
Abstract
Breast cancer metastasis is the main cause of cancer death in women, so far, no effective treatment has inhibited breast cancer metastasis. Sulforaphane (SFN), a natural compound derived from broccoli, has shown potential health benefits in many cancers. However, research on breast cancer metastasis is still insufficient. Here, we showed that SFN, including its two isomers of R-SFN and S-SFN, significantly inhibited TGF-β1-induced migration and invasion in breast cancer cells. Proteomic and phosphoproteomic analysis showed that SFN affected the formation of the cytoskeleton. Subsequent experiments confirmed that SFN significantly inhibited TGF-β1-induced actin stress fiber formation and the expression of actin stress fiber formation-associated proteins, including paxillin, IQGAP1, FAK, PAK2, and ROCK. Additionally, SFN is directly bound to RAF family proteins (including ARAF, BRAF, and CRAF) and inhibited MEK and ERK phosphorylation. These in vitro results indicate that SFN targets the RAF/MEK/ERK signaling pathway to inhibit the formation of actin stress fibers, thereby inhibiting breast cancer cell metastasis.
Similar content being viewed by others

Introduction
Breast cancer metastasis is the main cause of cancer death in women, especially triple-negative breast cancer (TNBC) is more aggressive and has a poorer prognosis than other types of breast cancer. So far, no effective treatment has inhibited breast cancer metastasis. Due to the high rate of metastasis, the mortality of young women (<45 years) with breast cancer, especially with TNBC type, has increased significantly recently1,2,3,4. The metastatic potential of cancer cells is coordinated through cell-cell adhesion, cell-matrix adhesion, protrusion, and contraction, all of which require proper cytoskeleton regulation and its dynamics5,6,7,8. Therefore, inhibiting the formation of the cytoskeleton is a promising strategy for the treatment of cancer metastasis.
Actin stress fibers are a higher-order cytoskeletal structure composed of cross-linked actin filament bundles, which play a vital role in cell migration and invasion. The actin stress fiber organization promotes cell stiffening and proliferation of preinvasive breast cancer cells9. Numerous studies have demonstrated that RhoA and Rho-kinase (ROCK) signaling pathway is involved in actin stress fiber formation10,11,12,13,14. Actin stress fibers connect to focal adhesions and focal adhesion-associated proteins, such as paxillin15,16, focal adhesion kinase (FAK)17,18, and Ras GTPase-activating-like protein (IQGAP1)19,20, which are key regulators of actin cytoskeleton dynamics.
RAF is a serine/threonine-protein kinase that can directly phosphorylate or promote protein phosphorylation by activating the downstream mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway21,22. The RAF/MEK/ERK cascade is involved in regulating the development and metastasis of multiple types of cancer, including lung cancer, liver cancer, and breast cancer23,24,25,26. RAF family members include ARAF, BRAF, and CRAF. Recent studies revealed that a dimerization-dependent mechanism drives ARAF catalytic activation and dimerization of ARAF promotes MAPK pathway activation and cell migration27.
Sulforaphane (SFN), a natural compound derived from broccoli/broccoli sprouts, has been proved to exhibit chemoprotective effects for various cancers, including prostate cancer, lung cancer, and colon cancer28,29,30. Multiple action mechanisms involved in the anticancer properties of SFN have been reported, including induction of apoptosis31,32, cell cycle arrest33,34, and inhibition of angiogenesis and metastasis35,36. Although SFN has shown potential health benefits in many cancers, there are few studies on the involvement of SFN in breast cancer metastasis. Whether SFN can inhibit breast cancer metastasis and its mechanism are still unclear.
In the present study, we aimed to investigate whether SFN has an inhibitory effect on migration and invasion of breast cancer cells. We show that SFN and its two isomers (R-SFN and S-SFN) inhibited transforming growth factor-β1 (TGF-β1)-induced migration and invasion in human TNBC cells. Further, the mechanism by which SFN suppresses migration and invasion was also investigated.
Results
Effect of SFN on the viability of MDA-MB-231 and MDA-MB-157 cells
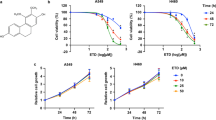
Many studies have demonstrated that TGF-β1 induced the migration and invasion of breast cancer cells37,38,39,40. However, the precise mechanism underlying TGF-β1-induced migration and invasion in breast cancer has not been fully elucidated. To determine the effect of SFN on TGF-β1-induced migration and invasion in breast cancer cells, we first measured the viability of cells after treatment with SFN (the chemical structure of SFN is shown in Fig. 1a) by using the CCK-8 assay. The previous report showed that the IC50 value of SFN varies greatly in the breast cancer cell lines41. Therefore, we treated the cells with various concentrations of SFN from 0.5 to 240 μM for 24 h and then cell viability was measured. As shown in Fig. 1b, in MDA-MB-231 cells, SFN has no significant effect on cell viability when the concentration is below 30 µM. However, at concentrations of 60, 120, and 240 µM SFN, significant toxicity of SFN was observed. In MDA-MB-157 cells, SFN showed no significant effect on cell viability when the concentration was lower than 7.5 µM (Fig. 1c). It is well known that when the compound produces a toxic response to cells, meaning that the compound can activate many intracellular signaling pathways. Avoiding toxicity to cells is crucial to selectively examine anti-migratory effects and mechanism(s) of SFN. Therefore, we used the concentration of 7.5, 15, and 30 µM for MDA-MB-231 cells and 1.9, 3.8, and 7.5 µM for MDA-MB-157 cells to study the effect of SFN on the migration and invasion in two human TNBC cell lines of MDA-MB-231 and MDA-MB-157.

a The chemical structure of SFN. b Effects of SFN at different concentrations on the viability of MDA-MB-231 cells. Data shown are mean ± SEM. n = 5, *P < 0.05; #P > 0.05. c Effects of SFN at different concentrations on the viability of MDA-MB-157 cells. Data shown are mean ± SEM. n = 5, *P < 0.05; #P > 0.05.
SFN inhibits migration and invasion of MDA-MB-231 and MDA-MB-157 cells
To investigate whether SFN inhibits breast cancer metastasis, we first examined the effect of SFN on the migration of MDA-MB-231 cells using a wound healing assay and recorded the real-time migration. As shown in Fig. 2a, b, TGF-β1 induced remarkable migration. However, after being treated for 24 h with SFN at the concentration of 7.5, 15, and 30 μM, the number of MDA-MB-231 cells migrated to the wound was markedly reduced (Fig. 2a). Quantitative analysis showed that SFN significantly inhibited cell migration, and as the concentration increased, the inhibitory effect became stronger (Fig. 2b). Similarly, SFN also showed a significant inhibitory effect on TGF-β1-induced invasion of MDA-MB-231 cells (Fig. 2e, f). In addition, SFN also significantly inhibited TGF-β1-induced migration and invasion of MDA-MB-157 cells (Fig. 2c, d, e, g).

a, c Representative images showing that wound width of MDA-MB-231 (a) and MDA-MB-157 (c) cells in the presence of SFN at different concentrations (7.5, 15, and 30 μM for MDA-MB-231 cells and 1.9, 3.8, and 7.5 μM for MDA-MB-157 cells) at 24 h by wound healing assay. Scale bar = 100 μm. b, d Statistical assay showing the effects of SFN on TGF-β1-induced migration of MDA-MB-231 (b) and MDA-MB-157 (d) cells at different concentrations at 24 h. Data shown are mean ± SEM. n = 5, **P < 0.05. e Representative images showing that invasion of MDA-MB-231 and MDA-MB-157 cells in the presence of SFN at different concentrations (7.5, 15, and 30 μM for MDA-MB-231 cells and 1.9, 3.8, and 7.5 μM for MDA-MB-157 cells) at 24 h by transwell assay. f, g Statistical assay showing the effects of SFN on TGF-β1-induced invasion of MDA-MB-231 (f) and MDA-MB-157 (g) cells at different concentrations at 24 h. Data shown are mean ± SEM. n = 5 or 6, **P < 0.05.
R-sulforaphane (R-SFN) and S-sulforaphane (S-SFN) are two isomers of SFN. Their chemical structures are shown in Fig. 3a. R-SFN is the naturally-occurring isomer, found in broccoli and other vegetables. S-SFN is a synthesized isomer. To investigate whether the conformational structure affects its function, we then compared the effects of two isomers of SFN, R-SFN, and S-SFN, on cell migration and invasion. As shown in Fig. 3b, c, R-SFN and S-SFN significantly inhibited TGF-β1-induced migration of MDA-MB-231 cells. In the transwell invasion assay, R-SFN and S-SFN also significantly inhibited TGF-β1-induced invasion of MDA-MB-231 cells (Fig. 3f, g). Similarly, R-SFN and S-SFN also inhibited TGF-β1-induced migration and invasion of MDA-MB-157 cells (Fig. 3d, e, f, h). These results showed that R-SFN and S-SFN have equally inhibitory effects on breast cancer cell migration and invasion.

a The 3D-conformer of R-SFN and S-SFN. The dotted circle shows the difference in structure between R-SFN (PubChem Identifier: CID 9577379. URL: https://pubchem.ncbi.nlm.nih.gov/compound/9577379#section=3D-Conformer) and S-SFN (PubChem Identifier: CID 10479733. URL: https://pubchem.ncbi.nlm.nih.gov/compound/S_-sulforaphane#section=3D-Conformer). b, d Representative images showing the wound width of MDA-MB-231 (b) and MDA-MB-157 (d) cells in the presence of R-SFN and S-SFN at 24 h by wound healing assay. Scale bar = 100 μm. c, e Statistical assay showing that the effects of R-SFN and S-SFN on TGF-β1-induced migration of MDA-MB-231 (c) and MDA-MB-157 (e) cells at 24 h. Data shown are mean ± SEM. n = 5 or 6, **P < 0.05; ***P < 0.01. f Representative images showing that invasion of MDA-MB-231 and MDA-MB-157 cells in the presence of R-SFN and S-SFN at 24 h by transwell assay. g, h Statistical assay showing that the effects of R-SFN and S-SFN on TGF-β1-induced invasion of MDA-MB-231 (g) and MDA-MB-157 (h) cells at 24 h. Data shown are mean ± SEM. n = 4 or 5, **P < 0.05; ***P < 0.001.
The inhibition of SFN on migration and invasion is highly correlated with the regulation of actin cytoskeleton
To investigate the potential mechanism by which SFN inhibited TGF-β1-induced migration and invasion of MDA-MB-231 cells, we performed quantitative proteomic and phosphoproteomic analysis. In total, 1669 normal proteins (Supplementary Table S1, <1% false discovery rate [FDR]) and 3198 phosphorylation sites in 1321 phosphoproteins were quantified (Supplementary Table S2, <1% FDR). The principal-component analysis (PCA) of normal proteins and phosphorylation sites verified the reproducibility of these results (Fig. 4b).

a The workflow of the mass spectrometry-based phosphoproteomic analysis. b Principal-component analysis of all identified normal proteins and phosphorylation sites shows that samples cluster tightly according to biology. The log2 ratios of normal proteins and phosphorylation sites in the control group was defaulted to 0.
Compared with the TGF-β1 treatment group, SFN treatment significantly changed the expression of 15 proteins (six downregulated and nine upregulated; Fig. 5a, c) and the phosphorylation level of 128 phosphorylation sites (37 downregulated and 91 upregulated; Fig. 5b, d) in 100 proteins. Next, to explore the possible cellular functions and signal pathways activated after SFN treatment, we used the String database (https://string-db.org/) to establish protein–protein interactions (PPI) of differentially expressed proteins and phosphoproteins and extracted the core network by combining the interaction score >0.7 and degree >5 (Fig. 5e). PPI analysis and the KEGG pathway enrichment of the extracted core network showed that the inhibitory effect of SFN on migration and invasion was highly correlated with the regulation of actin cytoskeleton (Fig. 5f).

a A volcano plot shows that 15 proteins (red dots) changed significantly (FDR <0.05, S0 of 1) upon SFN treatment. b A volcano plot shows that 128 localized phosphorylation sites (red dots) changed significantly (FDR <0.05, S0 of 1) upon SFN treatment. c Cluster analysis of differentially expressed proteins. d Cluster analysis of differentially expressed phosphorylation sites. e The core network of protein–protein interaction map of the altered proteins and phosphoproteins that are illustrated with color by clusters and size by degree. MAPK1, TP53, and PTK2 are in the top three degrees and served with a red border. The nodes involved in the regulation of the actin cytoskeleton are displayed by arrows. f KEGG pathways enrichment of core altered proteins and phosphoproteins.
SFN inhibits actin stress fiber formation and the expression of actin stress fiber formation-associated proteins
Proteomics analysis results indicated that the actin cytoskeleton played an important role in SFN inhibiting cell migration and invasion. The driving force for cell migration and invasion is provided by actin cytoskeleton polymerization, which induces membrane protrusion and pseudopod formation42,43. Therefore, we next evaluated the effect of SFN on actin stress fiber formation in breast cancer cells. The results showed that TGF-β1 induced actin stress fiber formation in MDA-MB-231 cells. However, when the cells were treated with SFN, TGF-β1-induced actin stress fiber formation was almost completely abolished (Fig. 6a, c). Similar phenomena were also observed in MDA-MB-157 cells (Fig. 6b, d).

a, b Representative images showing the effect of SFN, R-SFN, and S-SFN on TGF-β1-induced actin stress fiber formation in MDA-MB-231 (a) and MDA-MB-157 (b) cells. Yellow arrows label the cells with remarkable actin stress fiber formation. Scale bar = 20 μm. c, d Statistical assay showing the effect of SFN on TGF-β1-induced actin stress fiber formation in MDA-MB-231 (c) and MDA-MB-157 (d) cells. Ten fields of view were randomly selected and then the ratio of the cells with actin stress fiber formation in each group was calculated. At least 60 cells in three independent experiments were counted. Data shown are mean ± SEM.**P < 0.05.
Then the expression of actin stress fiber formation-associated proteins was examined. TGF-β1 treatment increased the expression of actin stress fiber formation-associated proteins, including paxillin, IQGAP1, FAK, PAK2, and ROCK. However, SFN treatment decreased the expression of those proteins, indicating that SFN inhibited TGF-β1-induced expression of paxillin, IQGAP1, FAK, PAK2, and ROCK. With the increase of SFN concentration, a more obvious inhibitory effect was observed, suggesting that SFN concentration-dependently inhibits actin stress fiber formation-associated proteins expression (Fig. 7a–d).

a, b Representative western blot results showing that the effects of SFN at different concentrations (7.5, 15, and 30 μM for MDA-MB-231 cells and 3.8, 7.5, and 15 μM for MDA-MB-157 cells) on TGF-β1-induced actin stress fiber formation-associated protein expression, including paxillin, focal adhesion kinase (FAK), Ras GTPase-activating-like protein (IQGAP1), Rho-kinase (ROCK), and p21-activated kinase 2 (PAK2) in MDA-MB-231 (a) and MDA-MB-157 (b) cells. c, d Statistical assay showing that the effects of SFN at different concentrations on TGF-β1-induced actin stress fiber formation-associated proteins expression in MDA-MB-231 (c) and MDA-MB-157 (d) cells. Data shown are mean ± SEM. n = 3, ∗P < 0.05 vs Cont; #P < 0.01 vs TGF-β1.
SFN inhibits the RAF/MEK/ERK signaling pathway by direct binding to RAF kinases
The core network of the PPI map of the altered proteins and phosphoproteins (Fig. 5e) showed ARAF may also be involved in actin cytoskeleton regulation. ARAF is one of the three members of the RAF serine/threonine kinase family, and the other two are BRAF and CRAF. RAF kinases activation can cause the phosphorylation of many proteins, including MEK and ERK phosphorylation. Previous reports demonstrated the RAF/MEK/ERK signaling pathway plays a pivotal role in cancer metastasis44,45. Therefore, we then examined whether SFN could regulate the MEK and ERK phosphorylation. As shown in Fig. 8a–c, SFN concentration-dependently inhibited TGF-β1-induced MEK and ERK phosphorylation, suggesting that SFN inhibited the function of RAF.

a Representative western blot results showing the effects of SFN at different concentrations (7.5, 15, and 30 μM) on TGF-β1-induced MEK and ERK phosphorylation. b, c Statistical assay showing the effects of SFN at different concentrations (7.5, 15, and 30 μM) on MEK and ERK phosphorylation, respectively. Data shown are mean ± SEM. n = 3, ∗P < 0.05 vs Control; #P < 0.05 vs TGF-β1. d–f Molecular docking showing that SFN directly binds to RAF family kinases, including ARAF, BRAF, and CRAF through H-bond. The dotted lines and the corresponding values present the H-bonds and their distances. pERK phosphorylated ERK, tERK total ERK, pMEK phosphorylated MEK, tMEK total MEK.
SFN is a small-molecule compound and is reported to be an inhibitor of histone deacetylase (HDAC)46,47. Recent studies have shown that several small-molecule HDAC inhibitors are also RAF kinase inhibitors48,49. Additionally, inhibition of HDAC6 activity, as well as the EGFR-RAS-RAF-MEK-ERK signaling pathway, may cooperatively reduce cell migration50. Therefore, we hypothesized that SFN may directly act on RAF family proteins to regulate the RAF-MEK-ERK signaling pathway. Then, we examined the molecular docking of SFN and RAF family proteins, including ARAF, BRAF, and CRAF. Our results showed that SFN could interact with ARAF, BRAF, and CRAF through hydrogen bonds. As shown in Fig. 8d, SFN can form two hydrogen bonds with the Thr34 and Lys36 of ARAF. SFN forms a hydrogen bond with BRAF at the position of Ser536 (Fig. 8e). SFN can form three hydrogen bonds with CRAF at the position of Lys399, Tyr340, and Tyr341 (Fig. 8f). These results suggest that SFN targets RAF serine/threonine kinases to inhibit the RAF/MEK/ERK signaling pathway, thereby inhibiting the migration and invasion of breast cancer cells.
Discussion
In this study, we demonstrated that SFN effectively suppressed the metastasis of TNBC cells, including cell migration and invasion. The results of mass spectrometry-based quantitative phosphoproteomic profiling, immunofluorescence, and western blot showed that the inhibitory effect of SFN on breast cancer cell migration and invasion is attributed to the reduction of actin stress fiber formation and its associated protein expression, including paxillin, FAK, IQGAP1, ROCK, and PAK2. Further, we demonstrated that SFN bound to the RAF family proteins, including ARAF, BRAF, and CRAF, subsequently inhibited phosphorylation of MEK and ERK. These in vitro results indicate that SFN suppresses metastasis of breast cancer cells by targeting the RAF/MEK/ERK pathway to inhibit actin stress fiber formation. Our results provide a novel insight into the role of SFN in breast cancer metastasis through the RAF/MEK/ERK signaling pathway.
Many studies have shown that SFN has anticancer properties by affecting different biological functions of tumor cells. SFN acts as an HDAC inhibitor in breast cancer cell lines and decreases the protein expression of ER, EGFR, and HER-251. SFN also induces cell cycle arrest and apoptosis in HT29 human colon cancer cells52. In addition, SFX-01, a stabilized formulation of SFN, has been reported to inhibit endocrine-resistant stem-like cells in ER-positive breast cancer53. A recent clinical study showed that RAF, MEK, pMEK, ERK, and pERK mRNA and protein expressions are higher in the axillary lymph node metastasis (ALNM) group compared to the non-ALNM group and normal group, suggesting that the RAF/MEK/ERK signaling pathway may be a higher correlation with breast cancer metastasis44. Here, we demonstrated that SFN inhibits the metastasis of TNBC cells through targeting the RAF/MEK/ERK signaling pathway to inhibit the formation of actin stress fiber.
The RAF/MEK/ERK signaling pathway plays a crucial role in diverse cellular processes, including cell proliferation, migration, and survival. RAS-RAF interaction is the first step during the process of RAF activation54. The activated RAF phosphorylates and activates MEK kinases, which in turn phosphorylate and activate ERK. Although the activation process of RAF is not completely understood, the interaction between RAS and RAF and RAF phosphorylation are essential for the activation of RAF. The structure of RAF kinases is divided into two functional regions as the regulatory domain and the kinase domain55. Although all RAF kinases have a high degree of sequence similarity, they are under different regulations and have their own functions. This may be the reason why our molecular docking results indicate that SFN binds to all molecules but the binding sites are different. Thr34 of the ARAF kinase is in a RAS binding domain56,57. Molecular docking results showed that the N atom of SFN can form a hydrogen bond with Thr34 of the ARAF kinase, suggesting that SFN may inhibit the activation of ARAF by blocking the binding of RAS and ARAF, thereby inhibiting the RAF/MEK/ERK signaling pathway. For BRAF, as shown in Fig. 8e, SFN forms a hydrogen bond with Ser536 of the BRAF kinase. Ser536 of BRAF is in the kinase domain, which is the MEK binding site56,58. The combination of SFN and BRAF may inhibit RAF to phosphorylate MEK. As shown in Fig. 8a, c, SFN treatment inhibited MEK phosphorylation. However, unlike ARAF and BRAF, CRAF provides three sites of Lys399, Tyr340, and Tyr341 in the kinase domain58 interacting with SFN through three hydrogen bonds. The phosphorylation of RAF plays an important role in the kinase activation and provides a conformation to interact with regulatory molecules such as MEK and 14-3-3 protein58. The phosphorylation of Ser338, Tyr340, and Tyr341 positively regulates the kinase activity of CRAF59. Our recent data showed that SFN inhibits TGF-β1-induced phosphorylation of Ser338 in CRAF (Unpublished data), indicating that SFN can affect the activity of CRAF kinase. In summary, SFN interacts with RAF kinases to inhibit RAF/MEK/ERK signaling pathway. The detailed function of the interaction between SFN and RAF kinases needs further study in the future.
In addition, we also examined the effects of the two isomers of SFN, R-SFN, and S-SFN, on breast cancer metastasis. Research suggests that the R-isomer may be more bioactive than the S-isomer. However, in our current study, R-SFN and S-SFN exhibited equivalent anti-metastasis activity. A principal mechanism of the chemopreventive activity of SFN is modulation of the carcinogen-metabolizing enzyme systems. R-SFN and S-SFN have different effects on the elevation of hepatic glutathione S-transferase and quinone reductase in different tissues. A study by AFA Razis et al. showed that R-SFN is a more effective inducer of the carcinogen-detoxification enzyme system than S-SFN in rat liver and lung60. So far, few studies have compared the effects of R-SFN and S-SFN, and the equivalent inhibitory mechanism of R-SFN and S-SFN on breast cancer metastasis is unclear. Based on previous reports, we speculate that R-SFN and S-SFN may have the same effect on the carcinogen-metabolizing enzyme system in breast cancer cells. As a result, they inhibited the migration and invasion of breast cancer cells to the same extent. P Kiełbasiński et al. synthesized some new enantiomeric fluorine-containing derivatives of SFN and examined the relationship between absolute configuration and biological activity, suggesting that absolute configuration of the fluorine-containing derivatives of SFN is not a key factor determining their activity61. In addition, both isomers are ligands of RAF family kinases and regulate migration and invasion of breast cancer cells through RAF/MEK/ERK signaling pathway. R-SFN and S-SFN showed similar binding affinity (Supplementary Table S3), suggesting that both isomers may exhibit equivalent anti-metastasis activity.
SFN, a natural compound derived from cruciferous vegetables, has attracted more and more attention in the adjuvant treatment and chemoprevention of breast cancer due to its lower toxicity and fewer side effects. In MDA-MB-231 cell lines, the combined treatment of SFN and Withaferin A is more effective than either compound alone62. The combination of SFN and Gemcitabine, or 5-fluorouracil, or doxorubicin showed a synergistic inhibitory effect in breast cancer cell growth63,64,65. These studies indicate that SFN is a promising anticancer regent in adjuvant therapy in breast cancer patients. Although not many clinical studies have investigated the effect of SFN on breast cancer, in vitro and the in vivo studies have been conducted to show that SFN has a potential clinical scenario as a promising antineoplastic and chemopreventive compound. Atwell et al. provided evidence for chemopreventive activity of SFN in human breast tissue from a clinical trial in 54 women. SFN decreased HDAC activity and Ki-67 level, which are malignancy and proliferation markers66. Another study on the preclinical and clinical evaluation of SFN for breast chemoprevention showed that SFN is distributed to breast epithelial cells in vivo and exerts pharmacological effects in these target cells, further supporting the role of SFN for breast chemoprevention67. In the future, clinical trials are needed to clarify the efficacy of SFN and its mechanism in the adjuvant therapy of breast cancer.
In this study, we provided in vitro evidence that SFN inhibits breast cancer cell metastasis by targeting the RAF/MEK/ERK pathway to attenuate actin stress fiber formation (Fig. 9). These results provide some new ideas and insights for exploring more effective treatment strategies for breast cancer metastasis, especially TNBC breast cancer. In addition, our data indicate that R-SFN and S-SFN have equal effects on migration and invasion of breast cancer cells. The current findings raise the possibility that the chemopreventive potential of the S-enantiomer may be underestimated. However, our study is limited to the results of in vitro studies, and the results of in vivo studies are needed to carefully evaluate whether SFN influences the metastasis of breast cancer patients.

The illustrations such as receptors, plasma membrane, and cell migration and invasion were obtained from Smart Servier Medical Art (https://smart.servier.com/).
Methods
Chemicals and reagents
SFN (a synthetic compound containing R- and S- isomers), R-SFN, and S-SFN were purchased from Abcam Laboratories (St. Paul, MN, USA) and resuspended in dimethyl sulfoxide (EMD Millipore, Darmstadt, Germany) to make a 100 mM stock stored at −20 °C. TGF-β1 was purchased from Fujifilm Wako Pure Chemical Corporation (Osaka, Japan) and resuspended in phosphate-buffered saline (PBS) containing 2 mg/mL bovine serum albumin to make a 40 μg/mL stock stored at 20 °C.
The following antibodies were used: anti-paxillin antibody (BD Biosciences, 711725, CA, USA), anti-GAPDH monoclonal antibody (Fujifilm, 014–25524, Osaka, Japan), anti-Rho-kinase II antibody (BD Biosciences, 610624, CA, USA), anti-IQGAP1 antibody (H-109, Santa Cruz, sc-10792, Dallas, USA), anti-FAK antibody (Sigma-Aldrich, 06–543, St. Louis, MO), anti-PAK2 antibody (Cosmo Bio Co., LTD, CSB-PA010537, Tokyo, Japan), anti-phospho-MEK1/2 (Ser217/221) (41G9) rabbit antibody (Cell Signaling, 9154 S, MA, USA), anti-MEK antibody (Cell Signaling, 9122 S, MA, USA), anti-phospho-p44/42 Erk1/2 (Thr202/Tyr204) antibody (Cell Signaling, 9101 S, MA, USA), anti-ERK antibody (Sigma-Aldrich, 06–182, St. Louis, MO). Secondary HRP-labeled antibodies (anti-mouse, W4021 and anti-rabbit, W4011) were purchased from Promega. Alexa Fluor-conjugated secondary antibodies for immunofluorescence imaging (Molecular Probes, Eugene, OR) were used in accordance with the manufacturer’s protocol.
Cell culture
Two human TNBC cell lines were used in this study. MDA-MB-231 (Ds Pharma Biomedical, Osaka, Japan) was cultured in L-15 medium containing glutamine supplemented with 15% FBS and 0.5% NaHCO3 in the presence of 50 U/ml penicillin and 50 mg/ml streptomycin at 37 °C with 95% air and 5% CO2. MDA-MB-157 (ATCC, Rockville, MD) was cultured in L-15 medium containing glutamine supplemented with 10% FBS in the presence of 50 U/ml penicillin and 50 mg/ml streptomycin at 37 °C without CO2. The cells were digested using trypsin/EDTA and planted into the new flask at a ratio between 1:3 and 1:6 to passage. We stocked the cells using Cellbanker 1 (Kurabo, Osaka, Japan) at −80 °C or liquid nitrogen. All cells were used in experiments during the linear phase of growth.
Cell viability assay
A cell counting kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan) was used to measure the cytotoxicity of SFN on MDA-MB-231 cells and MDA-MB-157 cells. Cells were seeded at a density of 1 × 104 cells per well in 100 μL of L-15 medium into a 96-well plate, and cultured overnight. Cells were treated with different concentrations of SFN (0–240 μM) and incubated for 24 h in five parallel wells. At the high concentration of 240 μM, we treated cells with an equal amount of DMSO as control. Next, added 10 µL of CCK-8 solution to each well and incubated for another 2 h. The absorbance was measured at a wavelength of 450 nm using a microplate reader (Bio-Rad Laboratories, Richmond, CA, USA). Cell viability was expressed as a percentage of that of the control (untreated) cells.
Wound healing assay
The cells were grown in 35-mm glass-based dishes (Iwaki, Japan). When cell confluence reached 90–100%, FBS-free L-15 medium (GE Healthcare Bioscience, Piscataway, NJ) supplemented with 0.5% NaHCO3 for MDA-MB-231 cells or without 0.5% NaHCO3 for MDA-MB-157 cells in the presence of 50 U/ml penicillin and 50 mg/ml streptomycin was changed. After starving treatment for 24 h, cells were wounded using 200 μL micropipette tips and then TGF-β1 (20 ng/mL) and SFN were added. Time-lapse recording of cell migration was performed under a fluorescence microscope with a phase-contrast model (KEYENCE, Osaka, Japan) with a chamber at 37 °C and 5% CO2. Time-lapse images were captured at 30 min intervals for 24 h under phase contrast with 10× magnification of microscope objective. The data were used for quantitative analyses of cell migration using NIH Image J software. We calculated the migration rate as follows: The migration rate (%) = (scratch width at 0 h − scratch width at 24 h)/scratch width at 0 h× 100%.
Transwell invasion assay
The invasion of MDA-MB-231 cells or MDA-MB-157 cells was determined using a corning invasion chamber 24-well plate (354480, Corning BioCoat, USA). The membrane was coated with extracellular matrigel matrix (BD Biosciences, CA, USA) at 37 °C for 30 min. Then serum-free cell suspension (1 × 106/well) was placed into the upper chamber. The lower compartment was filled with L-15 medium adding with TGF-β1 (20 ng/mL) in the absence or presence of SFN, R-SFN, and S-SFN. After being cultured for 24 h, uninvaded cells were removed. The invading cells on the underside of the membrane were fixed with formaldehyde (4%) and stained with crystal violet (0.1%). Cells were visualized under a bright-field microscope (KEYENCE, Osaka, Japan).
Digestion, stable isotope dimethyl labeling, and phosphopeptide enrichment
The protein extracts were precipitated with five volumes of ice-cold acetone/ethanol/acetic acid (v/v/v = 50/50/0.1) at −20 °C overnight. The precipitated protein sample was centrifuged at 15,000 g for 30 min and washed with acetone and 75% ethanol. The protein sample was re-dissolved into 8 M urea/50 mM HEPES (pH 8.0) solution and the protein concentration was determined by BCA assay. Then, the protein was reduced by 10 mM dithiothreitol (DTT) at 60 °C for 1 h and alkylated by 20 mM Iodoacetamide (IAA) in the darkness at room temperature for 30 min. Then the protein concentration was diluted to 1 mg/mL with 100 mM HEPES buffer, and digested by trypsin (enzyme/substrate, 1/25 (w/w)) overnight at 37 °C. Finally, the tryptic digests were isotopically labeled with dual mass-differentiate dimethyl groups as shown in Fig. 4a. The phosphopeptides were enriched by Ti4+-IMAC microspheres.
LC-MS/MS analysis and database searching
All samples were analyzed by Orbitrap Fusion™ Tribrid™ mass spectrometer coupled with an UltiMateTM 3000 RSLCnano System (Thermo Fisher, San Jose, CA). The samples were loaded on a C18 capillary trap column (200 μm i.d. × 4 cm) packed with C18 AQ beads (5 μm, 120 Å). 15 cm × 75 µm i.d. analytical column packed with C18 AQ beads (3 μm, 120 Å) was used for LC separation. The buffers used for online separation were 0.1% (v/v) formic acid in water and 0.1% (v/v) formic acid in acetonitrile (ACN), the flow rate was 200 nL/min for the nanoflow LC-MS/MS analysis. The gradient from 5 to 35% (v/v) ACN was performed in 90 min. The MS and MS/MS spectra were collected by high collision-induced dissociation (HCD) at 35% energy in a data-dependent mode that 1 MS scan from 400 to 2000 m/z with the mass resolution of 60,000 at m/z 400 in the Orbitrap analyzer followed by 40 MS/MS scans in the ion trap, respectively. The dynamic exclusion repeat count was 1 with a duration time of 30 s, and the charge state rejection function was enabled to reject the ions with single and “unassigned” charge states.
The obtained raw files were searched using MaxQuant (Version 1.5.5.1) with a Uniprot protein FASTA database of humans. Trypsin was selected as the specific enzyme with up to 2 miss cleavages. The oxidation of methionine (M) was set as variable modifications. Meanwhile, the carbamidomethylation of cysteine (C) was set as fixed modification. The false discovery rate (FDR) for both peptide and protein identification were set below 1%. The results were then imported into the MaxQuant associated software suite Perseus for further analysis.
Network analysis and KEGG enrichment of differentially expressed proteins
Digestion, stable isotope dimethyl labeling, phosphopeptide enrichment, and LC-MS/MS analysis are shown in the supplementary materials. Protein–protein interactions (PPI) of the differentially expressed proteins and phosphoproteins were predicted by STRING (https://string-db.org/) database with a filter of combined score >0.7 which was considered as high confidence68,69, and then visualized using Cytoscape v3.8.2. Topology parameters of the PPI network were calculated by NetworkAnalyzer. The core network was extracted according to the degree value (greater than the median degree value for all nodes in the network)70, followed by the cluster analysis with ClusterONE and KEGG pathways enrichment with String database69.
Actin stress fiber staining
Actin stress fiber staining were performed as described previously71. Briefly, breast cancer cells were cultured on 0.3% gelatin-coated glass coverslips. After treatment with TGF-β1 (20 ng/mL) for 24 h with or without SFN, cells were fixed in 2% paraformaldehyde in PBS for 10 min, permeabilized with 0.1% Triton X-100 in PBS for 2 min, and blocked in NanoBio blocker solution (Nano Bio-Tech Co., Ltd) diluted in PBS for 60 min at 25 °C. Cells were then incubated with rhodamine-phalloidin (1:100; Molecular Probes, Eugene, OR) and DAPI (Molecular Probes, Eugene, OR) for 60 min at 25 °C. Coverslips were washed with PBS, rinsed with deionized water, and mounted with PermaFluor aqueous mounting medium (Thermo Fisher Scientific, Waltham, MA). Specimens were observed using fluorescence microscopy (KEYENCE, Osaka, Japan) at 60× magnification of microscope objective. F-actin with a thick linear structure was observed in the whole cells, which means the actin stress fiber formation. Actin stress fiber formation was quantified by counting the number of the cells that contain a thick linear structure of actin stress fibers in the cell cortex or not within cells in at least ten random independent fields (n > 60) under a fluorescence microscope12,72.
Western blotting analysis
MDA-MB-231 or MDA-MB-157 cells were serum-starved for 24 h, then stimulated with TGF-β1 (20 ng/mL) and in the presence of SFN or vehicle control for 24 h. Cell lysates were prepared in RIPA cell lysis buffer (Wako, Osaka, Japan) supplemented with protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO), 0.2 mM Na3VO4, 10 mM NaF, 10 μM leupeptin, and 1 mg/ml aprotinin. The lysates were centrifuged, and the supernatants were boiled in sodium dodecyl sulfate (SDS) loading buffer. Proteins in the total cell lysate (10 μg) were separated using 10% SDS-polyacrylamide gel electrophoresis, transferred to Amersham Hybond-P PVDF membrane (GE Healthcare Life Sciences), and blocked in 5% nonfat milk in 0.05% Tween-20 phosphate-buffered saline (TBS-T) for 60 min at room temperature. The membranes were incubated with primary antibodies overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit antibodies, 1:5,000) for 1 h at room temperature. Immunoreactive bands were detected using a Supersignal West Pico Chemiluminescent Substrate kit (Thermo Fisher Scientific, Waltham, MA). GAPDH was used as a loading control. All blots were processed in parallel and derived from the same experiment (Uncropped blots are shown in Supplementary Fig. S1).
The following concentrations of primary antibodies were used: anti-paxillin antibody, 1:5,000; anti-IQGAP1 antibody, 1:1,000; anti-ROCK antibody, 1:500; anti-FAK antibody, 1:1,000; anti-PAK2 antibody, 1:1,000; anti-GAPDH antibody, 1:10,000; anti-phospho-ERK, 1:1,000; anti-ERK antibody, 1:1,000; anti-phospho-MEK antibody, 1:1,000; anti-MEK antibody, 1:1,000.
Molecular docking
The binding ability, sites, and interactions between SFN and RAF serine/threonine kinases were achieved and analyzed by classical molecular dynamics using DockThor database73 and visualized using Pymol 2.4. The 3D chemical structural formula of SFN was obtained from PubChem (http://pubchem.ncbi.nlm.nih.gov) and the crystal structures of putative RAF serine/threonine kinases were obtained from the Protein Data Bank (http: //www.rcsb.org). PDB code of ARAF: 1wxm; PDB code of BRAF: 6p3d; PDB code of CRAF: 3omv.
ProteomeXchange accession
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE74 partner repository with the dataset identifier PXD025639.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0.2. All experiments were performed on at least three independent occasions. Data shown represent mean ± SEM. The student’s t-test was used to evaluate the differences between the control group and the experimental group, and a one-way analysis of variance followed by Tukey’s test was used to evaluate the differences among multiple groups. P < 0.05 was considered statistically significant.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
Detailed data analysis can be found in the methods section. All relevant data are deposited into a public repository and accession codes are provided.
References
Huang, Z. et al. Breast cancer incidence and mortality: trends over 40 years among women in Shanghai, China. Ann. Oncol. 27, 1129–1134 (2016).
Saika, K. & Sobue, T. Epidemiology of breast cancer in Japan and the US. Jpn. Med. Assoc. J. 52, 39–44 (2009).
Momenimovahed, Z. & Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer. 11, 151–164 (2019).
Anders, C. K., Johnson, R., Litton, J., Phillips, M. & Bleyer, A. Breast cancer before age 40 years. Semin. Oncol. 36, 237–249 (2009).
Li, X. & Wang, J. Mechanical tumor microenvironment and transduction: Cytoskeleton mediates cancer cell invasion and metastasis. Int. J. Biol. Sci. 16, 2014–2028 (2020).
Ong, M. S. et al. Cytoskeletal proteins in cancer and intracellular stress: a therapeutic perspective. Cancers 12, 1–24 (2020).
Vallenius, T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 3, 130001 (2013).
Peng, J. M. et al. Actin cytoskeleton remodeling drives epithelial-mesenchymal transition for hepatoma invasion and metastasis in mice. Hepatology 67, 2226–2243 (2018).
Tavares, S. et al. Actin stress fiber organization promotes cell stiffening and proliferation of pre-invasive breast cancer cells. Nat. Commun. 8, 15237 (2017).
Hall, A. Rho GTpases and the actin cytoskeleton. Science 279, 509–514 (1998).
Ruiz-Loredo, A. Y., López, E. & López-Colomé, A. M. Thrombin promotes actin stress fiber formation in RPE through Rho/ROCK-mediated MLC phosphorylation. J. Cell. Physiol. 226, 414–423 (2011).
Xu, D. et al. Sphingosylphosphorylcholine induces stress fiber formation via activation of Fyn-RhoA-ROCK signaling pathway in fibroblasts. Cell. Signal. 24, 282–289 (2012).
Katoh, K., Kano, Y. & Ookawara, S. Rho-kinase dependent organization of stress fibers and focal adhesions in cultured fibroblasts. Genes Cells 12, 623–638 (2007).
Pellegrin, S. & Mellor, H. Actin stress fibers. J. Cell Sci. 120, 3491–3499 (2007).
Deakin, N. O. & Turner, C. E. Paxillin comes of age. J. Cell Sci. 121, 2435–2444 (2008).
Hu, Y. L. & Chien, S. Dynamic motion of paxillin on actin filaments in living endothelial cells. Biochem. Biophys. Res. Commun. 357, 871–876 (2007).
Schober, M. et al. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J. Cell Biol. 176, 667–680 (2007).
Fabry, B., Klemm, A. H., Kienle, S., Schäffer, T. E. & Goldmann, W. H. Focal adhesion kinase stabilizes the cytoskeleton. Biophys. J. 101, 2131–2138 (2011).
Watanabe, T., Wang, S. & Kaibuchi, K. IQGAPS as key regulators of actin-cytoskeleton dynamics. Cell Struct. Funct. 40, 69–77 (2015).
Kim, H., White, C. D. & Sacks, D. B. IQGAP1 in microbial pathogenesis: targeting the actin cytoskeleton. FEBS Lett. 585, 723–729 (2011).
Chang, F. et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 17, 1263–1293 (2003).
Roberts, P. J. & Der, C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 (2007).
Wen, Z. et al. Inhibition of lung cancer cells and Ras/Raf/MEK/ERK signal transduction by ectonucleoside triphosphate phosphohydrolase-7 (ENTPD7). Respir. Res. 20, 194 (2019).
de Luca, A., Maiello, M. R., D’Alessio, A., Pergameno, M. & Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 16, S17–S27 (2012).
Yang, S. & Liu, G. Targeting the RAS/RAF/MEK/ERK pathway in hepatocellular carcinoma. Oncol. Lett. 13, 1041–1047 (2017).
Li, L. et al. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC (Review). Oncol. Lett. 12, 3045–3050 (2016).
Mooz, J. et al. Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci. Signal. 7, ra73 (2014).
Martin, S. L., Kala, R. & Tollefsbol, T. O. Mechanisms for the inhibition of colon cancer cells by sulforaphane through epigenetic modulation of microRNA-21 and human telomerase reverse transcriptase (hTERT) down-regulation. Curr. Cancer Drug Targets 18, 97–106 (2017).
Traka, M. H., Melchini, A. & Mithen, R. F. Sulforaphane and prostate cancer interception. Drug Discov. Today 19, 1488–1492 (2014).
Zheng, Z. et al. Sulforaphane metabolites inhibit migration and invasion via microtubule-mediated Claudins dysfunction or inhibition of autolysosome formation in human non-small cell lung cancer cells. Cell Death Dis. 10, 259 (2019).
Geng, Y. et al. Sulforaphane induced apoptosis via promotion of mitochondrial fusion and ERK1/2-mediated 26s proteasome degradation of novel pro-survival bim and upregulation of bax in human non-small cell lung cancer cells. J. Cancer 8, 2456–2470 (2017).
Lin, K. et al. Sulforaphane-cysteine-induced apoptosis via phosphorylated ERK1/2-mediated maspin pathway in human non-small cell lung cancer cells. Cell Death Discov. 3, 17025 (2017).
Lewinska, A., Adamczyk-Grochala, J., Deregowska, A. & Wnuk, M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics 7, 3461–3477 (2017).
Suppipat, K., Park, C. S., Shen, Y., Zhu, X. & Lacorazza, H. D. Sulforaphane induces cell cycle arrest and apoptosis in acute lymphoblastic leukemia cells. PLoS ONE 7, e51251 (2012).
Shankar, S., Ganapathy, S. & Srivastava, R. K. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin. Cancer Res. 14, 6855–6866 (2008).
Liu, P. et al. Sulforaphane exerts anti-angiogenesis effects against hepatocellular carcinoma through inhibition of STAT3/HIF-1α/VEGF signalling. Sci. Rep. 7, 12651 (2017).
Pang, M. F. et al. TGF-β1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 35, 748–760 (2016).
Ma, M. et al. Mir-487a promotes TGF-β1-induced EMT, the migration and invasion of breast cancer cells by directly targeting MAGI2. Int. J. Biol. Sci. 12, 397–408 (2016).
Xie, F., Ling, L., van Dam, H., Zhou, F. & Zhang, L. TGF-β signaling in cancer metastasis. Acta Biochim. Biophys. Sin. 50, 121–132 (2018).
Padua, D. & Massagué, J. Roles of TGFβ in metastasis. Cell Res. 19, 89–102 (2009).
Kuran, D., Pogorzelska, A. & Wiktorska, K. Breast cancer prevention-is there a future for sulforaphane and its analogs? Nutrients 12, 1559 (2020).
Innocenti, M. New insights into the formation and the function of lamellipodia and ruffles in mesenchymal cell migration. Cell Adhes. Migr. 12, 401–416 (2018).
Yamaguchi, H. & Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 642–652, 2007 (1773).
Shao, G. L. et al. Correlation between Raf/MEK/ERKs signaling pathway and clinicopathological features and prognosis for patients with breast cancer having axillary lymph node metastasis. Technol. Cancer Res. Treat. 17, 1533034617754024 (2018).
Lee, S., Rauch, J. & Kolch, W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 21, 1102 (2020).
Rajendran, P. et al. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 10, 68 (2011).
Myzak, M. C., Karplus, P. A., Chung, F.-L. & Dashwood, R. H. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 64, 5767–5774 (2004).
Geng, A. et al. Discovery of novel phenoxybenzamide analogues as Raf/HDAC dual inhibitors. Bioorg. Med. Chem. Lett. 29, 1605–1608 (2019).
Chen, X. et al. Design, synthesis and biological evaluation of novel benzoylimidazole derivatives as Raf and histone deacetylases dual inhibitors. Chem. Pharm. Bull. 67, 1116–1122 (2019).
Williams, K. A. et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 288, 33156–33170 (2013).
Pledgie-Tracy, A., Sobolewski, M. D. & Davidson, N. E. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 6, 1013–1021 (2007).
Gamet-Payrastre, L. et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 60, 1426–1433 (2000).
Simões, B. M. et al. Targeting STAT3 signaling using stabilised sulforaphane (SFX-01) inhibits endocrine resistant stem-like cells in ER-positive breast cancer. Oncogene 39, 4896–4908 (2020).
Tran, T. H. et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 12, 1176 (2021).
Matallanas, D. et al. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2, 232–260 (2011).
Roskoski, R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 135, 239–258 (2018).
Roskoski, R. RAF protein-serine/threonine kinases: structure and regulation. Biochem. Biophys. Res. Commun. 399, 313–317 (2010).
Fischer, A. et al. Regulation of RAF activity by 14-3-3 proteins: RAF kinases associate functionally with both homo- and heterodimeric forms of 14-3-3 proteins. J. Biol. Chem. 284, 3183–3194 (2009).
Xiang, X., Zang, M., Waelde, C. A., Wen, R. & Luo, Z. Phosphorylation of 338SSYY341 regulates specific interaction between Raf-1 and MEK1. J. Biol. Chem. 277, 44996–45003 (2002).
Abdull Razis, A. F., Iori, R. & Ioannides, C. The natural chemopreventive phytochemical R-sulforaphane is a far more potent inducer of the carcinogen-detoxifying enzyme systems in rat liver and lung than the S-isomer. Int. J. Cancer 128, 2775–2782 (2011).
Kiełbasiński, P. et al. New enantiomeric fluorine-containing derivatives of sulforaphane: synthesis, absolute configurations and biological activity. Eur. J. Med. Chem. 76, 332–342 (2014).
Royston, K. J., Udayakumar, N., Lewis, K. & Tollefsbol, T. O. A novel combination of withaferin A and sulforaphane inhibits epigenetic machinery, cellular viability and induces apoptosis of breast cancer cells. Int. J. Mol. Sci. 18, 1092 (2017).
Milczarek, M. et al. Autophagic cell death and premature senescence: New mechanism of 5-fluorouracil and sulforaphane synergistic anticancer effect in MDA-MB-231 triple negative breast cancer cell line. Food Chem. Toxicol. 111, 1–8 (2018).
Mielczarek, L. et al. In the triple-negative breast cancer MDA-MB-231 cell line, sulforaphane enhances the intracellular accumulation and anticancer action of doxorubicin encapsulated in liposomes. Int. J. Pharm. 558, 311–318 (2019).
Hussain, A. et al. Sulforaphane inhibits growth of human breast cancer cells and augments the therapeutic index of the chemotherapeutic drug, gemcitabine. Asian Pac. J. Cancer Prev. 14, 5855–5860 (2013).
Atwell, L. L. et al. Sulforaphane bioavailability and chemopreventive activity in women scheduled for breast biopsy. Cancer Prev. Res. 8, 1184–1191 (2015).
Cornblatt, B. S. et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 28, 1485–1490 (2007).
Anitha, P., Anbarasu, A. & Ramaiah, S. Gene network analysis reveals the association of important functional partners involved in antibiotic resistance: a report on an important pathogenic bacterium Staphylococcus aureus. Gene 575, 253–263 (2016).
Jensen, L. J. et al. STRING 8 - A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416 (2009).
Wu, L. et al. Mechanism of action of Bu-fei-yi-shen formula in treating chronic obstructive pulmonary disease based on network pharmacology analysis and molecular docking validation. BioMed. Res. Int. 2020, 9105972 (2020).
Zhang, Y., Zhang, M., Lyu, B., Kishi, H. & Kobayashi, S. Omega-3 and omega-6 DPA equally inhibit the sphingosylphosphorylcholine-induced Ca2+-sensitization of vascular smooth muscle contraction via inhibiting Rho-kinase activation and translocation. Sci. Rep. 7, 36368 (2017).
Zhang, Y., Kishi, H., Morita, T. & Kobayashi, S. Paxillin controls actin stress fiber formation and migration of vascular smooth muscle cells by directly binding to the active Fyn. FASEB J. 35, e22012 (2021).
Santos, K. B., Guedes, I. A., Karl, A. L. M. & Dardenne, L. E. Highly flexible ligand docking: benchmarking of the DockThor Program on the LEADS-PEP protein-peptide data set. J. Chem. Inf. Modeling 60, 667–683 (2020).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Acknowledgements
We would like to thank Dr. Hiroko Kishi for providing the cell culture incubator. This work was partly supported by Grants-in-Aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grant Numbers 25893150, 15K18970, and 18K06875 to Y.Z., and 20K21845 and 21H02718 to T.M.) and National Natural Science Foundation of China (Grant Number 81702995 to J.L.).
Author information
Authors and Affiliations
Contributions
Y.Z. designed and performed the experiments, analysed data, funding acquisition, and wrote the manuscript; Q.L. and N.L. performed the experiments and generated the data; M.X. contributed to the writing of the manuscript; T.M. assisted with methodology and writing; J.L. performed the experiments, analysed data, and contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Lu, Q., Li, N. et al. Sulforaphane suppresses metastasis of triple-negative breast cancer cells by targeting the RAF/MEK/ERK pathway. npj Breast Cancer 8, 40 (2022). https://doi.org/10.1038/s41523-022-00402-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-022-00402-4
This article is cited by
-
An Insight on Synergistic Anti-cancer Efficacy of Biochanin A and Sulforaphane Combination Against Breast Cancer
Applied Biochemistry and Biotechnology (2024)
