
Abstract
Germline genetic variation has been suggested to influence the survival of breast cancer patients independently of tumor pathology. We have studied survival associations of genetic variants in two etiologically unique groups of breast cancer patients, the carriers of germline pathogenic variants in BRCA1 or BRCA2 genes. We found that rs57025206 was significantly associated with the overall survival, predicting higher mortality of BRCA1 carrier patients with estrogen receptor-negative breast cancer, with a hazard ratio 4.37 (95% confidence interval 3.03–6.30, P = 3.1 × 10−9). Multivariable analysis adjusted for tumor characteristics suggested that rs57025206 was an independent survival marker. In addition, our exploratory analyses suggest that the associations between genetic variants and breast cancer patient survival may depend on tumor biological subgroup and clinical patient characteristics.
Similar content being viewed by others


Introduction
Breast cancer is globally the leading cause of cancer-related mortality for women1. The average 5-year survival rate is 83–90% in the Western countries, substantially better than for many other cancers, but due to its high incidence, breast cancer still leads the statistics for cancer mortality in Europe and comes second in North America1,2. On an individual level, prognosis varies greatly, depending on both the inherent tumor biology and the stage of malignant progression at diagnosis. Women with early-stage, localized disease have a very good 5-year prognosis of ~97–99%, but for 10–15% of women, diagnosed with locally advanced disease, the expected 5- and 10-year survival rates range between 40–80% and 30–40%, respectively. Furthermore, the mortality associated with metastatic breast cancer is even higher, with median survival of <3 years2,3,4.
Currently, the prognosis of breast cancer patients is based on the tumor characteristics. Gene expression and copy number profiling5,6,7 or expression of marker proteins, like estrogen receptor (ER), progesterone receptor (PgR), human epidermal growth factor receptor 2, and marker of proliferation Ki-67, can be used to categorize breast cancers into biological subtypes with specific treatment recommendations and different estimates for patient survival8,9. Tumor grade is a histological measure of cellular growth pattern and the best individual prognostic factor10. The phase of tumor progression is approximated by clinical stage, which is based on the tumor size, as well as local and systemic spread of metastatic cells11. However, there is great variation in the survival rates between tumors with similar characteristics and stage. This variance has been suggested to have a heritable component, possibly consisting of genetic differences in metastatic potential and sensitivity to adjuvant therapy12,13,14,15,16,17,18. In addition, host factors, like tumor–microenvironment interaction, immune surveillance, and efficiency in drug metabolism may account for the genetic variability in breast cancer patient survival19,20,21.
Genetic determinants of patient prognosis and the treatment outcome prediction have been intensively sought using both candidate gene and genome-wide approaches, but only some of the discoveries have been successfully validated in subsequent studies. A meta-analysis of ten genome-wide association studies nominally validated 12 out of 45 earlier discoveries for survival from ER-positive or any breast cancer22. Most recently, the Breast Cancer Association Consortium (BCAC) reported two loci from chromosome 7, based on a well-powered meta-analysis of genome-wide association studies23.
In this study, we focused on the survival of women who carry germline pathogenic BRCA1 or BRCA2 variants. BRCA1 and BRCA2 are the two most important breast cancer susceptibility genes, with ~72 and 69% life-time risk of breast cancer and 44 and 17% risk of ovarian cancer, respectively24. BRCA1/2-deficient tumors have distinctive genomic aberration profiles, characterized by homologous recombination deficiency25, making them stand out as etiologically and phenotypically coherent groups of breast carcinomas. BRCA1 risk variants are associated with high grade and triple-negative breast cancer, which both predict poor outcome. BRCA2 risk variants predispose primarily to ER-positive breast cancer, but the risk of ER-negative breast cancer increases with age26,27. Meta-analyses on the survival of women with BRCA1/2 variants and breast cancer have suggested no significant difference in comparison to noncarriers with phenotypically similar tumors28,29,30. However, a recent study suggested ER-positive breast cancer as an adverse indicator for cases with BRCA2 variants, unlike for noncarriers27.
Results
Tumor characteristics
We investigated genetic survival associations in pathogenic variant carriers from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA), genotyped on the OncoArray31,32. Twenty-one independent studies participating in CIMBA had survival data available for germline carriers of pathogenic BRCA1 variants (n = 3008) and 15 studies for carriers of BRCA2 variants (n = 2,009; Supplementary Table 1). Primarily, we analyzed patient survival in relation to all-cause death, because this was most complete across the participating studies. As a sensitivity analysis, the discovered survival variants were also always assessed for breast cancer-specific death.
Data on tumor characteristics was available for about two thirds of patients (Table 1). The distribution of the tumor characteristics of BRCA1 and BRCA2 carriers agreed with previous reports, so that small, high-grade, and early-onset tumors had a relatively high frequency26,33. Tumors from BRCA1 carriers were mostly ER-negative, while those from BRCA2 carriers were largely ER-positive. Therefore, the main survival analyses were performed in parallel in the following patient groups: all BRCA1 carriers, BRCA1 carriers with ER-negative tumors, all BRCA2 carriers, and BRCA2 carriers with ER-positive tumors.
rs57025206 predicts survival of BRCA1 carriers with ER-negative breast cancer
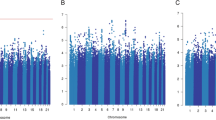
We analyzed the association of germline genetic variants with the overall survival after the first primary breast cancer diagnosis in carriers of pathogenic BRCA1 or BRCA2 variants, using Cox regression. The analyses were adjusted for age at breast cancer diagnosis and stratified by country group to account for underlying genetic and clinical differences between populations. A single variant, rs57025206 (minor allele frequency in European population, MAFEUR, 0.027; info score 0.97 for imputed variant), a nine base pair insertion in an intergenic region of 3p21.2, was identified as a highly significant survival marker for ER-negative breast cancer patients carrying BRCA1 pathogenic variants, with a hazard ratio (HR) of 4.37 (95% confidence interval (CI) 3.03–6.30, P = 3.1 × 10−9, Fig. 1). Furthermore, a multivariable analysis adjusted for tumor grade, size, PgR expression status, and lymph node involvement, suggested that the association is independent of these tumor characteristics, with rs57025206-associated HR: 6.19, 95% CI: 3.73–10.3. A similar trend was observed in the analysis of breast cancer-specific death, although the number of patients with available data was much smaller (Supplementary Table 2).

a Kaplan–Meier plot on the survival of ER-negative breast cancer patients carrying pathogenic BRCA1 variants, stratified by rs57025206. b Forest plot of HR associated with rs57025206 across country groups. (P.het: P-value against between-study heterogeneity). c Kaplan–Meier plot on the survival of ER-positive breast cancer patients carrying pathogenic BRCA1 variants, stratified by rs57025206.
The survival association was specific to women with ER-negative breast cancer, since in the analysis of all BRCA1 carriers the HR was attenuated (HR: 2.12, 95% CI: 1.55–2.91, Supplementary Fig. 1) and not seen in those with ER-positive breast cancer (n: 476, HR: 0.52, 95% CI: 0.17–1.60, Fig. 1c). In the analysis of BRCA2 carriers, rs57025206 was not associated with patient survival (HR: 0.94, 95% CI: 0.59–1.51).
The main analyses suggested nine further survival loci
We used a looser discovery threshold P-value, P < 5 × 10−7, than in the traditional genome-wide setting to characterize loci that potentially modify the patient survival and may form a basis for future research hypotheses. In the analysis of the overall survival of BRCA1 carriers, six variants exceeded the selected threshold (Table 2 and Supplementary Fig. 2). The risk magnitude was proportionally associated with the number of risk alleles for two variants, rs59010985 and rs537497819, whereas the other four variants followed a dominant inheritance pattern. The analysis of BRCA1 carriers with ER-negative breast cancer identified four further potential variants influencing survival (Table 2 includes also rs57025206, presented above). No variant exceeded the significance threshold in the analysis of BRCA2 carriers, alone. However, a meta-analysis of BRCA1 and BRCA2 carriers (see below) suggested germline variants associated with survival in both of these groups.
The reliability of the findings was estimated with a Bayesian false discovery probability (BFDP) and by assessing the between-strata (country group) heterogeneity. Ten out of the 11 BRCA1-associated survival variants had BFDP ≤ 0.33 for at least one of the three tested prior maximum effect sizes (Table 2). For these ten variants, there was little heterogeneity between countries (Supplementary Fig. 3, P against heterogeneity >0.1 for all variants). Moreover, the variant effect sizes were consistent in multivariable models adjusted for tumor pathologic characteristics and in the analyses of breast cancer-associated death (Supplementary Table 2), suggesting a consistent effect throughout the data.
Most of the identified survival variants complied with the proportional hazards assumption, suggesting a constant HR over time, but rs537497819 was especially associated with poor short-term prognosis, the effect leveling out after the first 5 years following the diagnosis (Table 2).
Meta-analysis suggested four variants with the consistent survival effect in BRCA1 and BRCA2 carriers
The correlation between variant effect sizes in the BRCA1 and BRCA2 survival analyses was quite low (Pearson’s R = 0.0065 [95% CI 0.0059–0.0072]), suggesting no overall trend for similar genetic survival effects in these patient groups. Nevertheless, a fixed-effects meta-analysis highlighted four candidate variants, which had consistent effect on patient survival in both groups (Table 3, Supplementary Table 3 and Supplementary Fig. 4).
Four variants may have age-dependent survival effects for BRCA2 carriers
The lack of positive findings in the BRCA2-specific analyses prompted us to consider the possibility that there may be confounding factors, so that the genetic survival associations would depend on tumor and patient characteristics. We had tumor pathology data available only for a subgroup of the study subjects, and lacked statistical power to investigate the interaction between genetic variants and tumor characteristics. However, the age at diagnosis was available for all cases, and to investigate potential age-dependent genetic survival effects, we included in the Cox regression model a covariate coded as the product of the variant genotype and diagnosis age (continuous). The model containing the interaction term was tested against a nested model without interaction. Likelihood-ratio test P-values (Table 4) were corrected for multiple testing using the Benjamini–Hochberg method and variants with false discovery rate (FDR) ≤ 0.33 were considered as potential discoveries. Four variants, all from the analysis of BRCA2 carriers, passed the threshold. For illustrative purposes, the HRs in Table 4 are presented separately for two age groups, even though the regression model suggested that the interaction between the variant survival effect and the diagnosis age is continuous (Supplementary Fig. 5). The variant effect sizes were consistent in multivariable models, including tumor pathologic characteristics, as well as in the analysis of breast cancer-specific death (Supplementary Table 4).
ER-negative breast cancer is more common for postmenopausal than premenopausal BRCA2 carriers26,33. To test whether the age-dependent survival association of the four variants (Table 4), was a hidden association with the tumor subtype, we analyzed the survival separately in age- and subtype-stratified subgroups, including only patients with data on ER expression available (Table 1). This analysis suggested that the ER status did not explain the age-dependent survival association. For two variants (rs2109815 and rs35431863), the age-dependent survival association was similar in ER-positive and ER-negative patient groups. For the other two variants (rs372812554 and rs11255420), the survival association did not vary by age of diagnosis in the ER-negative patient group, and the group resembled the younger ER-positive patient group (Supplementary Table 5).
The positional and eQTL analyses reveal potential target genes
None of the variants we found associated with survival was located on a protein-coding region. Therefore, we took two parallel approaches, positional and gene expression based, to identify potential target genes. A target gene was considered to have a strong positional evidence, if the survival variant or any regulatory variant in linkage disequilibrium was located on an experimentally validated enhancer of the target gene in a relevant tissue (mammary, ovarian, blood, and adipose)34, based on the 1000 genomes and Encode data35,36. This is indicated in the Table 5, column “Positional target tissue”. If no other evidence was available, the target gene was assumed the closest gene (no “Positional target tissue” mentioned in Table 5). For the gene expression-based approach, we used eQTL data from public databases covering various human tissues and cell types (Supplementary Data 1 and 2).
rs57025206 and its proxy variants in high linkage disequilibrium (R2 > 0.8 in the European population, green track named “lead variant and proxies with R2 > 0.8” in Fig. 2) are co-located with a cluster of IQCF-genes (IQCF1–6), expressed exclusively in the testis. These variants have eQTL associations with altogether five genes in different tissues, for example, with MIR135A in adipose tissue. However, the region does not contain annotated regulatory elements. Furthermore, it is not in physical contact with any transcription start site (TSS) in human mammary epithelial cells (HMEC), suggesting that this region does not contain any HMEC-specific enhancer (see, for example, position “g” in the heatmap of Fig. 2). Furthermore, the MAFs of these variants range from 2.2 to 3.2% in the European population, but between 12 and 27% globally, questioning whether the causal variant would be rs57025206 or any of the proxies with R2 > 0.8.

The heatmap visualizes the strength of association between two genomic loci in chromosome conformation capture data from human mammary epithelial cells. The graph has been created and the topologically associated domains (TAD) estimated with 3D genome browser, promoter.bx.psu.edu/hi-c/. The UCSC browser tracks visualize genomic annotation for the region. The three custom tracks indicate the positions of variants in linkage disequilibrium with rs57025206. The vertical dashed lines indicate the positions of the lead variant rs57025206 and three linked regulatory variants. UCSC genes track visualizes the positions of exons and introns for all genes on the region. The GeneHancer track displays the interactions of distant enhancers and gene transcription start sites (TSSs). The histone mark track shows the positions of H3K27Ac from the Encode project. In the heatmap, seven positions (a–e) have been marked in the heatmap to indicate the probability of physical interaction between variants linked with rs57025206 and their target genes based on positional and eQTL analysis (Supplementary Data 1 and 2) as follows: (a) RAD54L2 TSS and rs72945708 on enhancer GH03J051383 targeting RAD54L2. (b) GRM2 TSS and GRM2 eQTL variant rs72945708 on enhancer GH03J051383. (c) DCAF1 TSS and rs9824779 on enhancer GH03J051668 targeting DCAF1. (d) DCAF1 TSS and rs56942057 on enhancer GH03J051684 targeting DCAF1. (e) BAP1 TSS and BAP1 eQTL variant rs9824779 on enhancer GH03J051668. (f) BAP1 TSS and BAP1 eQTL variant rs60497133. (g) MIR135A TSS and MIR135A eQTL variants rs1476290 and rs1605067.
The rs57025206 haploblock (with D′ > 0.8) covers a larger genomic region with functional variants located on enhancers regulating several protein-coding genes (turquoise and green tracks in Fig. 2, Table 5 and Supplementary Data 1). For example, rs56942057 (MAFEUR 0.1%, MAFGLOBAL 4.6%, D′EUR 1.0, and D′GLOBAL 0.34) and rs9824779 (MAFEUR 0.1%, MAFGLOBAL 3.9%, D′EUR 1.0, and D′GLOBAL 0.32) are located in enhancers of DCAF1, GH03J051684, and GH03J051668, respectively. Both variants affect conserved sequences of transcription factor binding motifs. Furthermore, the variant positions and DCAF1 TSS are in physical contact in HMEC (positions “c” and “d” in Fig. 2). These two (rs56942057 and rs9824779) and rs60497133 were eQTL variants for BAP1 in thyroid (Supplementary Data 2), but in HMEC there was no contact between the variant positions and BAP1 TSS (Fig. 2, positions “e” and “f”), suggesting that the regulatory associations of these variants and BAP1 may not exists in mammary epithelial cells. A third potentially causal variant, rs72945708 (MAFEUR 2.0%, MAFGLOBAL 18%, D′EUR 0.95, and D′GLOBAL 0.71), is located on a RAD54L2 enhancer GH03J051383 and associated with the expression of GRM2 in adipose tissue. However, this variant does not change conserved sequence of any known transcription factor binding motif, and the topological association between the variant locus and RAD54L2 or GRM2 TSS cannot be seen in HMEC (positions “a” and “b” in Fig. 2, respectively). Thus, the plausible culprit gene of the rs57025206 locus is DCAF1, an E3 ubiquitin ligase substrate receptor targeting, e.g., TP53, ER-alpha, and EZH2.
We performed a similar target gene analysis for the 17 further survival candidate variants (Tables 2–4) using positional and eQTL data (Table 5 and Supplementary Data 1 and 2). For only one variant, rs59010985, there was a single clear target gene, GAS7, which was supported by both analyses. The majority of the other variants could be divided into three categories. Some variants had multiple potential target genes. For example, variants in linkage disequilibrium with rs4879914, detected in the BRCA1–BRCA2 meta-analysis, were eQTLs for several genes in white blood cells. Positional evidence supported the same array of target genes, in both mammary epithelial cells and white blood cells. For some other loci, like ASPH and CREB5, the target gene prediction was based merely on the regulatory variants located on active enhancers in relevant tissues, with no eQTL evidence. Furthermore, for six loci, the target gene was assumed the closest gene. One of the 17 survival variants was located in an intergenic region with no apparent target gene.
To detect common pathways or recurrent cellular or molecular functions, we performed a systematic literature review (Fig. 3 and Supplementary Table 6). Relevant literature was available for 26 out of the 40 target genes listed in Table 5, when searched from PubMed with the keyword combinations described in the “Methods” section. Twelve of the genes had previously been connected with breast cancer patient survival, based on either mRNA or protein expression, germline genetic variation, or copy number change in mammary tumors (Fig. 3 and Supplementary Table 6). Fourteen of the target genes were associated with proliferative and migratory capacity of mammary or other epithelial cells. This association was mediated via two primary mechanisms: regulation of MAPK/ERK pathway activity or modulation of cytoskeletal proteins. Target genes in two out of the four loci associated with the survival of BRCA1 carriers with ER-negative breast cancer (DCAF1, ZRANB1, and CTBP2) and two genes from the BRCA1–BRCA2 meta-analysis (ZNF644 and TFCP2L1) affect the regulation of chromatin state either directly or via polycomb repressor complex 2 (PCR2). Eight target genes have been suggested to affect the response to adjuvant chemotherapy, whereas three target genes have been associated with the response to adjuvant endocrine therapy. The latter included two of the four loci (ASPH and GATA3) associated with age-dependent survival effect in BRCA2 carriers. Seven genes from six loci had been suggested to be involved in the regulation of immune response. However, four of these genes were also associated with regulation of mammary cell differentiation.

For details and references, see Supplementary Table 6. PRC2 polycomb repressor complex 2, BC breast cancer, CNA copy number aberration.
Comparison of the results to survival associations from a general breast cancer population
None of the survival associations discovered in the BRCA1/2 carriers were detected in the BCAC data of unselected and familial non-BRCA1/2 breast cancer patients, when our results were compared to those of Escala-Garcia et al. (Supplementary Table 7)23.
Discussion
We studied germline genetic variants associated with the survival of breast cancer patients carrying pathogenic variants in the high-risk BRCA1 and BRCA2 genes. We identified one variant, rs57025206, which was highly significantly associated with poor survival of BRCA1 carriers with ER-negative breast cancer, with HR = 4.37, 95% CI 3.03–6.30, P = 3.1 × 10−9. Furthermore, we discovered 17 additional candidate loci, which could enhance the understanding of the biological processes modifying the survival of breast cancer patients and form a basis for future research. It was noteworthy that the single significant discovery was made in a subgroup of women with BRCA1 pathogenic variants and ER-negative breast cancer, and this was attenuated in the wider analysis of all BRCA1 carriers, because inclusion of the ER-positive breast cancer cases, where the association did not exist, diluted the effect in the combined analysis. Thus, the strength of the discovery analysis came from a phenotypically homogeneous group of cases with shared disease etiology, rather than a large number of study subjects.
Minor alleles of two rare variants, rs9824779 and rs56942057, are in complete linkage (D′ = 1.0, R2 = 0.036) with the rare, poor-survival, allele of rs57025206. These two variants affect conserved transcription factor binding sequences in enhancers targeting the DCAF1 TSS, and the enhancer–TSS interaction is present in HMEC (Fig. 2). Based on our analysis, these variants would be the best functional candidates causing the association signal, although their frequency was too low to allow survival analysis in the CIMBA data.
DCAF1 is an ubiquitin ligase substrate receptor, which recognizes and binds substrates for the CLR4 and HECT-type ubiquitin ligases, thus regulating the substrate half-life. Furthermore, the casein kinase-like and Lis1 homology domains of the DCAF1 protein have been shown to modulate histones by phosphorylation and deacetylation, respectively37,38. According to in vitro models, DCAF1 silencing reduced proliferation and colony formation of MCF7 breast cancer and DU145 prostate cancer cell lines39,40, whereas high DCAF1 expression blocked the expression of tumor suppressor genes via H2A phosphorylation, and induced a proliferative gene expression signature together with FOXM1 (refs 38,40). In mammary epithelial progenitor cells, DCAF1 is involved in the regulation of the Hippo pathway and ER-alpha, contributing to the maturation of luminal and basal lineages41.
The genomic locus of rs57025206, 3p21, is frequently deleted in mammary carcinomas, especially in ER-negative and high-grade tumors42,43,44. Subsequent research has not been able to name an unequivocal cancer driver gene, even though several candidates have been suggested, including BAP1 and MIR135A45,46, which were detected as eQTL variants in our target gene analysis (Supplementary Data 2). However, the topological data suggested that these eQTLs might represent tissue-specific regulatory associations not present in HMEC, and that if the rs57025206-related survival effect is mediated via BAP1 or MIR135A, it probably reflects host–tumor interactions.
To explore the molecular mechanisms possibly contributing to the survival of breast cancer patients with pathological BRCA1 and BRCA2 variants, we included in a target gene analysis and functional literature review 17 sub-genome-wide significant variants with reasonably low false discovery probability. Four variants came from the BRCA1–BRCA2 meta-analysis, four variants had age-dependent survival association for BRCA2 carriers, and the remaining nine variants were associated with the survival of BRCA1 carriers (Tables 2–4). With the selected thresholds for reporting, five to six loci are expected to be false discoveries, while the rest are likely to be true survival loci. Significant between-strata heterogeneity was not detected for any tested variant.
A literature review of the plausible target genes indicated that similar biological functions underscored the genetic survival associations of BRCA1 and BRCA2 carrier breast cancer patients (Supplementary Table 6 and Fig. 3). The most prevalent function was the regulation of epithelial cell proliferative and migratory capacities, suggesting that germline variants modifying the liability of phenotypic changes in epithelial cells could be key determinants for the outcome of breast cancer. Another interesting observation was the accumulation of genes regulating mammary epithelial cell differentiation and stemness on the loci associated with the survival of BRCA1 carriers with ER-negative breast cancer. In addition to DCAF1, described above, these candidate genes include ZRANB1 and CTBP2 located on 10q26.13. ZRANB1 is a deubiquitinase targeting, e.g., EZH2, a component of the PRC2 and direct regulator of BRCA1 activation47,48, whereas CTBP2 is a transcriptional corepressor priming target gene promoters, including BRCA1, for PRC2-dependent silencing49,50.
The functional review highlighted treatment response as another important mechanism contributing to the survival of BRCA1/2 pathogenic variant carriers. The GATA3 locus from the analysis of BRCA2 carriers, the TPM2/GBA2 locus from the BRCA1–BRCA2 meta-analysis, and five out of ten loci from the analyses of BRCA1 carriers have previously been reported to affect the response to adjuvant chemotherapy (Supplementary Table 6 and Fig. 3). Interestingly, KIF26B, GAS7, MIR135A, and CTBP2, all potential target genes of variants associated with the survival of BRCA1 carriers, have been suggested to modify the response to platinum-based chemotherapy51,52,53,54,55,56. Furthermore, CTBP2 has been shown to affect the sensitivity to PARP inhibitors by targeting the BRCA1 promoter for epigenetic silencing50. Platinum compounds and PARP inhibitors are effective agents for adjuvant therapy of BRCA1 carriers57,58. Therefore, the information on genetic variation in these potential response-modifier loci could add to design of targeted therapy trials.
In the analysis of BRCA2 carriers, we did not find any variants exceeding the discovery threshold. This may be partly explained by the smaller number of BRCA2 carriers, and consequently events, in comparison to BRCA1 carrier analysis. However, it was intriguing that with similar thresholds for P-value and false discovery probability, we discovered four variants with age-dependent survival effects. The majority of the BRCA2-related breast cancers are hormone receptor positive (Table 1), and rely on the endogenous supply of estrogen and possibly progesterone for proliferation59. The level of both of these steroid hormones decreases between the ages of 35 and 55 years60, consistent with the diagnosis age of the majority of the BRCA2 carrier cancers in these data (Table 1). The target gene functional review indicated that two loci with age-dependent survival effect in BRCA2 carriers, GATA3 and ASPH, are associated with differences in response to adjuvant endocrine treatment61,62, further supporting the hypothesis that in hormone receptor-positive breast cancer, the age-related differences in hormone secretion and sensitivity may affect the survival of the patients.
We can draw two conclusions from our analyses, which support the hypothesis that the tumor etiology and subtype influence the genetic survival associations. First, the strongest BRCA1 survival variants were not associated with the survival of BRCA2 variant carriers. Second, we did not find any corroboration for our discoveries from the BCAC data (Supplementary Table 7). BRCA1 and BRCA2 carrier tumors have characteristic mutation profiles, accompanied by homologous recombination deficiency, which distinguishes these from unselected breast carcinomas on a molecular level25. Notwithstanding, the mutations in these two genes are associated with different breast cancer subtypes26,27. Taken together, our results encourage accuracy in definition of the outcome of interest and stringency in collecting comparable study subjects, in order to improve the future survival studies.
Like many earlier genetic survival association studies, this study had a limited discovery power, despite the fact that the analyzed high-risk variant carrier cohorts were enriched with poor-prognosis cancers, and therefore contained more events than any equally sized cohort of unselected patients would contain. With two phenotypically coherent groups of breast cancer patients, which represent the largest collections of BRCA1 and BRCA2 pathologic variant carriers in the world, we were able to identify a single genome-wide significant locus, but were probably underpowered to detect loci with more modest effect sizes. We estimate that better coverage of treatment and pathology would have greatly improved our analyses, especially as the functional literature review suggested that the survival differences could be mediated by differential response to adjuvant treatment. Unfortunately, we did not have enough well-documented treatment data available to test the hypothesis. The retrospective nature of our data brought its own limitations. The data included a notable proportion of prevalent cases, enrolled into the participating studies after a prolonged time period after the initial breast cancer diagnosis. The choice of the overall survival as the basis of the analyses impairs the clinical interpretation of the results to some degree. However, most of the follow-up information came from registries, and therefore the all-cause death was the best available indicator for poor prognosis. We tried to overcome these shortcomings with appropriate analysis methods for the prevalent data, with posterior likelihood estimation and sensitivity analyses with breast cancer-specific death as an end point, as well as by testing for internal consistency.
In conclusion, we report a survival locus for BRCA1 carriers with ER-negative breast cancer. Furthermore, the results from the exploratory analyses suggest that the survival in women with breast cancer is influenced by a complex action between clinical characteristics, tumor biology, and the germline genetic landscape.
Methods
Study subjects
The study subjects included women of European ancestry diagnosed with invasive breast cancer before the age of 70 years, enrolled in studies participating in the CIMBA (Supplementary Table 1). A CIMBA study was included in the analyses if it provided sufficient amount of follow-up data, defined as at least 15 study subjects at risk during the time when five events occurred. The study-wise inclusion criterion was applied separately for the main analyses and the ER-specific subgroup analyses. This selection yielded survival data on 3008 women carrying pathogenic germline variants in BRCA1 from 21 studies and 2009 women carrying variants in BRCA2 from 15 studies. Tumor characteristics of the study subjects are provided in Table 1. The BRCA1 carriers were collected from 2664 families: 2391 families with one study subject, 220 families with two study subjects, and 53 families with more than two study subjects. The BRCA2 carriers were collected from 1713 families: 1486 families with one study subject, 182 families with two study subjects, and 49 families with more than two study subjects. All participating studies were approved by their appropriate ethics review boards, and all subjects provided informed consent.
Genotype data
The study subjects were genotyped with a custom-made array as a part of the OncoArray project31. Details on the variant selection and data quality control have been published elsewhere. In short, the genotyping array included a GWAS backbone (Illumina HumanCore) and potentially cancer-associated variants nominated by the six participating consortia. Genotyping of the CIMBA samples was conducted in six independent laboratories, which used the same HapMap reference samples and a common genotype-clustering file to ensure interlaboratory comparability31. The data were imputed using the 1000 genomes as a reference panel, as described previously32. In the analyses, we included variants with at least 60 carriers, corresponding to MAFs 0.01 and 0.015 for BRCA1 and BRCA2 carriers, respectively. This yielded data on ~9.7 million SNPs for BRCA1 carriers and 9.1 million SNPs for BRCA2 carriers.
Survival analysis
The patients were followed from the diagnosis of the first primary breast cancer until death of any cause and censored after 15 years or when lost from follow-up. Left truncation was applied to account for delayed study entry. In the BRCA1 carrier analysis, the number of person years was 16,056, the number of deaths 461, and the 15-year survival rate 0.66. The maximum number of study subjects at risk was 1336 and this was reached 2.8 years after the baseline. The BRCA2 carrier dataset covered 10,712 person years with 311 deaths leading to a 15-year survival rate of 0.65. Here, the highest number of study subjects at risk, 930, was reached 3.9 years after the baseline.
The genome-wide analysis of association between genetic variants and all-cause mortality was performed with Cox regression as implemented in the survival library of the R environment for statistical computing version 3.5.1 and 3.6.0 (refs 63,64,65,66). Linear per-allele risk model and dominant inheritance model were estimated in parallel. Analyses were adjusted for diagnosis age, allowing for variant–age interaction, and stratified by country group to account for population-specific genetic and clinical features (Supplementary Table 1). Likelihood-ratio test was used as a measure of significant association with alpha risk P < 5 × 10−7 (two-sided). The interaction model, i.e., Cox regression model including an interaction term, coded as a product of the number of the effect alleles (0, 1, 2) and diagnosis age (continuous), was tested against a nested model containing the variant and age without interaction. Robust variance estimation was used to account for relatedness of study subjects from the same families. Parallel genome-wide analyses were performed also within the biologically homogeneous patient groups: BRCA1 carriers with ER-negative tumors (1385 patients from 17 studies, see above the study-wise inclusion criterion for study-stratified analysis) and BRCA2 carriers with ER-positive tumors (1050 patients from 14 studies). The results from analysis of all BRCA1 were compared to results from analysis of all BRCA2 carriers with Pearson correlation, and combined using a fixed-effects meta-analysis as implemented in R library metafor67. For the meta-analysis, the standard errors were recalculated using the likelihood-ratio test statistic to avoid inflation caused by rare variants, as suggested previously23.
Using R-library powerSurvEpi68, we estimated that we had sufficient statistical power, with the selected alpha-risk (5 × 10−7) and beta-risk 0.2, to detect significant risk associated with common variants (MAF > 0.10), if the HR was >1.8, whereas for rare variants (MAF 0.03–0.10), the HR should be >2.5 for a discovery. Since we selected an alpha-level lower than the commonly accepted genome-wide significance threshold 5 × 10−8, we calculated a BFDP with R library gap for nominal variant effects and FDR for the interaction models to estimate the validity of our findings69,70,71,72. In the BFDP analysis, the prior discovery probability was set to 0.0001 and HR alternatively to 1.3, 1.8, or 2.5, as in Escala-Garcia et al.23, and according to the estimated thresholds for discovery from the power analyses. SNPs with BFDP or FDR less than one-in-three were considered as interesting discoveries.
We performed additional survival analyses for the newly discovered SNPs only. These included multivariable survival model adjusted for tumor pathologic characteristics and analysis of breast cancer-specific death. Complete pathologic data were available for 1104 (36.7%) BRCA1 and 743 (37.0%) BRCA2 carriers, and the data on breast cancer-associated death for 2066 (68.7%) BRCA1 and 1591 (79.2%) BRCA2 carriers. However, data on both pathology and cause of death were available only for 683 (22.7%) BRCA1 and 544 (27.1%) BRCA2 carriers. The pathologic covariates in the multivariable model were coded as follows: tumor ER expression: categorical—no expression/positive expression (not included in the analyses of ER-stratified patient groups); tumor PgR expression: categorical—no expression/positive expression; tumor grade: linear—1, 2, 3; tumor size: linear—1 = less than 2 cm in diameter, 2 = diameter between 2 cm and 5 cm, 3 = larger than 5 cm in diameter; and lymph node status: categorical—affected/not affected. The validity of the proportional hazards assumption was evaluated for all discovered variants.
Candidate gene identification
The newly discovered survival SNPs were characterized in silico utilizing data from the 1000 genomes35 and Encode36 projects as integrated in databases LDlink73,74, RegulomeDB75, and GeneCards76,77. We retrieved all short genomic variants linked with the discovered survival variants in the 1000 genomes European data with D′ > 0.8. The proxy variants were subjected to RegulomeDB analysis and all variants with scores 1a–2f were considered as regulatory variants potentially contributing to the survival signal (Supplementary Data 1). The variant positions were matched to locations of protein-coding genes and validated enhancer regions to identify the plausibly target genes. Furthermore, we retrieved eQTL genes from GTEx78,79 releases V6, V6p, V7, and Westra et al. blood eQTL dataset80 for all functional proxy SNPs (D′ > 0.8) and for all strongly linked proxy SNPs (R2 > 0.8) irrespective of their functional annotation (Supplementary Data 2). The topological chromatin status at the rs57025206 locus was investigated in Rao et al.81 data from HMEC using the 3D genome browser82.
For a functional summary, we performed a systematic literature search using the gene symbol and any of the keywords “breast cancer”, “mammary”, “estrogen”, “immune”, “leukocyte”, and “lymphocyte”. If none of these queries returned relevant publications, the search was continued with gene symbol and “cancer” or with gene symbol alone. This was the first stage of literature search. In the second stage, the queries consisted of the gene symbol and any of the recurrent functional terms or interacting proteins detected in the first stage (Fig. 3).
The survival associations of the candidate genes’ mRNA expression were tested in the KM plotter database for breast cancer (http://kmplot.com/analysis/)83, restricting the analysis in the relevant group of mammary tumors: ER-negative tumors for target genes from the analyses of BRCA1 carriers, ER-positive tumors for genes from BRCA2 analysis, and all tumors for genes from the meta-analysis. The best cutoff for the categorical survival analysis was selected automatically and results with FDR ≤ 5% were reported. Furthermore, the linear association between candidate genes’ mRNA expression and patient survival was tested in the METABRIC data (EGAD00010000434, 1302 breast cancer patients) using Cox regression. Expression data were available for 29 of the 39 candidate genes. For each of the candidate genes, the samples were split into three categories based on 33 and 67% percentiles of the expression values, and the categories analyzed for 10-year overall survival. The results are included in the functional summary in Supplementary Table 6.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All summary results will be made available on the CIMBA website upon publication (http://cimba.ccge.medschl.cam.ac.uk/). A subset of the genotype data that support the findings of this study is publically available via dbGaP: https://identifiers.org/dbgap:phs001321.v1.p1. Requests for data can be made to the CIMBA Data Access Coordination Committee. DACC approval is required to access data from the BCFR-ON, EMBRACE, GC-HBOC, HEBCS, HEBON, IHCC, IPOBCS, MCGILL, and OUH studies (Supplementary Table 1). The contact for data access requests is Lesley McGuffog (ljm26@medschl.cam.ac.uk), Data Manager, Department of Public Health and Primary Care, University of Cambridge. A full description of data supporting the findings of this study is available in figshare: https://doi.org/10.6084/m9.figshare.12613043 (ref. 84).
Code availability
All statistical analyses were performed within the R environment for statistical computing version 3.5.1 and 3.6.0, using libraries survival, gap, metafor, and powerSurvEpi, for Cox regression, BFDP, meta-analysis, and power estimation, respectively.
References
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953 (2019).
Holleczek, B., Jansen, L. & Brenner, H. Breast cancer survival in Germany: a population-based high resolution study from Saarland. PLoS ONE 8, e70680 (2013).
Simos, D., Clemons, M., Ginsburg, O. M. & Jacobs, C. Definition and consequences of locally advanced breast cancer. Curr. Opin. Support. Palliat. Care. 8, 33–38 (2014).
Sundquist, M., Brudin, L. & Tejler, G. Improved survival in metastatic breast cancer 1985-2016. Breast 31, 46–50 (2017).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009).
Wallden, B. et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genomics 8, 54–6 (2015).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Goldhirsch, A. et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 24, 2206–2223 (2013).
Coates, A. S. et al. Tailoring therapies–improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. 26, 1533–1546 (2015).
Elston, C. W. & Ellis, I. O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19, 403–410 (1991).
Cserni, G., Chmielik, E., Cserni, B. & Tot, T. The new TNM-based staging of breast cancer. Virchows Arch. 472, 697–703 (2018).
Heikkinen, T. et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin. Cancer Res. 15, 3214–3222 (2009).
Kiiski, J. I. et al. FANCM c.5101C>T mutation associates with breast cancer survival and treatment outcome. Int. J. Cancer 139, 2760–2770 (2016).
Ohmoto, A. & Yachida, S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 10, 5195–5208 (2017).
Fagerholm, R. et al. NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat. Genet. 40, 844–853 (2008).
Fagerholm, R. et al. The SNP rs6500843 in 16p13.3 is associated with survival specifically among chemotherapy-treated breast cancer patients. Oncotarget 6, 7390–7407 (2015).
Khan, S. et al. Polymorphism at 19q13.41 predicts breast cancer survival specifically after endocrine therapy. Clin. Cancer Res. 21, 4086–4096 (2015).
Jamshidi, M. et al. Germline variation in TP53 regulatory network genes associates with breast cancer survival and treatment outcome. Int. J. Cancer 132, 2044–2055 (2013).
Lindström, L. S. et al. Familial concordance in cancer survival: a Swedish population-based study. Lancet Oncol. 8, 1001–1006 (2007).
Hartman, M. et al. Is breast cancer prognosis inherited? Breast Cancer Res. 9, R39 (2007).
Verkooijen, H. M. et al. Breast cancer prognosis is inherited independently of patient, tumor and treatment characteristics. Int. J. Cancer 130, 2103–2110 (2012).
Pirie, A. et al. Common germline polymorphisms associated with breast cancer-specific survival. Breast Cancer Res. 17, 58 (2015).
Escala-Garcia, M. et al. Genome-wide association study of germline variants and breast cancer-specific mortality. Br. J. Cancer 120, 647–657 (2019).
Kuchenbaecker, K. B. et al. Risks of breast, ovarian, and contralateral breast cancer for brca1 and brca2 mutation carriers. JAMA 317, 2402–2416 (2017).
Nones, K. et al. Whole-genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann. Oncol. 30, 1071–1079 (2019).
Eerola, H. et al. Relationship of patients’ age to histopathological features of breast tumours in BRCA1 and BRCA2 and mutation-negative breast cancer families. Breast Cancer Res. 7, 465 (2005).
Vocka, M. et al. Estrogen receptor status oppositely modifies breast cancer prognosis in brca1/brca2 mutation carriers versus non-carriers. Cancers 11, 738 (2019).
Copson, E. R. et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study. Lancet Oncol. 19, 169–180 (2018).
Baretta, Z., Mocellin, S., Goldin, E., Olopade, O. I. & Huo, D. Effect of BRCA germline mutations on breast cancer prognosis: a systematic review and meta-analysis. Medicine 95, e4975 (2016).
van den Broek, AlexandraJ., Schmidt, M. K., van ‘t Veer, LauraJ., Tollenaar, RobA. E. M. & van Leeuwen, F. E. Worse breast cancer prognosis of BRCA1/BRCA2 mutation carriers: what’s the evidence? A systematic review with meta-analysis. PLoS ONE 10, e0120189 (2015).
Amos, C. I. et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 26, 126–135 (2017).
Milne, R. L. et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 49, 1767–1778 (2017).
Mavaddat, N. et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 21, 134–147 (2012).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
1000 Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
ENCODE Project Consortium. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Schabla, N. M., Mondal, K. & Swanson, P. C. DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J. Mol. Cell Biol. 11, 725–735 (2019).
Wang, X. et al. VprBP/DCAF1 regulates the degradation and nonproteolytic activation of the cell cycle transcription factor FoxM1. Mol. Cell. Biol. 37, e00609-16 (2017).
Lee, J. M. et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 48, 572–586 (2012).
Kim, K. et al. VprBP has intrinsic kinase activity targeting histone H2A and represses gene transcription. Mol. Cell 52, 459–467 (2013).
Britschgi, A. et al. The Hippo kinases LATS1 and 2 control human breast cell fate via crosstalk with ERα. Nature 541, 541–545 (2017).
Martinez, A. et al. Chromosome 3p allele loss in early invasive breast cancer: detailed mapping and association with clinicopathological features. Mol. Pathol. 54, 300–306 (2001).
Maitra, A. et al. High-resolution chromosome 3p allelotyping of breast carcinomas and precursor lesions demonstrates frequent loss of heterozygosity and a discontinuous pattern of allele loss. Am. J. Pathol. 159, 119–130 (2001).
Loo, L. W. et al. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res. 64, 8541–8549 (2004).
Zhang, W. et al. Loss of estrogen-regulated MIR135A1 at 3p21.1 promotes tamoxifen resistance in breast cancer. Cancer Res. 78, 4915–4928 (2018).
Linne, H. et al. Functional role of SETD2, BAP1, PARP-3 and PBRM1 candidate genes on the regulation of hTERT gene expression. Oncotarget 8, 61890–61900 (2017).
Zhang, P. et al. ZRANB1 is an EZH2 deubiquitinase and a potential therapeutic target in breast cancer. Cell Rep. 23, 823–837 (2018).
Yoo, K. H. & Hennighausen, L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int. J. Biol. Sci. 8, 59–65 (2012).
Kim, T. W. et al. Ctbp2 modulates NuRD-mediated deacetylation of H3K27 and facilitates PRC2-mediated H3K27me3 in active embryonic stem cell genes during exit from pluripotency. Stem Cells 33, 2442–2455 (2015).
May, T. et al. BRCA1 expression is epigenetically repressed in sporadic ovarian cancer cells by overexpression of C-terminal binding protein 2. Neoplasia 15, 600–608 (2013).
Pu, Y. et al. MiR-20a-5p represses multi-drug resistance in osteosarcoma by targeting the KIF26B gene. Cancer Cell Int. 16, 64 (2016).
Hung, F. & Chao, C. C.- Knockdown of growth-arrest-specific gene 7b (gas7b) using short-hairpin RNA desensitizes neuroblastoma cells to cisplatin: Implications for preventing apoptosis of neurons. J. Neurosci. Res. 88, 3578–3587 (2010).
Li, D., Zhang, B. & Hu, C. Oxaliplatin inhibits proliferation and migration of human hepatocellular carcinoma cells via GAS7C and the N-WASP/FAK/F-actin pathway. Acta Biochim. Biophys. Sin. 49, 581–587 (2017).
Yan, L. et al. miR-135a promotes gastric cancer progression and resistance to oxaliplatin. Oncotarget 7, 70699–70714 (2016).
Wang, D. P., Gu, L. L., Xue, Q., Chen, H. & Mao, G. X. CtBP2 promotes proliferation and reduces drug sensitivity in non-small cell lung cancer via the Wnt/β-catenin pathway. Neoplasma 65, 888–897 (2018).
Shi, H. et al. C-terminal binding protein-2 mediates cisplatin chemoresistance in esophageal cancer cells via the inhibition of apoptosis. Int. J. Oncol. 53, 167–176 (2018).
Robson, M. et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017).
Gerratana, L., Fanotto, V., Pelizzari, G., Agostinetto, E. & Puglisi, F. Do platinum salts fit all triple negative breast cancers? Cancer Treat. Rev. 48, 34–41 (2016).
Trabert, B., Sherman, M. E., Kannan, N. & Stanczyk, F. Z. Progesterone and breast cancer. Endocr. Rev. 41, 320–344 (2020).
Cutler, W. B. & Genovese-Stone, E. Wellness in women after 40 years of age: the role of sex hormones and pheromones. Dis. Mon. 44, 421–546 (1998).
Shimoda, M. et al. Endocrine sensitivity of estrogen receptor-positive breast cancer is negatively correlated with aspartate-β-hydroxylase expression. Cancer Sci. 108, 2454–2461 (2017).
Tominaga, N. et al. Clinicopathological analysis of GATA3-positive breast cancers with special reference to response to neoadjuvant chemotherapy. Ann. Oncol. 23, 3051–3057 (2012).
R Core Team. R: a Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2018).
Cox, D. R. Regression models and life-tables. J. Roy. Stat. Soc. Ser. B 34, 187–220 (1972).
Therneau, T. & Grambsch, P. M. in Modeling Survival Data: Extending the Cox Model (Springer, New York, 2000).
Therneau, T. A Package for Survival Analysis in S. 2.38. https://cran.r-project.org/web/packages/survival/index.html (2015).
Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
Qiu, W., Chavarro, J., Lazarus, R., Rosner, B. & Ma, J. powerSurvEpi: Power and Sample Size Calculation for Survival Analysis of Epidemiological Studies. 0.1.0. https://cran.r-project.org/web/packages/powerSurvEpi/index.html (2018).
Wakefield, J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet. 81, 208–227 (2007).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B 57, 289–300 (1995).
Zhao, J. H. Gap: genetic analysis package. J. Stat. Softw. 23, 1–18 (2007).
Zhao, J. H. gap: Genetic Analysis Package. 1.2.1. https://cran.r-project.org/web/packages/gap/index.html.
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Machiela, M. J. & Chanock, S. J. LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 34, 887–889 (2018).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Stelzer, G. et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinformatics 54, 1.30.1–1.30.33 (2016).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Wang, Y. et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Gyorffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010).
Muranen, T. A. et al. Metadata Supporting Association of Germline Variation with the Survival Of Women with Brca1/2 Pathogenic Variants and Breast Cancer. figshare. https://doi.org/10.6084/m9.figshare.12613043 (2020).
Acknowledgements
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians, and administrative staff who have enabled this work to be carried out. The CIMBA data management and data analysis were supported by Cancer Research—UK grants C12292/A20861 and C12292/A11174. Genotyping for the OncoArray was funded by the government of Canada through Genome Canada and the Canadian Institutes of Health Research (GPH-129344), the Ministère de l'Économie, de la Science et de l’Innovation du Québec through Génome Québec, the Quebec Breast Cancer Foundation for the PERSPECTIVE project, the US National Institutes of Health (NIH; 1 U19 CA 148065 for the Discovery, Biology and Risk of Inherited Variants in Breast Cancer (DRIVE) project and X01HG007492 to the Center for Inherited Disease Research (CIDR) under contract HHSN268201200008I), Cancer Research UK (C1287/A16563), the Odense University Hospital Research Foundation (Denmark), the National R&D Program for Cancer Control–Ministry of Health and Welfare (Republic of Korea; 1420190), the Italian Association for Cancer Research (AIRC; IG16933), the Breast Cancer Research Foundation, the National Health and Medical Research Council (Australia) and German Cancer Aid (110837). Detailed acknowledgements appear in the Supplementary note.
Author information
Authors and Affiliations
Consortia
Contributions
Study conception, interpretation of results, and writing group: T.A.M., S.K., R.F., M.K.S., A.C.A., G.C.-T., C.B., and H.N. Data analyses and drafting of the manuscript: T.A.M. The OncoArray core group: D.F.E., A.C.A., J.S., G.C.-T., J.M.C., L.M., M.K.S., P. S., J.D., and G.L. Provision of DNA samples and/or phenotypic data: K.A., D.F.E., M.Tischkowitz, L.S., kConFab Investigators, R.K.S., B.W., E.H., M.J.H., HEBON investigators, C.F.S., G.W., M.Thomassen, I.S.P., S.M.D., K.L.N., C.L., C.M.R., I.L.A., M.R.T., P.J., J.G., J.N.W., SWE-BRCA investigators, A.J., D.Y., E.M.J., M.C.S., G.C.-T., C.B., and H.N. All authors revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
H.N. has received an honorarium from AstraZeneca. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muranen, T.A., Khan, S., Fagerholm, R. et al. Association of germline variation with the survival of women with BRCA1/2 pathogenic variants and breast cancer. npj Breast Cancer 6, 44 (2020). https://doi.org/10.1038/s41523-020-00185-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-020-00185-6
