversaw and supervised this project, and provided guidance and support. All authors contributed to the discussion, overall data interpretation and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fung, J.L.F., Yu, M.H.C., Huang, S. et al. A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis. npj Genom. Med. 5, 37 (2020). https://doi.org/10.1038/s41525-020-00144-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-020-00144-x
This article is cited by
-
A novel CHD7 variant in a chinese family with CHARGE syndrome
Genes & Genomics (2024)
-
International Undiagnosed Diseases Programs (UDPs): components and outcomes
Orphanet Journal of Rare Diseases (2023)
-
Reanalysis of clinical exome identifies the second variant in two individuals with recessive disorders
European Journal of Human Genetics (2023)
-
Familial co-segregation and the emerging role of long-read sequencing to re-classify variants of uncertain significance in inherited retinal diseases
npj Genomic Medicine (2023)
-
Reanalysis of whole-exome sequencing (WES) data of children with neurodevelopmental disorders in a standard patient care context
European Journal of Pediatrics (2023)
