
Abstract
Anti-estrogen therapy is a key component of the treatment of both early and advanced-stage hormone receptor (HR)-positive breast cancer. This review discusses the recent emergence of several anti-estrogen therapies, some of which were designed to overcome common mechanisms of endocrine resistance. The new generation of drugs includes selective estrogen receptor modulators (SERMs), orally administered selective estrogen receptor degraders (SERDs), as well as more unique agents such as complete estrogen receptor antagonists (CERANs), proteolysis targeting chimeric (PROTACs), and selective estrogen receptor covalent antagonists (SERCAs). These drugs are at various stages of development and are being evaluated in both early and metastatic settings. We discuss the efficacy, toxicity profile, and completed and ongoing clinical trials for each drug and highlight key differences in their activity and study population that have ultimately influenced their advancement.
Similar content being viewed by others


Introduction
Anti-estrogen therapy targeting the estrogen-mediated signaling pathway is an essential component of treatment for both early and advanced-stage breast cancer expressing the estrogen receptor (ER) and/or progesterone receptor (PR)1,2. The ER is a steroid hormone nuclear receptor consisting of a DNA-binding domain (DBD), ligand-binding domain (LBD), and transcriptional activation function domains 1 (AF1) and 2 (AF2). Activated ER can interact with estrogen-responsive elements (EREs) within the DNA through its DBD or interactions with other transcription factors3. ER expression occurs in the normal ductal epithelium and invasive breast cancer, and immunohistochemistry can be used to semi-quantitatively measure the degree of ER and PR expression in tumor tissue4. Approximately 70% of all breast cancers exhibit ER and/or PR expression and, therefore, potentially sensitive to agents targeting the estrogen signaling pathway, also commonly referred to as “endocrine therapy” (ET)5.
For the past 30 years, ET for the treatment of ER-positive metastatic breast cancer (MBC) has generally included selective estrogen receptor modulators (SERMs, e.g., oral tamoxifen), aromatase inhibitors (AIs, e.g., oral anastrozole, letrozole, exemestane), and selective estrogen receptor degraders/downregulators (SERDs, e.g., intramuscular fulvestrant). Tamoxifen, AIs, or ovarian function suppression plus AIs are also effective in reducing recurrence risk when used as adjuvant therapy after primary surgical treatment of localized disease. Of note, AIs have demonstrated superior efficacy compared to tamoxifen, likely due to the agonist activity of tamoxifen, which limits its effectiveness6,7. Combination of CDK 4/6 inhibitors with ET has been shown to improve objective response rate (ORR), progression-free survival (PFS), and overall survival (OS) in ER-positive MBC, whether added to an aromatase inhibitor (AI) for first-line ET or fulvestrant as second-line ET after progression or relapse on an AI8,9,10,11,12,13. The CDK 4/6 inhibitor abemaciclib has also been shown to reduce recurrence risk when added to adjuvant AI therapy in those with localized disease at high risk of recurrence14. ET combined with agents targeting the PI3K-AKT-mTOR pathway, specifically the mTOR inhibitor everolimus and PI3K inhibitor alpelisib, in the metastatic setting has demonstrated improvements in PFS compared with ET alone15,16.
Although most ER-positive breast cancers benefit from ET, some exhibit primary intrinsic resistance, defined as disease progression within 6 months of initiating ET for MBC or relapse within 2 years of initiating adjuvant ET for early breast cancer (EBC). Secondary endocrine resistance, defined as progression ≥6 months after initiating ET for MBC, ultimately develops in most patients. Relapse while on adjuvant ET but after the first 2 years or within 1 year of completing adjuvant ET is also commonly characterized as acquired secondary resistance5,17. Secondary resistance to AI therapy is often associated with mutations in the ligand-binding domain of Estrogen Receptor 1 (ESR1) that confers ligand-independent activation of ERα18. ESR1 mutations occur in up to 50% of patients receiving AI therapy for MBC and in some receiving adjuvant ET and may be detected by blood using assays that identify circulating tumor DNA (ctDNA)19. ESR1 mutations often occur concurrently with other genomic alterations, which collectively are associated with a worse prognosis20. As described in the PADA-1 trial, among patients with baseline ESR1 mutations and on AI and CDK 4/6 inhibitor therapy for MBC, up to 27% can develop a rise in ESR1 mutation based on ctDNA at a median time of 15.6 months21. Other resistance mechanisms that may be implicated in primary or secondary resistance to ET include ESR1 loss, amplification, and translocation, and activating alterations in the PI3K-AKT-mTOR, RAS-MAPK, and CDK4/6-RB-E2F pathways, some of which may also contribute to resistance to CDK4/6 inhibitors18.
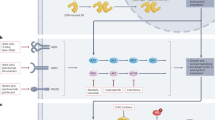
A new generation of novel anti-estrogen therapies was designed to circumvent some of these resistance mechanisms, especially acquired ESR1 mutations, and address limitations of current endocrine therapy, such as the agonist activity of tamoxifen and intramuscular administration of fulvestrant. These agents include variations of drug classes that already exist, including SERMs other than tamoxifen and novel orally administered SERDs. SERDs were initially identified as selective estrogen receptor down-regulators, but after studies confirmed that reduction in ER levels through proteasome-dependent degradation was responsible for their efficacy, they were termed degraders22. Novel anti-estrogen drug classes include complete estrogen receptor antagonists (CERANs), selective estrogen receptor covalent antagonists (SERCAs), and proteolysis-targeting chimerics (PROTACs) targeting ER. Each class of medication has a distinct mechanism of action, as illustrated in Fig. 1.

The binding of estrogen to the ligand-binding domain of ER induces an activating conformational change enabling its dimerization and intranuclear localization. Activated ER can interact with estrogen-responsive elements (EREs), allowing for gene transcription, which leads to cell survival and proliferation. Aromatase inhibitors (AIs). AIs, block estrogen production by inhibiting aromatase, which converts androgens to estrogens. Selective estrogen receptor modulators (SERMs). SERMs competitively inhibit the binding of estrogen to ER. SERM-bound ER dimers interact with chromatin at EREs of the DNA. In the breast, they are associated with co-repressors (CoR) which inhibit ER transcriptional activity, but in other organ tissues such as bone and endometrium, they are associated with co-activators (CoA), allowing for gene transcription. Selective estrogen receptor downregulators (SERDs). SERDs are pure ER antagonists. The SERD–ER complex is unable to translocate to the nucleus or undergo an open chromatin conformation that would allow transcription of ER-regulated genes. The SERD-ER complex subsequently undergoes proteosomal degradation. Proteolysis targeting chimerics (PROTACs): PROTACs are bifunctional molecules that consist of a ligand that binds to a target protein (ER) and another ligand that binds to the E3 ubiquitin ligase. The interaction results in ubiquitination and degradation of the target protein through the ubiquitin-proteasome complex. Complete estrogen receptor antagonists (CERANs). CERANs block both transcriptional activation domains (AF1 and AF2) of ER by recruiting nuclear receptor corepressors (N-CoR) to inactivate AF1 and directly inhibit AF2. Selective estrogen receptor covalent antagonists (SERCAs). SERCAs covalently bind to a cysteine residue (C530) on ER, resulting in ER inactivation and inhibition of gene transcription.
In this perspective, we will review novel anti-estrogenic agents being evaluated in breast cancer, including preliminary or final efficacy and safety results from some trials and ongoing and/or planned randomized phase II–III trials that will define whether they will have a potential role in the management of early and advanced stage breast cancer. The results of phase I trials evaluating various agents are summarized in Table 1, including the recommended phase II doses (RP2D) when used as monotherapy or in combination with CDK4/6 inhibitors. The efficacy of various novel agents in phase I or phase I–II trials are summarized in Table 2, which also includes the characteristics of the patient populations. Notably, several of these trial results have only been presented at national meetings in abstract form and published data in peer-reviewed journals is pending.
Results of completed randomized phase II–III trials
Randomized trials with results reported are summarized in Table 3, including those for metastatic and localized breast cancer. In all of these trials, only patients with ER-positive and HER2-negative diseases were included. For some of these agents, other randomized trials (Table 4) and/or non-randomized trials (Table 5) are ongoing.
Selective estrogen receptor modulators
SERMs display ER antagonist or agonist activity, depending on the cell type, through the recruitment of different co-activators and co-repressors. SERMs inhibit activating function domain 2 (AF2) of ER but allow for agonist signaling through activating function domain 1 (AF1) through other signaling pathways such as mTOR, PI3K, and MAPK. Tamoxifen was the first approved SERM and is now widely used in the adjuvant and metastatic settings for breast cancer based on randomized Phase III trials23,24. Evidence of superior efficacy of AIs and the side effect profile of tamoxifen has decreased the enthusiasm for this class though other SERMs are currently in development6,7. Raloxifene, another SERM, was as effective as tamoxifen in breast cancer prevention in high-risk women without increasing the risk of endometrial cancer in the National Surgical Adjuvant Breast and Bowel Project (NSABP) Study of Tamoxifen and Raloxifene trial25. Toremifene is a SERM with a structure and efficacy nearly identical to that of tamoxifen. The drug was initially developed to allow for an improved side effect profile though studies have not demonstrated any safety advantage for toremifene26,27. The SERM arzoxifene showed initial promising efficacy and favorable safety with antiestrogenic effects on both breast and endometrium, but phase III data found it to be inferior to tamoxifen, ending further clinical development28,29.
Lasofoxifene
Lasofoxifene is a next-generation non-steroidal SERM that differs from other SERMs based on its binding affinity, which is similar to 17β-estradiol, and strong preclinical data in ER-mutated breast cancer models which are resistant to AIs30. Lasofoxifene first demonstrated a reduction in the risk of both fractures and breast cancer in patients in the post-menopausal evaluation and risk-reduction with the Lasofoxifene (PEARL) trial31. Subsequently, in ESR1 mutant models, lasofoxifene was shown to inhibit tumor growth at primary and metastatic sites compared to fulvestrant30. ELAINE I (NCT03781063) assessed the efficacy of lasofoxifene vs. fulvestrant in 103 patients, both pre- and postmenopausal, with MBC who have ESR1 mutations and progressed on prior AI and CDK 4/6 inhibitors (Table 3). Results showed numerically improved PFS with lasofoxifene compared with fulvestrant (6.04 vs 4.04 months; hazard ratio 0.699, p = 0.138), though this did not reach statistical significance32. ELAINE II (NCT04432454) is an ongoing, non-randomized phase II study evaluating lasofoxifene in combination with abemaciclib (Table 5)33.
Selective estrogen receptor degraders
Although SERMs inhibit ER through changes in ER structure and cofactor recruitment and AIs effectively reduce estrogen levels, the presence of ER itself can allow tumor to escape from ET and activate the ER signaling pathway. Progression in ER-positive breast cancer ultimately results from ligand-independent activation either through direct mutation of ER or phosphorylation of ER or its coregulators through signaling pathways such as PI3K-AKT-mTOR. SERDs address some of these resistance mechanisms, unlike SERMs and AIs, as they function not only as competitive ER antagonists but also induce proteasome-dependent degradation of ER34. Fulvestrant is the prototype of the SERD class and is currently the only approved SERD for the treatment of ER-positive MBC. The promising efficacy of fulvestrant has fueled interest in the SERD approach and steered the advancement of numerous orally bioavailable SERDs.
Fulvestrant
Several randomized trials have established the efficacy of fulvestrant as a single agent and in combination with various biologic and targeted agents. A meta-analysis of 11 trials including 5808 patients found that fulvestrant 500 mg was superior to fulvestrant 250 mg, megestrol acetate, and anastrozole, with regard to PFS35. In the phase III FALCON trial, women with ER-positive MBC without prior ET were randomized to either fulvestrant or anastrozole. The primary endpoint of PFS was increased in the fulvestrant arm (16.6 months) compared to the anastrozole arm (13.8 months)36. In phase III randomized trials in patients with metastatic ER-positive MBC, fulvestrant has demonstrated increases in PFS when combined with targeted agents such as CDK 4/6 inhibitors, alpelisib, and everolimus11,12,13,16,37.
Particularly, limitations of fulvestrant include its intramuscular administration. This has prompted the search for alternative orally bioavailable SERDs, which are currently under evaluation in clinical trials for use in metastatic, adjuvant, and neoadjuvant settings. Herein, we describe the development, toxicity profile, and corresponding trial for each novel SERD.
Elacestrant (RAD1901)
Elacestrant is an orally bioavailable SERM/SERD hybrid that is furthest along in development at this time. The drug functions as a partial agonist at lower doses and as an antagonist at higher doses. As receptor occupancy increases, degradation occurs, resulting in the inhibition of ESR1 signaling38. Elacestrant first demonstrated anti-tumor activity in breast cancer patient-derived xenograft (PDX) models, including those harboring ESR1 mutations39,40. These preclinical studies formed the basis for a Phase I study of elacestrant monotherapy in patients with heavily pretreated ER-positive MBC. The drug demonstrated antitumor activity and tolerability, and the trial established the RP2D at 400 mg once daily (Table 1), with nausea, fatigue, vomiting, anorexia, and arthralgias being the most common adverse effects41. Objective response was observed in 19.4% of patients, of whom at least half had prior fulvestrant (54%), CDK 4/6 inhibitors (52%), and ESR1 mutations (50%, Table 2).
The ensuing phase III EMERALD trial (NCT03778931) included postmenopausal patients with ER-positive, HER2-negative MBC with prior CDK 4/6 inhibitor therapy, 1–2 lines of ET, and ≤1 chemotherapy (Table 3). A total of 477 patients were randomly assigned to elacestrant 400 mg orally once daily or standard-of-care (SOC) endocrine monotherapy, which included either fulvestrant or an AI. Approximately 48% of patients had detectable ESR1 mutations. The results revealed prolonged PFS in the intention-to-treat (ITT) population receiving elacestrant with 12-month PFS rates of 22.3% vs. 9.4% in patients on elacestrant versus SOC with a hazard ratio (HR) of 0.70 (0.55–0.88). A greater magnitude of benefit was observed in the subgroup of patients with tumors harboring ESR1 mutations with HR 0.55 (0.39-0.77). Notably, the absolute PFS benefit in the study was small (2.8 vs. 1.9 months in the overall population), and this was attributed to rapid progression in the majority of patients in both treatment arms, after which the PFS curves diverged. An interim OS analysis (149 deaths) performed at the time of the prespecified final PFS analysis revealed a trend favoring elacestrant in the overall population (HR 0.75, 95% CI: 0.54–1.04, p = 0.08) and ESR1 mutant population (HR 0.59, 95% CI: 0.36–0.96, p = 0.03), but not the ESR1 non-mutant population (HR 0.92, 95% CI: 0.59–1.42, p = 0.69). Final OS analysis is expected when approximately 50% of the study population has died (239 deaths). Regarding safety, 27% of patients on elacestrant experienced a Grade 3/4 AE, such as nausea, back pain, and increased ALT, compared with 20.5% on the SOC arm. There were no treatment-related deaths42.
EMERALD was the first phase III trial evaluating an oral SERD against SOC endocrine therapy in patients with MBC and previous treatment with CDK 4/6 inhibitor. The higher magnitude of response in the subset with ESR1 mutations highlights the potential use of ESR1 as a predictive biomarker for this drug and other novel anti-estrogen agents. In January 2023, the U.S. Food and Drug Administration (FDA) approved elacestrant for patients with ER-positive, HER2-negative, and ESR1 mutated MBC following at least 1 line of ET. Elacestrant is also currently being evaluated in combination with abemaciclib in patients with brain metastases (NCT04791384) and in the presurgical setting by assessing change in Ki67 (NCT04797728, Table 5)43,44.
Giredestrant (GDC-9545)
Giredestrant is another orally bioavailable SERD that first demonstrated antitumor activity as a single agent and in combination with a CDK 4/6 inhibitor in PDX models45. A subsequent phase I a/b study (NCT03332797) evaluated giredestrant monotherapy (30 mg oral daily) and combination therapy (100 mg daily) with palbociclib in postmenopausal patients with ER-positive MBC who had disease recurrence while on adjuvant ET for ≥24 months or progression after prior ET for ≥6 months and ≤2 lines of therapy (Table 1). Drug tolerance and clinical activity were observed as a single agent and in combination with palbociclib. Most common AEs with giredestrant monotherapy included fatigue, arthralgias, and nausea, and only 5% of patients had Grade 3 AEs. Of note, 7% had bradycardia, but these were Grade 1–2 events. Among patients on combination therapy, 57% had Grade ≥3 AEs, the most common of which was neutropenia. Thirteen percent of 85 patients had Grade 1 asymptomatic bradycardia46,47,48. In terms of efficacy, ORR was 20% in 41 patients on single-agent giredestrant (Table 2) and 38% in 44 patients on giredestrant and palbociclib combination. CBRs were 54% and 81% in 41 patients on monotherapy and 48 on combination, respectively. Paired pre- and on-treatment biopsies from 21 patients illustrated consistent downregulation of ER, PR, Ki67, and ER pathway activity as measured by gene expression analysis on Cycle 2 Day 8. Thirty-four of 36 patients (94%) with detectable baseline ctDNA ESR1 level had a decrease after 4 weeks of therapy47,49.
The encouraging activity of giredestrant in Phase I studies has led to several phase II/III studies in the metastatic and early-stage settings. acelERA (NCT04576455) was a randomized phase II study evaluating the efficacy and safety of giredestrant versus physician’s choice of ET in postmenopausal and premenopausal women on ovarian function suppression (OFS) with ER-positive, HER2-negative, advanced/MBC who have received 1–2 prior lines of systemic therapy, at least one of which was ET (Table 3). Interim analysis in 303 patients with a median follow-up of 7.89 months showed no significant improvements in PFS in the overall population, although there was a non-significant trend for benefit in the ESR1 mutant subgroup (median PFS 5.3 vs. 3.5 months; HR 0.60 [CI: 0.35–1.03], p = 0.06)50.
persevERA (NCT04546009) is an ongoing Phase III double-blind, placebo-controlled, randomized trial evaluating the efficacy and safety of giredestrant and palbociclib versus letrozole and palbociclib in patients with ER-positive, HER2-negative MBC in the first line setting (Table 4)51. The Phase III randomized evERA trial (NCT05306340), which evaluates the efficacy of giredestrant and everolimus compared with exemestane and everolimus, is also ongoing (Table 4)52. A randomized umbrella trial (NCT04802759) is assessing the efficacy of giredestrant in combination with CDK 4/6 inhibitors, ipatasertib, inavolisib, everolimus, and samuraciclib among others (Table 4)53.
In the early-stage setting, the phase II coopERA BC trial (NCT04436744) randomized postmenopausal women with untreated ER-positive early breast cancer (EBC) and baseline Ki67 ≥ 5% to receive preoperative giredestrant versus anastrozole for a 14-day window-of-opportunity phase followed by 16 weeks of continued ET in addition to palbociclib (Table 3). The primary endpoint of Ki67 change from baseline to Week 2 was higher with giredestrant (mean reduction in Ki67 of 80%) compared with anastrozole (mean reduction of 67%)54. Final analyses in 221 patients showed that Ki67 suppression at surgery remained higher in the giredestrant arm (81% versus 74%). ORR was similar in both arms (50% giredestrant versus 49% anastrozole)55. The randomized phase III lidERA trial (NCT04961996) will evaluate adjuvant giredestrant vs. physician’s choice of ET for at least 5 years in patients with medium- and high-risk ER-positive EBC. The primary endpoint is invasive disease-free survival (IDFS) with a target enrollment of 4100 patients (Table 4)56.
Amcenestrant (SAR-439859)
Amcenestrant is another oral SERD that was investigated in several clinical trials, but after a recent phase III study comparing amcenestrant plus palbociclib with letrozole plus palbociclib demonstrated an advantage for the letrozole plus palbociclib arm, Sanofi decided to end clinical development of amcenestrant, and other ongoing studies were discontinued57,58.
Camizestrant (AZD9833)
Camizestrant is an oral SERD that showed tumor growth suppression in PDX models, including those with ESR1 mutations59. The phase I SERENA-1 trial (NCT03616587) investigated camizestrant as monotherapy and in combination with palbociclib in postmenopausal and premenopausal women on OFS with advanced HR-positive BC after ≥1 ET and ≤2 chemotherapies (Table 1). In the monotherapy dose escalation phase, at dose levels from 25 to 450 mg daily, 3 patients experienced dose-limiting toxicities (DLTs), including Grade 3 QTc prolongation, Grade 3 vomiting, and a combination of Grade 2 visual disturbance, headache, and gait disturbance, all of which resolved with dose reduction. No Grade 4 or 5 AEs were reported. Most common TRAEs included visual disturbances, bradycardia, nausea, and fatigue, among others. The 75 mg dose was subsequently established as the RP2D60. In a heavily pretreated population, camizestrant demonstrated clinical activity as monotherapy with ORR of 10% and CBR of 35.3% across all dose levels and CBR of 53.3% and PFS of 11.1 months in patients on the 75 mg dose (Table 2). When studied in combination with palbociclib, the toxicity profile was overall similar to camizestrant monotherapy with two DLTs (Grade 3 QTc prolongation and Grade 2 visual disturbances), both of which resolved with dose interruption and reduction61. Updated analyses of the dose expansion cohort of camizestrant 75 mg daily and palbociclib in 48 patients revealed an ORR of 6.3% and CBR of 50%62. The trial is also evaluating the drug in combination with abemaciclib, everolimus, and capivasertib63.
Several additional studies with camizestrant are ongoing or planned in MBC (Table 4). SERENA-2 (NCT04214288) is a randomized phase II trial comparing efficacy and safety of three dose levels of camizestrant vs. fulvestrant in a population that has progressed after at least 1 ET64. There are two ongoing phase III randomized trials in the first-line metastatic setting. SERENA-4 (NCT04711252) is comparing camizestrant in combination with palbociclib versus AI and palbociclib65. SERENA-6 (NCT04964934) is enrolling patients who have received first-line AI and CDK 4/6 inhibitor (palbociclib or abemaciclib) for at least 6 months without progression and are monitored regularly for the presence of ESR1 mutations via ctDNA analysis; those with detectable ESR1 mutations without disease progression are randomized to either continue AI and CDK 4/6 inhibitor or switch ET to camizestrant and continue the same CDK 4/6 inhibitor66.
In the window-of-opportunity SERENA-3 trial (NCT04588298), postmenopausal women with newly diagnosed ER-positive EBC will be randomized to receive 75 mg or 150 mg oral camizestrant for 5–7 days prior to surgery (Table 5). The study will evaluate the drug’s effect on ER expression in pre- and on-treatment tumor samples67.
Imlunestrant (LY-3484356)
Imlunestrant is an oral SERD that demonstrated promising efficacy in the preclinical setting with potent inhibition of ESR1 wildtype and mutant xenograft tumors. Synergistic effects were observed when combined with abemaciclib, everolimus, and alpelisib68. The Phase I/II EMBER-1 trial (NCT04188548) is evaluating the drug as a single agent and in combination with alpelisib, abemaciclib, everolimus, trastuzumab or abemaciclib and trastuzumab in postmenopausal and premenopausal women on OFS who have advanced ER-positive BC and endometrial endometrioid cancer (Table 5)69. Data from the dose escalation and dose expansion cohort of 114 patients on imlunestrant monotherapy demonstrated a favorable safety profile and encouraged anti-tumor activity (Table 1). No DLTs were observed. Most treatment-emergent adverse events (TEAEs) were Grade 1 and included nausea, diarrhea, fatigue, and arthralgia. Grade ≥3 TRAEs occurred in 7 patients. In terms of efficacy, the ORR was 8%, and CBR was 42% (Table 2). Complete clearance or decline in ESR1 ctDNA levels was observed for 73% of 44 patients with baseline ESR1 mutations. While the median PFS for the overall population was 4.3 months, for patients on second-line imlunestrant, it was 6.5 months70. EMBER-3 (NCT04975308) is a phase III randomized study of imlunestrant monotherapy, investigator’s choice ET, or imlunestrant plus abemaciclib in patients with ER-positive MBC previously treated with ET (Table 4)71.
In the early-stage setting, EMBER-2 (NCT04647487) is investigating the biological effects of pre-operative imlunestrant by evaluating changes in ER expression (Table 5)72. A phase III trial, EMBER-4, is being planned to evaluate the benefit of adjuvant imlunestrant vs. standard adjuvant ET in patients with ER-positive EBC, with prior adjuvant ET for 2–5 years and an increased risk of recurrence (Table 4)70.
Other agents with no data from randomized trials
Other agents that have undergone phase I or phase I–II evaluation with safety data (Table 1) and preliminary efficacy data (Table 2) are summarized here, some of which include agents that are further being evaluated in ongoing randomized (Table 4) and non-randomized (Table 5) clinical trials.
Selective estrogen receptor degraders (SERDs)
Rintodestrant (G1T48)
Rintodestrant is a novel orally bioavailable SERD that has demonstrated potent tumor inhibition in animal models with tamoxifen resistance and ESR1 mutations73. A phase I study (NCT03455270) evaluated rintodestrant in pre- and postmenopausal women with HR-positive MBC after progression on ET. In the dose escalation phase, the drug demonstrated target engagement on 18F-fluoroestradiol positron emission tomography (FES-PET), a tolerable side effect profile, and antitumor activity in heavily pretreated patients (Table 1)74. In the dose expansion portion, among 67 patients with a median of 2 prior lines of therapy, ORR was 5% and CBR 30% (Table 2). The activity was observed regardless of ESR1 or PIK3CA mutation status. Most common TRAEs included hot flashes, fatigue, nausea, diarrhea, and vomiting, most of which were Grade 1–2. Serious TRAEs included 1 patient with Grade 5 cerebral hemorrhage and another with Grade 2 upper abdominal pain. Two patients discontinued treatment due to TRAEs. No DLTs were observed75. Part 3 of this trial is assessing rintodestrant 800 mg daily with palbociclib in a population with prior ET but no prior CDK 4/6 inhibitor therapy. Preliminary data in 40 patients revealed an ORR of 5% and a CBR of 60%. The combination was well tolerated, with most common AEs related to the known safety profiles of palbociclib and rintodestrant76.
ZN-c5
ZN-c5 is a novel, small-molecule SERD with high oral bioavailability. In preclinical studies, the drug resulted in tumor growth inhibition which was enhanced when combined with CDK 4/6 inhibitors or PI3K inhibitors. ZN-c5 also showed increased efficacy in ESR1 PDX models when compared to fulvestrant77. 565TiP is a phase I/II study (NCT03560531) evaluating ZN-c5 as monotherapy and in combination with palbociclib in postmenopausal and premenopausal women on OFS with advanced ER-positive BC with a prior response on ET for at least 6 months. Results from 45 evaluable subjects in the dose escalation and expansion cohorts with single agent ZN-c5 demonstrated no DLTs, with the most common TRAEs including hot flashes, nausea, and fatigue; grade 3 TEAEs included abdominal pain, hypertension, hyponatremia, pain in extremities, and GGT increase (Table 1). With regard to efficacy in this population, the ORR was 5% and CBR 38% (Table 2). Phase II testing of Zn-c5 monotherapy and phase I testing of combination with palbociclib are in progress (Table 5)78. Recruitment for 564TiP, a phase 1b trial (NCT04514159) of ZN-c5 combined with abemaciclib in patients without prior CDK 4/6 inhibitors, is ongoing as well (Table 5)79.
D-0502
D-0502 is another orally bioavailable SERD with anti-tumor activity in PDX models, including those with ESR1 mutations80. A Phase I trial (NCT03471663) is investigating D-0502 as monotherapy and in combination with palbociclib to identify the RP2D in postmenopausal and premenopausal women on OFS who have HR-positive MBC. In the dose escalation portion, no DLTs and a favorable safety profile were observed. The most common AEs included nausea, vomiting, diarrhea, fatigue, alanine aminotransferase elevation, and neutropenia (Table 1). Preliminary efficacy results showed ORR of 5% and CBR of 36% in 22 patients on monotherapy (Table 2). Among 13 patients on D-0502 plus palbociclib, ORR and CBR were 15% and 77%, respectively. Future efficacy results from the dose expansion cohort will be informative81.
Borestrant (ZB-716)
Borestrant is a boronic acid-modified fulvestrant with oral bioavailability. It has demonstrated the downregulation of ER in endocrine-resistant breast cancer cells and superior tumor inhibition when compared to fulvestrant in PDX models82. ENZENO (NCT04669587) is an ongoing first-in-human study evaluating the safety and tolerability of ZB-716 as a single agent and in combination with palbociclib in patients with ER-positive MBC (Table 5)83.
Proteolysis targeting chimerics (PROTACs)
PROTACs are bifunctional hybrids that simultaneously bind to a specific target protein, such as ER, and an E3 ubiquitin ligase resulting in ubiquitination and degradation of the target protein ER through the ubiquitin-proteasome system84. As their mechanism of action is catalytic, they are able to promote protein degradation even at low exposure levels. PROTAC technology has been adapted to target ER with several PROTACs in development, the furthest along of which is ARV-47185. In PDX models with and without ESR1 mutations, oral daily administration of ARV-471 resulted in tumor regression86. A Phase I first-in-human study of ARV-471 enrolled postmenopausal patients with ER-positive MBC that had progressed on ≥2 lines of ET and ≥1 CDK 4/6 inhibitor; the drug was tolerated well with no DLTs, and the most common AEs were nausea, fatigue, and vomiting (Table 1). Patients were heavily pretreated with a median of 4 prior therapies; all had received prior CDK 4/6 inhibitor, and 80% had previous fulvestrant. Of 47 evaluable patients, the CBR was 40% (Table 2)87. The phase II dose escalation portion of this study is ongoing. A Phase I/II clinical trial (NCT04072952) of the combination of ARV-471 and palbociclib in this patient population is also ongoing (Table 5)88.
Complete estrogen receptor antagonists (CERANs)
The estrogen receptor includes two distinct transcriptional activation domains, AF1, which is activated by signaling pathways such as mTOR, PI3K, and MAPK, among others, and AF2, which is activated by the estrogen ligand itself. Activation of AF1 and AF2 both lead to gene transcription and cell proliferation. CERANs block both AF1 and AF2 transcriptional activation domains of ER. CERANs directly inhibit AF2 and recruit nuclear receptor corepressors (N-CoR) to inactivate AF1. This differs from SERMs which inhibit AF2 but allow agonist signaling via AF1 through other signaling pathways89.
OP-1250 is an orally bioavailable CERAN that also acts as a SERD-inducing ER degradation. In preclinical studies, OP-1250 demonstrated blockade of both wild-type and mutant ER, inhibition of estrogen-stimulated proliferation in breast cells as well as receptor degradation. In xenograft models, OP-1250 resulted in shrinkage of breast tumors expressing both wild-type and mutant ER90. Nonclinical studies have also demonstrated activity in mutant ESR1 tumors in the brain91. A phase I/II first-in-human study (NCT04505826) is evaluating the safety and tolerability of OP-1250 in postmenopausal and premenopausal women on OFS who have HR-positive MBC with progression on prior ET. No DLTs were observed, and most TEAEs were Grade 1–2, with the most common being nausea, fatigue, and constipation (Table 1). Phase 1b dose expansion and Phase 2 efficacy evaluation are ongoing. Preliminary data in 40 subjects with a median of 3 prior lines of therapy demonstrated anti-tumor activity (ORR 9%, CBR 21%) and drug tolerability. In the cohort within the anticipated RP2D range, ORR was 18% (2/11), and CBR was 38% (3/8) (Table 2)92.
Selective estrogen receptor covalent antagonists (SERCAs)
SERCAs inactivate ER by engaging a unique cysteine residue that is not present in other hormone receptors93. HRB-6545 is a first-in-class SERCA that covalently binds to a cysteine residue at position 530 of both wild-type and mutant ER proteins. The novel drug was found to antagonize both wild-type and mutant ER in in vitro studies. In xenograft models, the small molecule showed superior antitumor activity compared to fulvestrant94. In Phase I/II study (NCT03250676) of single-agent H3B-6545, pre- and postmenopausal women with previously treated locally advanced or metastatic HR-positive BC tolerated the drug well, and no DLTs were observed (Table 1). Of note, 35% of patients experienced Grade 1 asymptomatic sinus bradycardia and 5% with Grade 2 symptomatic bradycardia without requiring intervention. Other common AEs included nausea, fatigue, diarrhea, glomerular filtrate rate (GFR) decrease, hemoglobin decrease, and lymphocyte decrease. Serious AEs were reported in 21% of patients and led to treatment discontinuation in 13% of patients. In the evaluable population of 94 patients who had a median of 3 prior lines of therapy, and most were previously treated with CDK 4/6 inhibitor, preliminary analyses showed an ORR of 16.4%, CBR of 39.7%, and median PFS of 3.8 months (Table 2). The response was seen in patients with visceral metastases, heavily pretreated disease, and ESR1 mutations95,96,97. H3B-6545 is also being studied in combination with palbociclib in patients with HR-positive MBC with 2 or more prior therapies (NCT04288089, Table 5)98.
A new generation of anti-estrogen therapies, including SERMs other than tamoxifen and novel orally administered SERDs, and novel agents such as CERANs, SERCAs, and PROTACs targeting ER are being actively developed, driven primarily by the quest to develop agents that circumvent mechanisms of primary and secondary resistance to ET. Results thus far have been mixed, with statistically significant but clinically modest benefits observed with the oral SERD elacestrant when used in ET-resistant disease, especially when associated with ESR1 mutations, and clear failures with the oral SERD amcenestrant when used as first-line or second-line ET. Preliminary results with other oral SERDs, such as giredestrant have also suggested some potential benefits in patients with tumors harboring ESR1 mutations. At least 3 oral SERDs thus far, including giredestrant, imlunestrant, and camizestrant, are being evaluated in phase III trials in metastatic and/or early breast cancer. Other novel agents, including CERANs, SERCAs, and PROTACs, are in the early phases of clinical development, with some expected to be further evaluated in phase III trials. Although efficacy data from Phase III trials will guide their incorporation into clinical practice, the optimal sequencing and combinations of these drugs with other agents will pose additional opportunities for drug development. Key factors that will influence their impact on practice include drug tolerability, efficacy combined with or after targeted therapies such as CDK 4/6 inhibitors, mTOR inhibitors, and PI3K inhibitors, activity in patients with ESR1 mutations, and differential ability to cross the blood-brain barrier. These considerations will impact whether these novel therapies will exceed existing ET options, including tamoxifen, aromatase inhibitors, and fulvestrant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Burstein, H. J. et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: ASCO Clinical Practice Guideline focused update. J. Clin. Oncol. 37, 423–438 (2019).
Burstein, H. J. et al. Endocrine treatment and targeted therapy for hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer: ASCO Guideline Update. J. Clin. Oncol. 39, 3959–3977 (2021).
Heldring, N. et al. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 87, 905–931 (2007).
Allison, K. H. et al. Estrogen and progesterone receptor testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists guideline update. Arch. Pathol. Lab. Med. 144, 545–563 (2020).
Hanker, A. B., Sudhan, D. R. & Arteaga, C. L. Overcoming endocrine resistance in breast cancer. Cancer Cell 37, 496–513 (2020).
Group, A. T. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 365, 60–62 (2005).
Francis, P. A. et al. Tailoring adjuvant endocrine therapy for premenopausal breast cancer. N. Engl. J. Med. 379, 122–137 (2018).
Hortobagyi, G. N. et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 375, 1738–1748 (2016).
Finn, R. S. et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936 (2016).
Goetz, M. P. et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35, 3638–3646 (2017).
Sledge, George W. et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2− advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 35, 2875–2884 (2017).
Slamon, D. et al. Ribociclib plus fulvestrant for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in the phase III randomized MONALEESA-3 trial: updated overall survival. Ann. Oncol. 32, 1015–1024 (2021).
Turner, N. C. et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 373, 209–219 (2015).
Harbeck, N. et al. Adjuvant abemaciclib combined with endocrine therapy for high-risk early breast cancer: updated efficacy and Ki-67 analysis from the monarchE study. Ann. Oncol. 32, 1571–1581 (2021).
Yardley, D. A. et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv. Ther. 30, 870–884 (2013).
André, F. et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase III SOLAR-1 trial. Ann. Oncol. 29, viii709 (2018).
Cardoso, F. et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 31, 1623–1649 (2020).
Razavi, P. et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 34, 427–438.e426 (2018).
Brett, J. O., Spring, L. M., Bardia, A. & Wander, S. A. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 23, 85 (2021).
Chandarlapaty, S. et al. Prevalence of ESR1 mutations in cell-free dna and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2, 1310–1315 (2016).
Bidard, F.-C. et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 23, 1367–1377 (2022).
McDonnell, D. P., Wardell, S. E., Chang, C.-Y. & Norris, J. D. Next-generation endocrine therapies for breast cancer. J. Clin. Oncol. 39, 1383–1388 (2021).
Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365, 1687–1717 (2005).
Davies, C. et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 381, 805–816 (2013).
Leung, S. C. Y. et al. Analytical validation of a standardised scoring protocol for Ki67 immunohistochemistry on breast cancer excision whole sections: an international multicentre collaboration. Histopathology 75, 225–235 (2019).
Vogel, C. L., Johnston, M. A., Capers, C. & Braccia, D. Toremifene for breast cancer: a review of 20 years of data. Clin. Breast Cancer 14, 1–9 (2014).
Hong, J. et al. A prospective, randomized study of Toremifene vs. tamoxifen for the treatment of premenopausal breast cancer: safety and genital symptom analysis. BMC Cancer 20, 663 (2020).
Deshmane, V., Krishnamurthy, S., Melemed, A. S., Peterson, P. & Buzdar, A. U. Phase III double-blind trial of arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. J. Clin. Oncol. 25, 4967–4973 (2007).
Jackson, L. R., Cheung, K. L., Buzdar, A. U. & Robertson, J. F. Arzoxifene: the evidence for its development in the management of breast cancer. Core Evid. 2, 251–258 (2008).
Lainé, M. et al. Lasofoxifene as a potential treatment for therapy-resistant ER-positive metastatic breast cancer. Breast Cancer Res. 23, 54 (2021).
LaCroix, A. Z. et al. Breast cancer incidence in the randomized PEARL trial of lasofoxifene in postmenopausal osteoporotic women. J. Natl Cancer Inst. 102, 1706–1715 (2010).
Goetz, MP. et al. In European Society of Medical Oncology. S808–S869 (Annals of Oncology).
Damodaran, S., Plourde, P. V., Moore, H. C. F., Anderson, I. C. & Portman, D. J. Open-label, phase 2, multicenter study of lasofoxifene (LAS) combined with abemaciclib (Abema) for treating pre- and postmenopausal women with locally advanced or metastatic ER+/HER2− breast cancer and an ESR1 mutation after progression on prior therapies. J. Clin. Oncol. 40, 1022 (2022).
McDonnell, D. P. & Wardell, S. E. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr. Opin. Pharmacol. 10, 620–628 (2010).
Cope, S., Ouwens, M. J., Jansen, J. P. & Schmid, P. Progression-free survival with fulvestrant 500 mg and alternative endocrine therapies as second-line treatment for advanced breast cancer: a network meta-analysis with parametric survival models. Value Health 16, 403–417 (2013).
Robertson, J. F. R. et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet 388, 2997–3005 (2016).
Kornblum, N. et al. Randomized phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer resistant to aromatase inhibitor therapy: results of PrE0102. J. Clin. Oncol. 36, 1556–1563 (2018).
Wardell, S. E., Nelson, E. R., Chao, C. A., Alley, H. M. & McDonnell, D. P. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr.-Relat. Cancer 22, 713–724 (2015).
Garner, F., Shomali, M., Paquin, D., Lyttle, C. R. & Hattersley, G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-Cancer Drugs 26, 948 (2015).
Bihani, T. et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER+ breast cancer patient-derived xenograft models. Clin. Cancer Res. 23, 4793–4804 (2017).
Bardia, A. et al. Phase I study of elacestrant (RAD1901), a novel selective estrogen receptor degrader, in ER-positive, HER2-negative advanced breast cancer. J. Clin. Oncol. 39, 1360–1370 (2021).
Bidard, F.-C. et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor–positive, human epidermal growth factor receptor 2–negative advanced breast cancer: results from the randomized phase III EMERALD trial. J. Clin. Oncol. https://doi.org/10.1200/jco.22.00338.
NCT04791384 Phase Ib/II Trial of Abemaciclib and Elacestrant in Patients With Brain Metastasis Due to HR+/Her2- Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT04791384?term=elacestrant&cond=Breast+Cancer&draw=2&rank=1 (2022).
NCT04797728 Elacestrant in Preoperative Setting, a Window of Opportunity Study (ELIPSE). https://clinicaltrials.gov/ct2/show/NCT04797728?term=elacestrant&cond=Breast+Cancer&draw=2&rank=5 (2022).
Liang, J. et al. GDC-9545 (Giredestrant): a potent and orally bioavailable selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ breast cancer. J. Med. Chem. 64, 11841–11856 (2021).
Jhaveri, K. L. et al. Safety and activity of single-agent giredestrant (GDC-9545) from a phase Ia/b study in patients (pts) with estrogen receptor-positive (ER+), HER2-negative locally advanced/metastatic breast cancer (LA/mBC). J. Clin. Oncol. 39, 1017–1017 (2021).
Lim, E. et al. A phase Ib study to evaluate the oral selective estrogen receptor degrader GDC-9545 alone or combined with palbociclib in metastatic ER-positive HER2-negative breast cancer. J. Clin. Oncol. 38, 1023–1023 (2020).
Jhaveri, K. et al. Abstract PD7-05: A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD), GDC-9545, in postmenopausal women with estrogen receptor-positive (ER+) HER2-negative (HER2-) metastatic breast cancer. Cancer Res. 80, PD7-05–PD07-05 (2020).
Turner, N. C. et al. Abstract PD13-07: Activity and biomarker analyses from a phase Ia/b study of giredestrant (GDC-9545; G) with or without palbociclib (palbo) in patients with estrogen receptor-positive, HER2-negative locally advanced/metastatic breast cancer (ER+/HER2- LA/mBC. Cancer Res. 82, PD13–07 (2022).
Martin Jimenez, M. et al. in European Society of Medical Oncology. S88–S121 (Annals of Oncology).
Turner, N. C. et al. persevERA Breast Cancer (BC): Phase III study evaluating the efficacy and safety of giredestrant (GDC-9545) + palbociclib versus letrozole + palbociclib in patients (pts) with estrogen-receptor-positive, HER2-negative locally advanced or metastatic BC (ER+/HER2– LA/mBC). J. Clin. Oncol. 39, TPS1103 (2021).
NCT05306340 A Study Evaluating the Efficacy and Safety of Giredestrant Plus Everolimus Compared With Exemestane Plus Everolimus in Participants With Estrogen Receptor-Positive, HER2-Negative, Locally Advanced or Metastatic Breast Cancer (evERA Breast Cancer). https://clinicaltrials.gov/ct2/show/NCT05306340 (2022).
NCT04802759 A Study Evaluating the Efficacy and Safety of Multiple Treatment Combinations in Participants With Breast Cancer. https://www.clinicaltrials.gov/ct2/show/NCT04802759 (2022).
Hurvitz, S. A. et al. LBA14 Neoadjuvant giredestrant (GDC-9545) + palbociclib (palbo) vs anastrozole (A) + palbo in post-menopausal women with oestrogen receptor-positive, HER2-negative, untreated early breast cancer (ER+/HER2– eBC): interim analysis of the randomised, open-label, phase II coopERA BC study. Ann. Oncol. 32, S1285–S1286 (2021).
Fasching, P. A. et al. Neoadjuvant giredestrant (GDC-9545) plus palbociclib (P) versus anastrozole (A) plus P in postmenopausal women with estrogen receptor–positive, HER2-negative, untreated early breast cancer (ER+/HER2– eBC): final analysis of the randomized, open-label, international phase 2 coopERA BC study. J. Clin. Oncol. 40, 589–589 (2022).
Bardia, A. et al. Abstract OT2-11-09: Lidera breast cancer: a phase III adjuvant study of giredestrant (GDC-9545) vs physician’s choice of endocrine therapy (ET) in patients (pts) with estrogen receptor-positive, HER2-negative early breast cancer (ER+/HER2- EBC). Cancer Res. 82, OT2-11-09–OT12-11-09 (2022).
Bardia, A. et al. AMEERA-5: a randomized, double-blind phase 3 study of amcenestrant plus palbociclib versus letrozole plus palbociclib for previously untreated ER+/HER2– advanced breast cancer. Therap. Adv. Med. Oncol. 14, 17588359221083956 (2022).
Press Release: Sanofi provides update on amcenestrant clinical development program. https://www.sanofi.com/en/media-room/press-releases/2022/2022-08-17-05-30-00-2499668 (2022).
Scott, J. S. et al. Abstract 5674: Discovery of AZD9833, an oral small molecule selective degrader of the estrogen receptor (SERD). Cancer Res. 80, 5674–5674 (2020).
Hamilton, E. P. et al. A phase I dose escalation and expansion study of the next generation oral SERD AZD9833 in women with ER-positive, HER2-negative advanced breast cancer. J. Clin. Oncol. 38, 1024–1024 (2020).
Baird, R. et al. Abstract PS11-05: Updated data from SERENA-1: a phase 1 dose escalation and expansion study of the next generation oral SERD AZD9833 as a monotherapy and in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. Cancer Res. 81, PS11-05–PS11-05 (2021).
Oliveira, M. et al. Serena-1: updated analyses from a phase 1 study (parts C/D) of the next-generation oral SERD camizestrant (AZD9833) in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. J. Clin. Oncol. 40, 1032–1032 (2022).
NCT03616587 Study of AZD9833 Alone or in Combination in Women With Advanced Breast Cancer. (SERENA-1). https://clinicaltrials.gov/ct2/show/NCT03616587 (2022).
Oliveira, M. et al. Abstract OT-09-02: A randomized, open-label, parallel-group, multicenter phase 2 study comparing the efficacy and safety of oral AZD9833 versus fulvestrant in women with advanced ER-positive HER2-negative breast cancer (SERENA-2). Cancer Res. 81, OT-09-02 (2021).
André, F. et al. Abstract OT2-11-06: SERENA-4: a phase III comparison of AZD9833 (camizestrant) plus palbociclib, versus anastrozole plus palbociclib, for patients with ER-positive/HER2-negative advanced breast cancer who have not previously received systemic treatment for advanced disease. Cancer Res. 82, OT2-11-06–OT12-11-06 (2022).
Bidard, F.-C. et al. Abstract OT2-11-05: SERENA-6: a phase III study to assess the efficacy and safety of AZD9833 (camizestrant) compared with aromatase inhibitors when given in combination with palbociclib or abemaciclib in patients with HR+/HER2- metastatic breast cancer with detectable ESR1m who have not experienced disease progression on first-line therapy. Cancer Res. 82, OT2-11-05–OT12-11-05 (2022).
Robertson, J. F. et al. Abstract OT-09-05: a randomized, pre-surgical study to investigate the biological effects of AZD9833 doses in women with ER-positive HER2-negative primary breast cancer (SERENA-3. Cancer Res. 81, OT-09-05 (2021).
Bhagwat, S. V. et al. Abstract 1236: preclinical characterization of LY3484356, a novel, potent and orally bioavailable selective estrogen receptor degrader (SERD). Cancer Res. 81, 1236–1236 (2021).
Lim, E. et al. Abstract OT-09-03: EMBER: a phase 1a/b trial of LY3484356, a novel, oral selective estrogen-receptor degrader (SERD), in advanced ER+ breast cancer and endometroid endometrial cancer. Cancer Res. 81, OT-09-03 (2021).
Jhaveri, K. L. et al. A phase 1a/b trial of imlunestrant (LY3484356), an oral selective estrogen receptor degrader (SERD) in ER-positive (ER+) advanced breast cancer (aBC) and endometrial endometrioid cancer (EEC): monotherapy results from EMBER. J. Clin. Oncol. 40, 1021–1021 (2022).
Jhaveri, K. et al. Abstract OT2-11-01: EMBER-3: a randomized phase 3 study of LY3484356, a novel, oral selective estrogen receptor degrader vs investigator’s choice of endocrine therapy of either fulvestrant or exemestane, in patients with estrogen receptor-positive, human epidermal growth factor receptor 2-negative, locally advanced or metastatic breast cancer previously treated with endocrine-based therapy. Cancer Res. 82, OT2-11-01–OT12-11-01 (2022).
NCT04647487 A Study of LY3484356 in Women With Breast Cancer Before Having Surgery (EMBER-2). https://clinicaltrials.gov/ct2/show/NCT04647487 (2022).
Wardell, S. E. et al. Abstract 5641: effects of G1T48, a novel orally bioavailable selective estrogen receptor degrader (SERD), and the CDK4/6 inhibitor, G1T38, on tumor growth in animal models of endocrine resistant breast cancer. Cancer Res. 77, 5641–5641 (2017).
Dees, E. C. et al. 340P—dose-escalation study of G1T48, an oral selective estrogen receptor degrader (SERD), in postmenopausal women with ER+/HER2- locally advanced or metastatic breast cancer (ABC). Ann. Oncol. 30, v121–v122 (2019).
Aftimos, P. et al. Abstract PS12-04: rintodestrant (G1T48), an oral selective estrogen receptor degrader in ER+/HER2- locally advanced or metastatic breast cancer: Updated phase 1 results and dose selection. Cancer Res. 81, PS12-04 (2021).
Maglakelidze, M. et al. Rintodestrant (G1T48), an oral selective estrogen receptor degrader, in combination with palbociclib for ER+/HER2– advanced breast cancer: phase 1 results. J. Clin. Oncol. 39, 1063–1063 (2021).
Samatar, A. A. et al. Abstract 4373: discovery of ZN-c5, a novel potent and oral selective estrogen receptor degrader. Cancer Res. 80, 4373–4373 (2020).
Kalinksy, K. et al. Abstract P1-17-02: ZN-c5, an oral selective estrogen receptor degrader (SERD), in women with advanced estrogen receptor-positive (ER+)/human epidermal growth factor receptor 2 negative (HER2-) breast cancer. Cancer Res. 82, P1-17-02–P11-17-02 (2022).
Keogh, G. P. et al. 564TiP A phase Ib dose-escalation study of ZN-c5, an oral selective estrogen receptor degrader (SERD), in combination with abemaciclib in patients with advanced estrogen receptor (ER)+/HER2- breast cancer. Ann. Oncol. 32, S618–S619 (2021).
Wang, Y., Shi, Z., Jiang, Y. & Dai, X. Abstract 5776: pharmacologic and PK/PD study of D-0502: an orally bioavailable SERD with potent antitumor activity in ER-positive breast cancer cell lines and xenograft models. Cancer Res. 78, 5776–5776 (2018).
Osborne, C. et al. Abstract PS11-26: A phase 1 study of D-0502, an orally bioavailable SERD, for advanced or metastatic HR-positive and HER2-negative breast cancer. Cancer Res. 81, PS11-26 (2021).
Guo, S. et al. ZB716, a steroidal selective estrogen receptor degrader (SERD), is orally efficacious in blocking tumor growth in mouse xenograft models. Oncotarget 9, 6924–6937 (2018).
NCT04669587 ER+/HER2- Locally Advanced or Metastatic Breast Cancer (ENZENO Study) (ENZENO). https://clinicaltrials.gov/ct2/show/NCT04669587 (2022).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Lin, X., Xiang, H. & Luo, G. Targeting estrogen receptor α for degradation with PROTACs: a promising approach to overcome endocrine resistance. Eur. J. Med. Chem. 206, 112689 (2020).
Flanagan, J. et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 79, P5-04-18–P05-04-18 (2019).
Hamilton, E. et al. Abstract PD13-08: first-in-human safety and activity of ARV-471, a novel PROTAC® estrogen receptor degrader, in ER+/HER2- locally advanced or metastatic breast cancer. Cancer Res. 82, PD13-08 (2022).
Hamilton, E. P. et al. ARV-471, an estrogen receptor (ER) PROTAC degrader, combined with palbociclib in advanced ER+/human epidermal growth factor receptor 2–negative (HER2-) breast cancer: Phase 1b cohort (part C) of a phase 1/2 study. J. Clin. Oncol. 40, TPS1120 (2022).
Hodges-Gallagher, L., Sun, R., Myles, D. C., Harmon, C. L. & Kushner, P. J. Abstract P5-05-02: preclinical development of OP-1250, an oral complete estrogen receptor antagonist (CERAN) that shrinks ER-positive breast tumors in xenograft models. Cancer Res. 80, P5-05-02–P05-05-02 (2020).
Hodges-Gallagher, L. et al. Abstract PS18-16: the complete estrogen receptor antagonist OP-1250 shrinks tumors in xenograft models and has favorable preclinical pharmacokinetic attributes. Cancer Res. 81, PS18-16 (2021).
Hodges-Gallagher, L. et al. Abstract LB122: the complete estrogen receptor antagonist (CERAN) OP-1250 shrinks ER+ brain metastases in an intracranial xenograft tumor model expressing mutant ESR1. Cancer Res. 81, LB122 (2021).
Patel, M. et al. Abstract P1-17-12: preliminary data from a phase I/II, multicenter, dose escalation study of OP-1250, an oral CERAN/SERD, in subjects with advanced and/or metastatic estrogen receptor (ER)-positive, HER2-negative breast cancer. Cancer Res. 82, P1-17-12–P11-17-12 (2022).
Puyang, X. et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERαWT and ERαMUT breast CancerSERCA H3B-5942 for treatment of ERαWT and ERαMUT breast cancer. Cancer Discov. 8, 1176–1193 (2018).
Smith, P. G. et al. Abstract DDT01-04: discovery and development of H3B-6545: A novel, oral, selective estrogen receptor covalent antagonist (SERCA) for the treatment of breast cancer. Cancer Res. 77, DDT01-04 (2017).
Hamilton, E. P. et al. Phase I dose escalation of H3B-6545, a first-in-class highly selective ERα covalent antagonist (SERCA), in women with ER-positive, HER2-negative breast cancer (HR+ BC). J. Clin. Oncol. 37, 1059–1059 (2019).
Hamilton, E. P. et al. Phase I/II study of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. J. Clin. Oncol. 39, 1018 (2021).
Hamilton, E. P. et al. Abstract P1-17-10: H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer—a phase II study. Cancer Res. 82, P1-17-10–P11-17-10 (2022).
Johnston, S. R. D. et al. Phase 1b study of H3B-6545 in combination with palbociclib in women with metastatic estrogen receptor–positive (ER+), human epidermal growth factor receptor 2 (HER2)-negative breast cancer. J. Clin. Oncol. 39, e13025 (2021).
Author information
Authors and Affiliations
Contributions
R.P. wrote the initial paper. P.K., A.T., and J.A.S. reviewed and significantly contributed to the final paper.
Corresponding author
Ethics declarations
Competing interests
Author Joseph Sparano is an Associate Editor of npj Breast Cancer. J.A.S. reports a consulting/advisory role for Genentech/Roche, Novartis, AstraZeneca, Celgene, Lilly, Celldex, Pfizer, Prescient Therapeutics, Juno Therapeutics, Merrimack, Adgero Biopharmaceuticals, Cardinal Health, GlaxoSmithKline, CStone Pharmaceuticals, Epic Sciences, Daiichi Sankyo, BMSi. A.T. reports a consulting/advisory role for Puma Biotechnology, Immunomedics, AstraZeneca, Novartis, Eisai, and Roche/Genentech. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, R., Klein, P., Tiersten, A. et al. An emerging generation of endocrine therapies in breast cancer: a clinical perspective. npj Breast Cancer 9, 20 (2023). https://doi.org/10.1038/s41523-023-00523-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-023-00523-4
