
Abstract
The human commensal fungus Candida albicans can attach to epithelia or indwelling medical devices and form biofilms, that are highly tolerant to antifungal drugs and can evade the immune response. The cell surface protein Pga59 has been shown to influence adhesion and biofilm formation. Here, we present evidence that Pga59 displays amyloid properties. Using electron microscopy, staining with an amyloid fibre-specific dye and X-ray diffraction experiments, we showed that the predicted amyloid-forming region of Pga59 is sufficient to build up an amyloid fibre in vitro and that recombinant Pga59 can also adopt a cross-β amyloid fibre architecture. Further, mutations impairing Pga59 amyloid assembly led to diminished adhesion to substrates and reduced biofilm production. Immunogold labelling on amyloid structures extracted from C. albicans revealed that Pga59 is used by the fungal cell to assemble amyloids within the cell wall in response to adhesion. Altogether, our results suggest that Pga59 amyloid properties are used by the fungal cell to mediate cell-substrate interactions and biofilm formation.
Similar content being viewed by others


Introduction
Fungal infections have emerged over the past decades as a serious threat for human health1,2,3. Among all human fungal pathogens, Candida albicans is the most frequently encountered species in hospitals. Though a normal resident of the human gastrointestinal or genital tracts, C. albicans can under certain circumstances proliferate and cause superficial benign infections such as oral or vaginal thrush. However, the combination of local factors (broad-spectrum antibiotics), underlying diseases (chemotherapy-induced mucosal erosion, neutropenia) and invasive procedures (abdominal surgery, central venous catheter) favours translocation of C. albicans across the mucosa and dissemination into the bloodstream4. Recent epidemiological surveys have reported that invasive candidiasis accounts for 75% of all invasive fungal infections5,6,7 and Candida species are now recurrently found among the five leading causes of healthcare-associated bloodstream infections8,9,10. The invasive candidiasis mortality rate (>40%) in immunocompromised patients is higher than that of any bacterial sepsis11,12,13. Because of the increase of hospitalised patients with serious underlying diseases, especially those undergoing treatment for haematological malignancies or hospitalised in ICUs, systemic candidiasis has become a life-threatening challenge for human health.
C. albicans is well known for its ability to form biofilms on medical devices, a process that contributes to relapse or persistence of candidiasis despite antifungal treatments14. Indeed, C. albicans biofilms are crucial for pathogenesis by reducing the sensitivity of the fungus to both antifungal agents and the host immune system11. The molecular processes allowing C. albicans biofilms to avoid antifungal toxicity are mechanistically complex. The resistance towards antifungal drugs is largely due to their trapping by the biofilm extracellular matrix (ECM) and, to some extent, the upregulation of efflux pumps15. Upregulation of both ABC and MFS transporters contributes to the cellular export of antifungal components and thus avoids their toxicity. Biofilms are embedded in the ECM, which protects them from environmental insults, creating a steric hindrance that prevents drug penetration inside the biofilm16. In addition, ergosterol production is decreased in mature biofilm as compared to planktonic cells rendering the microbial community less susceptible to azoles and polyenes than planktonic cells17. The acquisition of new knowledge on the establishment of fungal biofilm will allow the development of innovative strategies to counter the threat posed by these microbial communities.
Biofilm formation is a process tightly regulated in C. albicans that occurs in response to several environmental clues such as surface contact, CO2 levels or nutrient starvation18,19,20. This biological process is orchestrated by a « core » of nine transcription factors that finely regulate lipid metabolism, filamentation and above all adhesion21,22. Adhesion of C. albicans to a substrate is mediated by several polypeptides that belong to three families of adhesins: Als, Hwp1 and Iff/Hyr23. Some proteins among these families, namely Als1, Als2, Als3, Als5, Hwp1, Hwp2 and Eap1, were described to impact biofilm formation23. Except for Als2, all adhesins of C. albicans involved in biofilm establishment contain β-aggregation-prone sequences suggesting that they may be assembled as amyloid structures24,25,26. Amyloid fibres correspond to filamentous protein aggregates with an organised structure. The assembly of an amyloid fibre usually consists of two steps: first, amyloids are nucleated by the homomultimerisation of a protein through the amyloid-forming region. Subsequently, the amyloid fibre is elongated by the stacking of hundreds of monomers on top of each other. Structurally, amyloid fibres are characterised by a high β-sheet content as well as by the presence of the typical cross-β arrangement, which is a hallmark of such quaternary structures27. The cross-β structure is characterised by two reflections detected at 4.7 Å and ~10 Å by X-ray diffraction, which respectively corresponds to the inter-strand and inter-sheet spacing27.
Amyloid structures have long been thought to be harmful to living organisms. However, recent reports have demonstrated that some proteins in the amyloid form can acquire new properties that could be useful and beneficial in the cellular context, a phenomenon called functional amyloids28. For instance, the curli system in E. coli consists of amyloid fibres using the proteins CsgA and CsgB as substrates. These fibres extended in the extracellular matrix of the bacteria are involved in biofilm formation and surface colonisation29. Another example is the S. cerevisiae pyruvate kinase, Cdc19, which forms amyloid fibres aggregating into stress granules under different types of stress. This phenomenon protects the protein from degradation and facilitates the resumption of the cell cycle once the stress is over30.
Results based on electron microscopy, as well as heterologous expression of C. albicans proteins in S. cerevisiae, have revealed that Als1, Als3, Als5 and Eap1 genuinely adopt an amyloid-like structure22,23. Over the last decade, knowledge gathered mainly on Als1 and Als5 from C. albicans has tended towards a model in which these adhesins are unfolded by shear forces generated by liquid flow25, unmasking the amyloid-forming regions and allowing lateral and homotypic amyloid interactions to occur in the C. albicans cell wall, thus creating a very dense area of adhesins, called amyloid nanodomain31,32. Finally, these nanometre-sized nanodomains create adhesive patches on the cell surface, enabling cell–cell interactions through adhesins Ig-like domain32. Pga59 is another adhesin found in C. albicans that belongs to the Hwp1 family. Originally, Pga59 was identified by a computational screen that aimed to find all GPI-anchored proteins in several fungi species, including C. albicans33. The proteins found in the in silico analysis, with no functional name, were given the acronym Pga for predicted GPI-Anchored protein associated with a number i.e. Pga5933. Pga59 is a cell wall-associated protein harbouring both N- and O-glycosylation that was originally described to maintain cell wall integrity in C. albicans34. Previous results from our laboratory have shown that PGA59 is highly expressed in C. albicans biofilms34, and that overexpression of PGA59 has a positive impact on biofilm formation by increasing the dry weight of the microbial community35. Furthermore, atomic force microscopy experiments have revealed that PGA59 overexpression is also associated with higher adhesion forces on the cell surface, resulting in increased adhesion to a plastic substrate upon PGA59 upregulation35. However, the molecular mechanisms underlying Pga59 function in biofilm formation remain to be understood.
In this study, we have shown that Pga59 is assembled into amyloid structures upon adhesion, and we have demonstrated that Pga59 amyloid formation has a positive impact on cell adhesiveness as well as on biofilm establishment in C. albicans. In addition to the work on Als proteins, our study provides further strong evidence that amyloid structures play a role in fungal cell adhesion to a substrate. Furthermore, we describe for the first time that Pga59-based amyloid structures have a direct impact on biofilm formation in C. albicans, and thus constitute a promising avenue for the development of new antifungal strategies.
Results
The D1 domain of Pga59 triggers β-amyloid assembly in vitro
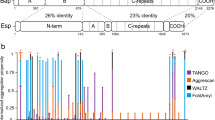
Using the in silico AMYLPRED2 tool36,37, we identified three potential amyloid-forming domains in the Pga59 mature sequence, D1 from position 28 to 35 (28IATTVVTI35), D2 from 48 to 56 (48VTTGVTTVT56) and D3 from 61 to 66 (61TYTTYC66) (Fig. 1a). Further analysis with TANGO, one of the programmes run by AMYLPRED2, showed that D1 was the only domain presenting a high β-aggregation potential (Fig. 1a). Within D1, the highest β-aggregation potential (~74%) was predicted for two valines at positions 32 and 33 (Fig. 1a). A synthetic eight amino acid-long peptide overlapping this sequence (residues 28 to 35) was obtained, as well as peptides incorporating mutations of either V32 or V33 or both into asparagine residues (V32N, V33N and V32,33N, respectively). To confirm the amyloidogenic properties of the D1 sequence, transmission electron microscopy (TEM) observation of the negatively stained synthetic peptides was performed after incubation at 37 °C for 16 h. The WT peptide of the D1 domain formed a large micrometre-sized aggregate with organised regions exhibiting a fibrillar morphology (Fig. 1b). The single mutant peptides V32N and V33N were still able to adopt a fibrillar morphology reminiscent of an amyloid fibre. However, the peptide in which both valines had been substituted for asparagines was unable to adopt an elongated fibrillar morphology (Fig. 1b). To determine if the fibrillar aggregates observed in TEM corresponded to amyloid structures, we performed in vitro Thioflavin T (ThT) assays on the same set of synthetic peptides. The WT peptide displayed a very high fluorescence intensity (~30k arbitrary units (AU)) generated by amyloid structures stained with ThT (Fig. 1c). The V32N mutation had a limited impact, reducing the fluorescence intensity by twofold to ~15k AU while the V33N mutation enhanced it by 1.4-fold to ~44k AU (Fig. 1c). However, the fluorescence intensity of the V32,33N peptide upon ThT staining was significantly reduced to a value of 987 AU, i.e. more than 30 times less than to the WT (Fig. 1c). As the WT D1 peptide of Pga59 self-assembles in fibrillar quaternary structures that are positive to ThT, we sought to determine if these aggregates are genuine amyloids exhibiting a typical cross-β signature. Hence, we used X-ray fibre diffraction to investigate the amyloid nature of the D1 WT peptide. The X-ray diffraction pattern showed two main reflections at 4.7 and 10 Å (Fig. 1d). Such a pattern is characteristic of the so-called cross-β arrangement, one of the structural hallmarks of amyloid fibrils27. This clearly indicates that the Pga59 D1 domain is assembled into amyloid fibres made of β-strands in its aggregated state. The cross-β pattern reveals a repetition of β-strands that are parallel to the main fibril axis and separated by 4.7 Å. The β-sheets are perpendicular to the fibril axis with a spacing of 10 Å. Taken together, these results revealed that the D1 domain of Pga59 is sufficient to craft a cross-β amyloid fibre and that the presence of valine at positions 32 and 33 is critical for this process.

a The graph shows the TANGO β-aggregation potential of each amino acid in the Pga59 mature region. The amino acid composition of the amyloid-forming regions D1, D2 and D3 is indicated below the graph. Pga59 is sketched under the graph. The black rectangle represents Pga59 full-length protein, and the numbers above indicate the amino acid position encompassing D1, D2 and D3 domains. The SPER and SPGPI regions correspond to the signal peptides for translocation into the endoplasmic reticulum (ER) and anchorage to a preformed GPI lipid attached to the ER membrane, respectively. The grey boxes named D1, D2, and D3 represent the amyloid-forming domains in Pga59 mature region predicted by TANGO. b, c Wild type and mutant peptides of Pg59 were resuspended in DMSO prior to their dilution in Tris buffer. After 16 h at 37 °C, peptides were analysed with transmission electron microscopy and ThT staining. b D1 peptides were dropped on electron microscopy grids and negatively stained with uranyl acetate. Peptides’ characteristics are indicated above each micrograph. Scale bar: 500 nm. c ThT intensity of each peptide was recorded at 496 nm. The histogram shows the quantification of data from three independent experiments. Values are displayed as the averages ± SD. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001. The significance of the ThT fluorescence differences were tested using Student’s t-test. d X-ray diffraction pattern of the quaternary structures formed by the WT D1 peptide. Source data are provided as a Source Data file.
Recombinant full-length Pga59 is assembled as an amyloid structure
We next examined if the ability of the D1 domain to form an amyloid structure is conserved with the full-length Pga59 protein. To do so, we produced Pga59 and Pga59 V32,33N (in which both valine residues were replaced by asparagines) fused to an N-terminal His-tag (His6-Pga59 and His6-Pga59V32,33N, respectively) in Escherichia coli SHuffle T7 and purified the recombinant Pga59 proteins (rPga59) by affinity chromatography. We chose to work with the double mutant since the amyloid assembly was blocked only in the D1V32,33N peptide (Fig. 1b, c). Both versions of the rPga59 were purified from bacterial inclusion bodies (Fig. 2a) and their purity was assessed by MALDI-TOF mass spectrometry. For His6-Pga59, the mass analysis revealed the presence of a single intense peak at 9, 839 Da, which corresponds to the expected mass of the WT recombinant protein. Regarding His6-Pga59V32,33N, a single intense peak was observed at the expected mass of 9, 869 Da (Supplementary Fig. 1). The mass analysis demonstrates that both WT and mutant rPga59 have been purified to a high degree. This result allows us to propose that the gel migration of His6-Pga59V32,33N, noticeably slower than that of His6-Pga59, is most likely due to charge variations affecting the migration of the mutant protein compared to the WT. Subsequently, a single signal was detected in the elution fraction by an α-His antibody, confirming that polypeptides eluted from the affinity chromatography matrix were genuinely either His6-Pga59 or His6-Pga59V32,33N (Fig. 2b). To evaluate the ability of rPga59 to adopt an amyloid shape, we conducted ThT assays and TEM. Following refolding, both the WT and the mutant version of rPga59 were incubated at 37 °C and subsequently either treated with ThT or negatively stained with uranyl acetate and observed by electron microscopy. His6-Pga59 displayed a strong ThT signal (~25k AU, Fig. 2c) and adopted elongated fibrillary structures (purple arrow, Fig. 2d). On the other hand, His6-Pga59V32,33N produced a 2.5-fold weaker ThT signal (~10k AU) and no fibrillary aggregates were observed (Fig. 2c, d). These results suggest that the amyloid assembly capabilities of His6-Pga59V32,33N are greatly reduced compared to His6-Pga59. X-ray diffraction was also used to confirm the amyloid nature of His6-Pga59, and typical signatures of β-amyloid fibre were observed for His6-Pga59, with an X-ray diffraction pattern showing reflections at 4.7 and 10 Å (Supplementary Fig. 2). In conclusion, ThT staining, TEM and X-ray diffraction measurements demonstrate that E. coli-produced Pga59 is assembled as a typical cross-β amyloid structure in vitro.

HIS6-Pga59 and HIS6-Pga59V32,33N proteins were expressed in Escherichia coli and purified by affinity chromatography. rPga59 proteins were analysed with Coomassie blue staining (a) and western blot using an α-His antibody (b). Fractions loaded on the gel correspond to the flow through (FT), the wash (W) and the elution (E). (M): Molecular weight markers. Sizes are indicated on the left. c ThT fluorescence intensity of each recombinant proteins analysed at 496 nm. The histogram shows the quantification of data from three independent experiments. Values are displayed as the averages ± SD. (**) p < 0.01. The significance of the ThT fluorescence differences was tested using Student’s t-test. d Wild type (left) and mutated (right) rPga59 were negatively stained with uranyl acetate and analysed with transmission electron microscopy. An isolated amyloid fibre formed by the WT protein is pinpointed on the micrograph with an arrow. Scale bar: 100 nm. Source data are provided as a Source Data file.
Heterologous expression of Pga59 triggers amyloid structure assembly in the budding yeast cell wall
We next sought to determine whether amyloid fibre assembly of Pga59 is a biological process relevant in yeast. We first expressed HA3-Pga59 and HA3-Pga59V32,33N in S. cerevisiae to avoid any background due to amyloid production by other proteins in C. albicans such as Als1 or Als5, among others23. Using immunofluorescence staining, we showed that both the WT HA3-Pga59 and the HA3-Pga59V32,33N mutant proteins were properly localised at the cell surface of S. cerevisiae, while no signal was observed in yeast cells expressing the empty vector (Fig. 3a). We also confirmed that both proteins were produced at similar levels (Fig. 3b). The molecular weight of Pga59 is higher when expressed in S. cerevisiae as protein glycosylation occurs in yeast and not in E. coli. We triggered cell adhesion by incubating S. cerevisiae cells expressing either HA3-Pga59 or HA3-Pga59V32,33N with BSA-coated magnetic beads, and assessed the formation of amyloid structures using ThT staining in vivo. S. cerevisiae cells expressing HA3-Pga59 displayed a ThT-fluorescent signal at the cell surface, as indicated by colocalization with a cell wall marker, namely Concanavalin A, Alexa FluorTM 594 conjugate (Fig. 3c). In contrast, S. cerevisiae cells expressing either the empty vector or HA3-Pga59V32,33N did not display a cell surface-localised ThT signal. Here, the ThT fluorescence was restricted to weak cytoplasmic foci that did not colocalize with the Concanavalin A signal (Fig. 3c). Altogether, these data suggest that Pga59 is involved in the assembly of amyloid structures in the fungal cell wall. Interestingly, the replacement of valines by asparagines at positions 32 and 33 in Pga59, shown above to impair the formation of amyloid structures in vitro, also prevents Pga59-induced assembly of parietal amyloid structures in S. cerevisiae.

a S. cerevisiae strains expressing HA3-Pga59, HA3-Pga59V32,33N or the empty vector as a negative control (EV), were cultivated in a YPD medium. The cellular location of HA3-Pga59 and HA3-Pga59V32,33N upon cell adhesion was assessed by immunofluorescence microscopy using an α-HA antibody. Right panels: Fluorescent signals observed with an α-mouse Texas Red-X conjugate as a secondary antibody. Left panels: Nomarski. b Pga59 levels were assessed in each genetic background by western blot (α-HA antibody, top) using α-tubulin (bottom) as a loading control. The chemiluminescent signals were obtained using a monoclonal α-mouse HRP secondary antibody. Left: Molecular weight standards (M). c amyloid assembly was assessed in the indicated strains by ThT staining (yellow channel) upon adhesion triggered by BSA-coated magnetic beads (arrows in the left panels). The cell wall of S. cerevisiae was stained with the Concanavalin A, Alexa Fluor 594 conjugate (red channel). Yellow and red channels were merged on the right part of the panel. Scale bar: 5 μm. The data presented here are representative of three independent experiments. Source data are provided as a Source Data file.
Amyloid structures formed upon adhesion in C. albicans involve Pga59
Based on the observations that Pga59 can self-assemble as amyloid fibres in vitro and trigger the formation of amyloid structures in the cell wall of S. cerevisiae, we aimed to assess the contribution of Pga59 in amyloidogenesis in C. albicans. To this aim, the WT strain, the pga59ΔΔ knockout and the two reintegrants PGA59/pga59Δ and pga59V32,33N/pga59Δ strains grown to mid-logarithmic phase were incubated with ThT and treated with BSA-coated magnetic beads to trigger adhesion38. Cells untreated with magnetic beads were used as a negative control for adhesion. Cells were then observed with an epifluorescence microscope to record the fluorescence generated by ThT bound on amyloid structures. Regardless of the genetic background, non-adherent cells (no magnetic beads) were poorly fluorescent, suggesting that amyloids are not formed in planktonic cells without adhesion. However, upon adhesion, Thioflavin T fluorescence dramatically increased in the WT, suggesting that amyloid structures are crafted in response to adhesion (Fig. 4a). In pga59ΔΔ mutant cells, there was a sharp reduction of the ThT fluorescence signal as compared to the WT (Fig. 4a), suggesting the involvement of Pga59 in the formation of adhesion-triggered amyloid structures. Accordingly, the reintegration of the PGA59 WT allele in the knockout strain resulted in a strong ThT-fluorescent signal. In contrast, the reintegration of the mutant expressing the pga59V32,33N allele did not restore a strong ThT-fluorescent signal, suggesting that this form of Pga59 is unable to trigger the production of cell surface amyloids in these conditions (Fig. 4a, right panels). We checked by RT-qPCR that PGA59 and pga59V32,33N are expressed at the same level, confirming that the phenotypes are not caused by differences in gene expression (Supplementary Fig. 3a). Further, we showed by western blot analysis and epifluorescence microscopy that the protein levels of GFP-Pga59 and GFP-Pga59V32,33N were similar, and that both proteins were properly located at the cell surface, respectively (Supplementary Fig. 3b, c). Taken together, these results demonstrate that Pga59 is involved in amyloid assembly upon adhesion. Furthermore, the Pga59V32,33N mutant protein that is unable to form amyloid fibres in vitro is also dysfunctional in vivo, possibly because of its inability to trigger amyloid structure formation in response to adhesion. To get a better insight into the cellular distribution of the amyloid structures, we observed the cells at a higher magnification (100X). The WT strain, as well as the reintegrant expressing the WT PGA59 allele, displayed a diffuse ThT fluorescence signal in the cytoplasm as well as a strong signal at the cell surface (Fig. 4b). This ThT cell surface signal was dependent on the presence of a functional Pga59 as the knockout mutant, as well as the strain expressing the pga59V32,33N allele, lost the ability to form cell surface amyloids (Fig. 4b). Results presented here clearly show that C. albicans produces cell surface amyloids upon adhesion in a Pga59-dependent manner.

a Amyloid structures formation in C. albicans was monitored by using ThT staining. WT, pga59Δ/pga59Δ, PGA59/pga59Δ and pga59V32,33N/pga59Δ strains were cultured in a YPD medium. Adhesion was triggered by adding magnetic beads to the culture medium. Fungal cells without magnetic beads were used as a negative control (no adhesion, left panels). The ThT signal was recorded in the yellow channel while cells were observed by Nomarski (with a 20X objective). Scale bar: 25 μm. b Amyloid assembly was monitored with ThT staining (yellow channel) at higher magnification (with a 100X objective). Amyloid structures found in the cell wall are indicated with white arrows. Scale bar: 3 μm.
Pga59 is used by C. albicans to craft amyloid fibres
Given that GFP-Pga59 colocalized with the ThT signal at the cell wall of C. albicans upon adhesion, we performed cell fractionation experiments to determine whether Pga59 is used by fungal cells to form amyloid material. We took advantage of the protocol published by Krynduskin et al. to extract amyloid structures from C. albicans39. Cells expressing either untagged Pga59, GFP-Pga59 or GFP-Pga59V32,33N were treated with BSA-coated magnetic beads to trigger the adhesion process. Cells which had not been treated with magnetic beads were used as a negative control as we had previously shown that Pga59 amyloid production was almost absent in non-adherent cells. Cells from the different strains and adhesion conditions were successively: (i) disrupted with glass beads, (ii) subjected to ultracentrifugation on a sucrose pad to enrich for amyloids and (iii) the resulting amyloids structures were submitted to an immunogold labelling procedure and the samples observed with a transmission electron microscope. As expected, no amyloid was found in samples obtained from non-adherent cells (Fig. 5a). However, amyloid structures were observed in the samples from both adherent WT cells and GFP-Pga59 expressing strains (Fig. 5a, upper and middle panels, respectively). No amyloid structure was observed on material extracted from adherent cells expressing GFP-pga59V32,33N (Fig. 5a, lower panels). All samples were submitted to immunolabelling with gold-labelled anti-GFP antibodies. A fraction of the amyloid materials extracted from cells expressing the GFP-PGA59 allele was positive to the immunogold labelling (black dots of 10 nm on the amyloid structures), which indicates that GFP-Pga59 was used by C. albicans to assemble these amyloid structures (Fig. 5a, middle panels). The specificity of the immunogold labelling was confirmed as no gold particles were associated with amyloid structures extracted from the WT strain expressing the untagged version of Pga59 (Fig. 5a, upper panels). To verify that aggregates resulting from the cell fractionation are genuine amyloids, we stained each protein sample with Thioflavin T. Extracts from adherent cells expressing either the untagged or the GFP-tagged version of Pga59 yielded a ThT fluorescence signal of 2000 AU and 1900 AU respectively, while mixtures coming from either non-adherent cells or cells expressing the GFP-tagged mutant version of Pga59 presented a weak ThT signal (Fig. 5b). Taken together, these results reveal that Pga59 is a component of the amyloid structures crafted by C. albicans in response to adhesion. The data also pinpoints the fact that replacing valine residues at positions 32 and 33 with asparagines tremendously reduces the ability of the fungus to produce adhesion-triggered amyloids.

a Adhesion of C. albicans strains expressing either GFP-Pga59, GFP-Pga59V32,33N or the empty vector was triggered with magnetic beads and non-adherent cells were used as a negative control. Amyloid structures were enriched using a cell fractionation protocol developed to extract amyloids from yeast crude extracts. The resulting fibres were subsequently dropped on a copper grid, labelled with both the α-GFP antibody and gold particles prior to their negative staining with uranyl acetate. Micrographs are representative of the amyloid content in each condition. Scale bar: 200 nm. The right panels are a 2.5X enlargement of the adhesion columns from the left part (scale bar: 80 nm). Pga59 positive fibres are indicated with red arrows (right part). b The presence of amyloid structures after the cell fractionation procedure was assessed with ThT staining. Quantification of the ThT fluorescence in each condition is reported on the histogram (n = 3). The average values ± SD are used to present the data. (*) p < 0.05, (***) p < 0.001. The significance of the ThT fluorescence differences were tested using Student’s t-test. Source data are provided as a Source Data file.
The amyloid property of Pga59 positively impacts adhesion and biofilm formation in C. albicans
Proteins with amyloid properties have been described in the literature to be involved in the adhesion process between microbial cells and their substrate25. To evaluate if Pga59 is used by C. albicans to trigger a cell-substrate adhesion mechanism, we performed adhesion assays on both magnetic beads and Caco-2 cells. Firstly, mid-logarithmic cells were incubated with BSA-coated magnetic beads and then the size of cellular aggregates was recorded with an Opera Phenix microscope. The WT strain was able to form huge cell aggregates tens of micrometres wide (Fig. 6a). Yeast cell aggregates came in the form of irregularly contoured dark structures in the focal plane, surrounded by empty areas (light grey). The size of cell aggregates formed by the pga59Δ/pga59Δ strain was greatly reduced, suggesting that adhesion forces between cells were weaker (Fig. 6a). Complementing the KO mutant with the WT PGA59 allele restored its ability to form huge cell aggregates. Interestingly, cells expressing the mutant pga59V32,33N in which amyloid assembly is blocked behaved like the KO strain and produced very small cell aggregates (Fig. 6a). We then performed adhesion assays on Caco-2 cells with the same set of strains. A monolayer of Caco-2 cells was infected by C. albicans, and after the adhesion time, non-adherent cells were washed away, and adherent cells were counted. Representative pictures of C. albicans adhered at the surface of the Caco-2 monolayer (forming germ tubes and filaments) are shown in Fig. 6b. On average, 58 ± 6 cells per area have been counted in the WT context. This number significantly dropped to 21 ± 5 and 19 ± 5 in the KO mutant or the strain expressing the pga59V32,33N allele respectively. In cells complemented with the WT allele, the average number of cells per area was determined to be 66 ± 10. These results clearly show that Pga59 full amyloid capabilities are required for cells to properly adhere to biotic surfaces.

a Adhesion to abiotic surfaces was assessed by cultivating the indicated strains with magnetic beads. The formation of cellular aggregates was assessed by transmission microscopy (Nomarski). Scale bar: 200 μm. b The indicated strains were cultivated on a monolayer of Caco-2 cells to monitor adhesion on biotic surfaces. After 1 h of adhesion, C. albicans cells were washed and observed under the microscope (Nomarski). Scale bar: 10 μm. c C. albicans adhered on Caco-2 cells were counted on 200 µm2 areas. The experiment was repeated three times. Significance was assessed using Student’s t-test. (****) p < 0.0001. d Biofilms of the indicated overexpression strains were grown in a continuous flow microfermentor in a GHAUM medium in the presence or absence (negative control) of 50 μg mL−1 doxycycline. After 40 h, the biofilms were detached from the spatula, dried for 24 h, and weighed. The histogram presents the quantification of data derived from three independent experiments. Values are the average of each condition ± SD. (**) p < 0.01. The significance of the dry mass differences between the conditions was assessed using Student’s t-test. Source data are provided as a Source Data file.
The involvement of Pga59 in biofilm formation has already been shown. Indeed, in a continuous flow microfermentor model, overexpression of Pga59 was shown to lead to thicker biofilms than the wild type35. In the same experimental setting, the pga59 knock-out strain forms a biofilm twice lighter than that formed by cells expressing Pga59 (Supplementary Fig. 4), thus confirming the implication of Pga59 in this process. To investigate the impact of Pga59 amyloid properties on biofilm establishment, we assessed the biofilm formation capacity in a continuous flow fermentor system of strains overexpressing either PGA59 or pga59V32,33N. Inducing the overexpression of PGA59 led to a significantly higher dry biofilm biomass (170 ± 27 mg) as compared to the same strain in uninduced conditions (100 ± 9 mg) (Fig. 6c). In contrast, when pga59V32,33N was overexpressed, the dry biofilm biomass dropped to 67 ± 6 mg compared to the identical strain without overexpression of pga59V32,33N (105 ± 15 mg) (Fig. 6c). The overexpression of PGA59 is associated to an increase of 70% in fungal biofilm production while the overexpression of pga59V32,33N is linked to a decrease of 37% of the biofilm biomass both compared to strains without overexpression of PGA59 and pga59V32,33N respectively.
Overall, these results show that amyloid properties of Pga59 are important for adhesion to both abiotic surfaces and human cells, as well as biofilm establishment in C. albicans.
The Pga59-derived D1 peptide containing the V32,33N mutation impairs amyloid formation upon adhesion and biofilm formation
Since the V32,33N mutation in the D1 domain of Pga59 impairs amyloid assembly in adherent cells and during biofilm establishment, we hypothesised that treating WT cells with the Pga59 peptide harbouring the V32,33N mutation would impact these biological phenomena. To assess the effect of the peptide on adhesion, 5 μM of either the WT or the mutant version of the D1 Pga59 peptide were added to cells adhered to BSA-coated magnetic beads together with 25 μM of ThT and incubated for 1 h. No peptide was added to ThT in the negative control. Then, amyloid assembly was monitored in each condition by visualisation of ThT staining with an epifluorescence microscope (Fig. 7a). Upon addition of the WT peptide, C. albicans formed aggregates even bigger than in the untreated condition. In contrast, the mutant peptide reduced the size of cellular aggregates suggesting reduced adhesion forces between cells compared to untreated cells or cells treated with the WT D1 peptide (Fig. 7a). Amyloid structures were produced to the same extent in both untreated conditions and cells treated with the WT D1 peptide (Fig. 7a). However, yeasts treated with the V32,33N peptide exhibited a weak ThT signal upon adhesion. This result clearly shows that amyloid formation was impaired when adherent cells were incubated with the mutant peptide. As the V32,33N peptide reduced the amyloid assembly and aggregation capabilities of adherent cells, we then tested the effect of this peptide on biofilm formation in TPP® six-well culture plates. About 5 μM of either the WT or the mutant version of the D1 Pga59 peptide were added to the wells after the adhesion step, and the biofilms were allowed to form for 48 h at 37 °C. Incubation with the WT peptide resulted in increased biofilm biomass (109.7 mg) compared to untreated cells (90.33 mg, Fig. 7b). Interestingly, the biofilm biomass dropped significantly to 78.67 mg when cells were treated with the V32,33N peptide (Fig. 7b). Overall, the V32,33N peptide reduces amyloid assembly upon adhesion, and hence impacts biofilm establishment by lowering the biomass of the fungal community.

a Amyloid assembly upon adhesion was assessed by incubating a WT strain of C. albicans with magnetic beads, ThT and either the Pga59 WT D1 peptide (middle panels) or the V32,33N mutant version (bottom panels). Adherent cells without peptide treatment were used as a control (top panels). The ThT fluorescence was observed with an epifluorescence microscope. Scale bar: 25 μm. The experiment was performed three times. b Biofilms of the WT C. albicans strain were grown in TPP® six-well culture plates in RPMI medium in the presence of either the Pga59 WT peptide or the V32,33N peptide. Biofilms incubated without Pga59 peptides were used as a control. After 48 h, biofilms were washed, dried, and subsequently weighed. For each experimental condition, values are average ± SD. (**) p < 0.01. The quantification of three independent experiments is presented on the histogram. The significance of the results was evaluated using a Student’s t-test. Source data are provided as a Source Data file.
Discussion
In the present work, we have investigated the molecular mechanisms underlying Pga59 function regarding biofilm formation in C. albicans. Pga59 belongs to the Hwp1 family of adhesins comprised of 12 proteins characterised by a conserved 42 amino acid-long region, the molecular function of which has not been uncovered yet23,34. Results from several members of this family, namely Hwp1, Hwp2, Eap1 and Rbt1 have revealed the presence of amyloid-forming regions within the 42 amino acid domains, suggesting a role of this specific domain in amyloid formation20,40.
Here, an in silico analysis revealed that the Pga59 42 amino acid domain contains three potential amyloid-forming regions, only one of which exhibits β-aggregation potential. Using an in vitro approach, we demonstrated that Pga59 can be assembled into β-amyloid fibres. Furthermore, we showed that this phenomenon is relevant in cellulo where Pga59-dependent amyloid structures are crafted upon adhesion. Importantly, we also demonstrated that amyloid structures assembled in a Pga59-dependent manner is important for fungal cell adhesion and effective biofilm formation.
Over the past decade, functional amyloids involved in adhesion mechanisms have been described in microorganisms. For instance, the Fap and Sbp amyloid systems, from Pseudomonas aeruginosa and Staphylococcus epidermidis, respectively, are ECM residents and are involved in cell–cell interactions for biofilm establishment41,42. Recently, the Bap system from Staphylococcus aureus was also described to trigger cellular adhesion and positively impact biofilm formation43. Under acidic pH and low calcium concentration, Bap is released from the cell wall and assembles as an amyloid-like aggregate to stimulate the formation of bacterial biofilm43,44. Other examples include C. albicans Als1 and Als5 adhesins that are clustered in specific regions of the cell wall, called amyloid nanodomains, to support cell–cell interactions. Blocking amyloid nanodomains assembly by expressing an Als1 mutant unable to form amyloid structures results in reduced cell–cell interactions45. Als1 and Als5 are large proteins with a common architecture: an N-terminal Ig-like domain which provides adhesion capabilities to a substrate, a Thr-rich T domain containing the amyloid-forming region required to form the amyloid structures, a TR domain that generates hydrophobic interactions between adhesins and substrates, a stalk region, and a C-terminal GPI anchor23. These domains cooperate to establish amyloid nanodomains at the yeast cell surface, while adhesiveness is provided by the Ig-like and the TR domains. Pga59 is a protein about ten times smaller than Als adhesins and shares no sequence similarity with Als proteins except in the amyloid-forming regions. These topological differences suggest that although Als proteins and Pga59 are involved in adhesion processes, they rely on different molecular mechanisms. Moreover, regarding the Hwp1 family of adhesins, Hwp1, Hwp2 and Rbt1 have been implicated in cell–cell aggregation and biofilm formation. While the amyloid-forming regions are conserved between Pga59, Hwp1, Hwp2 and Rbt1, the effector domains allowing Hwp1, Hwp2 and Rbt1 adhesins to adhere on a surface are not conserved in Pga59, making its adhesion mechanism unclear23. Further work is needed to decipher the adhesion mechanism of Pga59 to its substrate.
Several adhesins have already been described in the literature as having an impact on biofilm formation in C. albicans23. Therefore, the relevance of Pga59 in relation to other adhesins in biofilm formation must be discussed. Biofilm establishment in C. albicans is a biological process that includes four steps: (i) the adhesion of yeast cells to a surface, (ii) the initiation step where yeast cells start to filament, (iii) the maturation phase to constitute an intricate network of yeast cells encased within an ECM and finally (iv) the dispersion to release yeast cells from the biofilm and colonise new area46. Als1 is under the control of the Efg1 transcription factor and hence its expression occurs during filamentation at the maturation step47. Moreover, immunofluorescence microscopy clearly shows that Als1 is mainly located at the hyphae cell wall47. Hwp1, Hwp2 and Als3 are hyphae-specific proteins expressed once the filamentation is engaged during the maturation step20,48,49. Thus, in a fungal biofilm context, these adhesins were suggested to maintain microbial community cohesion by making cell–cell connections. However, in the present report, we showed that adhesion properties provided by Pga59 through cell surface amyloid formation start to occur prior to filamentation (Fig. 4). It is thus suggested that amyloid structures crafted by using Pga59 contribute to the initial adhesion of fungal cells to the substrate which chronologically would happen before the intervention of Als1, Hwp1, Hwp2 and Als3 adhesins. Further studies will be required to validate this statement.
To date, Als1 and Als5 are the only proteins described in C. albicans to form amyloid nanodomains and trigger adhesion in response to shear forces generated by a liquid flow31. Yet, the fact that Pga59 colocalized with amyloid structures upon adhesion leaves open the possibility for the existence of amyloid nanodomains constituted of Pga59 adhesins (Fig. 4). The β-aggregation-prone amyloid core sequence IVIVATT from Als1 is the minimal sequence to trigger amyloid links between homologous adhesins45. Here we showed that the IATTVVTI octapeptide from Pga59 is necessary and sufficient to form fibrillar structures24,25. Although the amyloid core sequences of Als1 and Pga59 are different, they are mainly comprised of isoleucines, threonines and valines, which are the residues most often encountered in amyloid-forming cores24,25. Therefore, the similar biochemical properties of both sequences suggest that Pga59 could behave as Als1 and generate amyloid nanodomains upon adhesion.
Amyloid structures extracted from adherent C. albicans are crafted in a Pga59-dependent manner and contain Pga59. In vitro, amyloid fibres are made up of several hundred monomers of the same protein50. However, in bacteria, some functional amyloids incorporate several proteins. For instance, the curli fibre of E. coli is mainly composed of CsgA but also includes the minor curli subunit CsgB51,52. Furthermore, a fibrous structure produced within the ECM of Streptococcus mutans biofilms is positive to immunogold labelling using antibodies targeting WapA and SMU_63c, suggesting that both proteins are assembled in an amyloid aggregate by the bacteria53. To date, the protein composition of Pga59-dependent amyloid structures found in the C. albicans cell wall upon adhesion is still unclear. The behaviour of the pga59 knock-out strain, as well as the strain producing Pga59V32,33N, let us suggest that Pga59 is not the only protein found in cell surface amyloids of adherent cells. Indeed, although the loss of either Pga59 itself or its ability to form amyloid structures greatly reduced the ThT staining on adherent cells, a weak ThT signal persisted (Fig. 4). Amyloid nanodomains are thought to be exclusively constituted by adhesins homotypic interactions. However, most of the work performed on amyloid nanodomains relied on heterologous expression of C. albicans Als adhesins in S. cerevisiae. Thus, the composition of amyloid nanodomains in the C. albicans cell wall is still not fully elucidated and it is thus possible that Pga59 is assembled within amyloid nanodomains with other adhesins at the cell surface of C. albicans upon adhesion.
The assembly of amyloid-like materials in the ECM is a common mechanism used by bacterial species (i.e., E. coli, P. aeruginosa) to form a biofilm. Here, we have demonstrated that the formation of amyloid materials positively impacts biofilm establishment in C. albicans. Cells expressing Pga59V32,33N, a mutant version of Pga59 unable to form Pga59-dependent amyloid structures in vivo, produce significantly thinner biofilms compared to a WT strain. Using atomic force microscopy, we had previously shown that overexpression of Pga59 is associated with higher adhesion forces35. Altogether, these results let us speculate on a model where Pga59 assembled as an amyloid structure creates adhesion forces which are beneficial for biofilm establishment.
Amyloid structures are directly involved in the pathophysiology of several human diseases54. Anti-amyloid compounds inhibiting the assembly of amyloid fibres are thought to be effective molecules to fight diseases where amyloid structures contribute to the pathology55. Regarding fungal biofilms, an amyloid inhibitory peptide targeting the amyloid-forming region of Als5 was previously shown to greatly reduce C. albicans biofilm formation in vitro38. Cells expressing pga59V32,33N showed a tremendous reduction in their abilities to both produce amyloid structures and to adhere on surfaces compared to strains expressing the wild-type version of PGA59 (Figs. 4–6). Adherent cells treated with the V32,33N mutant peptide showed reduced amyloid formation as well as smaller cellular aggregates compared to C. albicans cells treated with the Pga59 WT peptide (Fig. 7a). Additionally, biofilm biomass was lowered when cells were incubated with the Pga59 mutant peptide compared to untreated cells or cells cultivated with the Pga59 WT peptide (Fig. 7b). Adding the mutant peptide weakens adhesion forces between cells most probably by reducing amyloid assembly upon adhesion, and this negatively impacts biofilm formation. Thus Pga59-based peptides harbouring the V32,33N mutation could constitute a promising avenue to develop new antifungal strategies by lowering amyloid assembly and thus impairing the adhesion process and biofilm formation. Further, as Als5-derived peptides have been shown to inhibit cell–cell adhesion38, blocking amyloid assembly with a combination of peptides targeting several amyloid proteins important for adhesion could lead to a stronger strategy to block biofilm establishment of the pathogenic yeast C. albicans.
Methods
Yeast strains and culture conditions
C. albicans and S. cerevisiae strains used in this study are listed in Table 1. Strains were routinely cultivated in YPD medium (1% yeast extract, 2% peptone and 2% glucose) at 30 °C or SD minimal medium (0.67% yeast nitrogen base without amino acids, 2% glucose) with supplements (arginine 20 μg mL−1, uridine 40 μg mL−1, histidine 20 μg mL−1). C. albicans strains were transformed using the lithium acetate-PEG protocol56. Briefly, an overnight culture was diluted to OD600 = 0.2 in 50 mL YPD and grown at 30 °C until OD600 = 0.6–0.8. Cells were rinsed first in ice-cold 10X TE (100 mM Tris pH 7.5/10 mM EDTA), then in ice-cold 1X TE/100 mM lithium acetate, resuspended in 200 μL 1x TE/100 mM lithium acetate and incubated for 1 h on ice; 50 μL of competent cells were mixed with 1 μg DNA, 50 μg of salmon-sperm DNA and 300 μL of a 40% PEG 3000/1X TE/100 mM lithium acetate solution. The transformation mix was then incubated O/N at 30 °C; cells were then heat-shocked for 15 min at 44 °C and washed with 500 μL of SD medium; cells were pelleted by centrifugation and resuspended either in 300 μL of SD medium and plated on selection plates for prototrophic selection, or in 1 mL YPD, the cells incubated at 30 °C for 3–4 h before plating on YPD medium supplemented with 200 μg mL−1 nourseothricin. S. cerevisiae strains were routinely transformed with the lithium method57. In short, cells were grown as described above for C. albicans, but rinsed in water and resuspended in 1 mL of water. One hundred μL of the cells were used per transformation as follows: cells were pelleted by centrifugation and resuspended in 360 μL transformation mix (30% PEG 3000; 100 mM LiAc, 50 µg salmon-sperm DNA; up to 1 μg DNA). Cells were heat-shocked at 42 °C for 40 min, pelleted, resuspended in 300 μL of water and plated on SC-Ura medium (0.67% yeast nitrogen base without amino acids, Dropout mix without uracil and 2% glucose). For biofilm formation in a continuous flow fermentor system, C. albicans cells were cultivated in GHAUM medium (SD minimal medium supplemented with arginine 20 μg mL−1, uridine 40 μg mL−1, histidine 20 μg mL−1 and methionine 40 μg mL−1) with or without 50 μg mL−1 of doxycycline.
DNA constructs
Primers used to generate the DNA constructs are listed in Table 2. The mutant ORF PGA59V32,32N was created by fusion PCR using CIpSAT1::PGA59 as a template34. Primers A and B and primers C and D were used to amplify two overlapping fragments with the Q5 High-Fidelity DNA polymerase (NEB) (98 °C-30s; (98 °C-10 s; 59 °C-45 s; 72 °C-60 s) x36; 72 °C-2 min), and the fusion PCR (98 °C-30 s; (98 °C-10 s; 59 °C-45 s; 72 °C-90 s) x36; 72 °C-2 min) was performed by using 300 ng of each fragment as templates and primers A and D. Similarly, a PCR fusion was performed to engineer the GFP-tagged PGA59V32,32N mutant using pGEM-T::(PGA59-GFP, ARG4) as a template34; two overlapping fragments were obtained with primers E and B and primers F and C, respectively, using the Q5 High-Fidelity DNA polymerase as above. The fusion PCR was performed as before, using primers E and H. Then, PGA59V32,32N and GFP-PGA59V32,32N were cloned in the KpnI and SalI sites of CIpSAT2 to generate CIpSAT2::PGA59V32,33N and CIpSAT2::GFP-PGA59V32,33N, respectively.
For expression of HA3-Pga59 and HA3-Pga59V32,33N proteins in the cell wall of S. cerevisiae, PGA59 (primers G and H) and PGA59V32,32N (primers G and H) were PCR-amplified from CIpSAT1::PGA59 and CIpSAT1::PGA59V32,33N, respectively, with the Q5 High-Fidelity DNA polymerase (98 °C-30 s; (98 °C-10 s; 59 °C-45 s; 72 °C-45 s) x36; 72 °C-2 min). We then used the Gateway recombinase-based cloning system (Thermo Fisher Scientific) to introduce the fragments in pBC54258, according to the manufacturer’s instructions. The resulting vectors have been named pBC542(URA3 + PGA59) and pBC542(URA3 + PGA59V32,33N).
Prediction of amyloidogenic domains in Pga59 and other related proteins
The amyloidogenic domains in Pga59 full-length primary sequence were predicted using the AMYLPRED2 (http://aias.biol.uoa.gr/AMYLPRED2/) and TANGO (http://tango.crg.es) tools36,37. The primary sequence of Pga59 was obtained from Uniprot (https://www.uniprot.org/) with the following accession number: Q5AF39.
In vitro assembly of fungal amyloid fibres
In order to investigate amyloid fibres formation in vitro, synthetic octapeptides corresponding to the wild type (NH2-IATTVVTI-COOH) and mutant versions V32N (NH2-IATTNVTI-COOH), V33N (NH2-IATTVNTI-COOH) and V32,33N (NH2-IATTNNTI-COOH) of the Pga59 D1 domain were purchased from Thermo Fisher Scientific. Peptides were resuspended at 100 μM in DMSO. Next, each peptide was diluted at 5 μM in a buffer composed of 20 mM Tris-HCl pH 7.4, 150 mM NaCl, 40 μM thioflavin T and incubated at 37 °C for 16 h.
For rHIS6-Pga59 and rHIS6-Pga59V32,33N purified from Escherichia coli, 5 μM of recombinant protein was incubated at 37 °C for 16 h in the same solution used to trigger the aggregation of D1 peptides.
Expression and purification of rPga59
PGA59 codon-optimised synthetic gene lacking the SPER (signal peptide) and SPGPI (propeptide used to post-translationally modify a protein with a GPI anchor) coding regions were produced by GeneArt Gene Synthesis (Thermo Fisher Scientific). NdeI and XhoI restriction sites added at the N- and C-terminal ends, respectively, were used to excise the gene from the initial pMAT vector; the fragment was inserted into the pET28a(+) expression vector previously linearised with the same sites and the ligation was transformed into One Shot TOP10 Chemically Competent E. coli cells (Thermo Fisher Scientific) according to the manufacturer’s instructions. The pET28a(+)-PGA59 construct was verified by sequencing. The pET28a(+)-PGA59V32,33N vector was produced by GeneArt Gene Synthesis (Thermo Fisher Scientific). Both plasmids were transformed in E. coli SHuffle® T7 Express lysY Competent cells according to the manufacturer’s instructions, and selected on LB containing 30 mg L−1 kanamycin. For HIS6-Pga59 and HIS6-Pga59V32,33N production, 50 mL of overnight bacterial cultures were inoculated into 2.5 L baffled flasks containing 1 L of LB supplemented with 30 mg L−1 of kanamycin. Cultures were grown by shaking at 220 rpm at 30 °C until OD600 reached 0.6. PGA59 expression was induced with 1 mM IPTG at 37 °C for 4 h. Bacterial cells were sedimented by centrifugation at 3315×g for 20 min, weighted and stored at −80 °C. Cell pellets were thawed on ice and subsequently resuspended in a 50 mM Tris-HCl pH 8400 mM NaCl, 0.1% Triton X-100, 1 mM DTT lysis buffer supplemented with cOmpleteTM Mini EDTA-free protease inhibitor (Roche) at a ratio of 3 mL/g of pellet. One mg mL−1 of lysozyme was added to the mixture to allow cell lysis at 4 °C for 45 min. Then, 50 μg mL−1 of DNase I was added and lysates were incubated for 45 min at 4 °C, followed by centrifugation at 27,000×g for 15 min. The resulting pellets were washed two times for 1 h at 4 °C with 25 mL of lysis buffer containing 1% of Triton X-100 and dissolved overnight at 4 °C with 25 mL of lysis buffer containing 6 M of guanidine hydrochloride. HIS6-Pga59 and HIS6-Pga59V32,33N polypeptides were then purified on a 1 mL Protino® Ni-NTA column (Macherey-Nagel) using the \({{{\ddot{\mathrm A}}}}\)KTA Express chromatography system at 4 °C, and a linear gradient of imidazole to a final concentration of 0.5 M. The presence of both peptides was assessed at different steps of the purification by loading 10 μL of rPga59 onto a 4–20% gradient Criterion™ gel (Bio–Rad), using 10 μL of Multicolour Low Range protein ladder (Euromedex) as a molecular weight marker, and either staining 30 min with InstantBlue® Coomassie protein (Abcam) followed by a water rinse, or transferring onto a PVDF membrane with an iBlot2 (Thermo Fisher Scientific) using programme 0 (step 1: 20 V for 1 min, step 2: 23 V for 4 min, step 3: 25 V for 2 min); the membrane was blocked with 5% skimmed milk in 1x PBS for 1 h at room temperature. After rinsing in 1X PBS, the membrane was incubated for 1 h with the A7058 anti-His-tag antibody (Sigma-Aldrich) diluted 1/1000 in 1x PBS. The membrane was then washed, treated with Clarity® Western ECL Substrate (Bio-Rad) and observed using the ChemiDoc MP imaging system (Bio-Rad). Uncropped and unprocessed scans of the gel and the blot are given in the Source Data file. The fractions that contained rPga59 peptides were dialysed against a 20 mM Tris-HCl pH8, 150 mM NaCl, 4 M guanidine hydrochloride solution supplemented with a glutathione redox couple (10 mM reduced, 1 mM oxidised) using a 3 mL Slide-A-Lyzer G2 Dialysis Cassette, 3.5 K MWCO (Thermo Fischer Scientific). Dialysis was performed at 4 °C overnight under gentle agitation. The dialysis process was repeated 3 times by decreasing the quantity of guanidine hydrochloride from 4 to 0 M. The protein concentrations in the refolded fractions were estimated at 280 nm with a spectrophotometer.
Thioflavin T assays
Amyloid fibres made either of Pga59 peptides or rHIS6-Pga59, or directly extracted from C. albicans, were stained with Thioflavin T (ThT), a dye specific for amyloid structures, as follows. Amyloid fibres were diluted (5 μM for peptides and rHIS6-Pga59; 100 μg of amyloid mixtures enriched from C. albicans) in 300 μL Tris buffer (20 mM Tris-HCl pH8 and 150 mM NaCl) containing 40 μM of ThT (stock solution: 3.3 mM in ethanol). Subsequently, 100 μL of each mixture was deposited in a black flat-bottom 96-wells plate (Greiner) and incubated for 16 h at 37 °C. Then the fluorescence intensity was monitored with a TECAN Infinite 200 Pro spectrophotometer using an excitation wavelength of 440 nm and emission wavelength of 496 nm at 37 °C to evaluate amyloid fibril formation.
For in vivo ThT staining, C. albicans strains were grown overnight in a YPD medium at 30 °C. Then, cultures were diluted to an OD600 of 0.3 in 4 mL of fresh YPD medium and allowed to grow at 30 °C for an additional 4 h. At the mid-logarithmic phase, C. albicans strains were washed twice with sterile PBS and resuspended in 1 mL of SD medium. Subsequently, 108 yeast cells were incubated with 106 magnetic beads treated overnight with 1 mg/mL−1 heat-denatured BSA on a benchtop rotator for 1 h to trigger adhesion or left untreated (negative control)24,59. Then, all fungal cultures were treated with 25 μM of ThT and incubated at room temperature on a benchtop rotator for 1 h. For testing the effect of WT and mutant peptides on cell adhesion, 5 μM of either NH2-IATTVVTI-COOH or NH2-IATTVVTI-COOH were added during the incubation with ThT.
S. cerevisiae BY4742 strains expressing either the WT allele of Pga59 (CEY679), the Pga59V32,33N mutant (CEY680) or the empty vector (VIF205) were grown in SC-Ura medium at 30 °C. Overnight cultures were diluted in 4 mL of fresh SC-Ura medium to an OD600 of 0.5 and incubated with agitation at 30 °C for 4 h. S. cerevisiae cultures were centrifuged at 200 × g for 3 min and resuspended in 1 mL of SC-Ura medium. Then, cells were simultaneously treated with BSA-coated magnetic beads, 25 μM ThT and 50 μg mL−1 Concanavalin A, Alexa Fluor 594 Conjugate. After 1 h of incubation on a benchtop rotator at room temperature, cells were washed two times with SC-Ura medium and observed by epifluorescence microscopy.
Fungal cells (C. albicans and S. cerevisiae) were observed with an ApoTome microscope (Carl Zeiss) equipped with both OlympusTM 40X X-Apo (0.95 NA) and OlympusTM 100X X-Apo (1.45 NA) objectives. Pictures were recorded with a Nikon DXM1200F digital camera. Thioflavin T signals were recorded with the blue filter, while the Concanavalin A, Alexa Fluor 594 Conjugate fluorescence was recorded with the red filter.
The size of C. albicans cellular aggregates in each genetic background upon adhesion was evaluated in Nomarski at a magnification of 20X with the Opera Phenix microscope (Perkin Elmer).
Negative staining
Ten microlitres of rHIS6-Pga59 samples or D1 synthetic peptides were spotted on glow discharged formvar/carbon grids (FCF200-Cu, DELTA microscopies), negatively stained with 2% uranyl acetate, pH 4.2, analyzed with an FEI Tecnai T12 120 kV transmission electron microscope (FEI Company) equipped with a Gatan Ultrascan US4000 CCD detector.
Immunogold labelling
C. albicans cells expressing either GFP-PGA59 (CEC56), GFP-pga59V32,33N (CEC6218) or the empty vector (CEC832) were grown in YPD medium. Overnight cultures were diluted to an OD600 of 0.3 in 1 L of YPD medium and subsequently incubated under agitation at 30 °C. Yeasts were centrifuged and resuspended in 50 mL of SD medium. Fungal cells were then treated with BSA-coated magnetic beads for 1 h at room temperature to trigger adhesion or left untreated (negative controls). After incubation, cells were centrifuged and stored at −80 °C. Amyloid structures were extracted from C. albicans as described by ref. 39 Briefly, the pellets were resuspended in 4 mL lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.1% Nonidet P-40, 1 mM PMSF, and a cOmplete protease inhibitor tablet (Roche)); then 600 μL were distributed in ten 1.5 mL screwcap tubes and 200 μL of 0.5 mm glass beads were added. Subsequently, fungal cells were mechanically disrupted six times for 1 min in a Bullet Blender bead beater (Next Advance) at full speed. The whole-cell extract was first incubated for 10 min with 0.1 mg mL−1 of RNAse A at room temperature, and then with 0.5% of Triton X-100 at 4 °C for 10 min. Samples were centrifuged at 2000 × g for 10 min at 4 °C and supernatants were loaded on a 40% sucrose pad. The protein mixtures were then ultracentrifuged at 200,000 × g, 4 °C for 2 h. Pellets were resuspended in the amyloid-prion resuspension buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 2% SDS, 5 mM DTT and 5% glycerol) and loaded onto an acrylamide gel. Following the migration, amyloids were eluted from the gel with Amyloid-prion buffer R (Tris-HCl pH 7.4, DTT 5 mM and SDS 0.4%). Ten microlitres of enriched amyloid structures were spotted on glow discharged formvar/carbon grids and processed throughout an immunogold labelling procedure as described in Mourer et al.60. Briefly, electron microscopy grids were incubated for 5 min in a droplet of blocking solution (1X PBS, 1% BSA), and then in a solution of 1X PBS, 1% BSA containing the α-GFP antibody (Santa Cruz, Clone B-2 diluted 200 times) for 1 h. After five washes in 1X PBS, 0.1% BSA and five washes in 1X PBS, grids were incubated for 20 min in a solution composed of 1X PBS, 1% BSA and a 1/50 dilution of 10 nm gold particles conjugated to protein A. Grids were washed several times in 1X PBS and incubated in 1X PBS, 1% glutaraldehyde. Finally, grids were washed with distilled water, incubated with 4% uranyl acetate for 30 min, rinsed five times in a droplet of water, and incubated in 2% lead citrate for 2 min. Grids were then rinsed five times in water and dried for at least 24 h at room temperature. Samples were observed with an FEI Tecnai T12 120 kV transmission electron microscope (FEI Company) equipped with a Gatan Ultrascan US4000 CCD detector.
Adhesion assay
CEC369 (WT), CEC373 (pga59Δ/pga59Δ), CEC832 (pga59Δ/pga59Δ CIpSAT1-PGA59) and CEC6217 (pga59Δ/pga59Δ CIpSAT1-PGA59V32,33N) were used to test the involvement of Pga59 on cellular adhesion as described in ref. 61 as follows. Caco-2 (colon carcinoma) epithelial cells were kindly provided by the Dynamics of Host-Pathogen Interactions Unit, Institut Pasteur. A monolayer of Caco-2 cells was cultivated on coverslips deposited in each well of a TPP® 24-well plate (Merck) in DMEM medium (Gibco) supplemented with 10% FBS for 5 days at 37 °C with 5% CO2. Caco-2 cells monolayers were washed with sterile PBS and then infected with 2 × 105 fungal cells per mL diluted in RPMI medium. After 1 h, non-adherent C. albicans cells were washed off three times with sterile PBS. The remaining adherent cells were cross-linked to Caco-2 cells by overnight incubation at 4 °C with Histofix®. Following the chemical fixation, coverslips were washed with PBS and mounted on a microscope slide with a droplet of ProLongTM Gold antifade (Thermo Fisher Scientific) mounting medium. Fifty random pictures per sample showing an area of 200 μm × 200 μm were recorded in Nomarski on an ApoTome microscope. The number of cells was counted and averaged in each genetic background. The results presented here are representative of three independent experiments. The statistical analysis was performed on GraphPad Prism 9 using a Student’s t-test.
Immunofluorescence
S. cerevisiae cells transformed with pBC542_HA3-Pga59 (CEY679) pBC542_HA3-Pga59V32,33N (CEY680) or pBC524 (VIF205) were grown in SC-Ura medium. About 80 × 106 cells were fixed with 4.6% formaldehyde for 40 min at room temperature, washed two times with PBS and then resuspended in 1 mL of PBS. Twenty microlitres of yeast cells were dropped microscope slides previously coated with 0.1% on poly-l-Lysine (Sigma-Aldrich) and were allowed to adhere to the substrate for 30 min at room temperature. After several washes with 1X PBS, cells were covered with 20 μL of 1X PBS, 0.5% BSA for 5 min. The samples were successively incubated with 20 μL of: (i) a monoclonal α-HA antibody (working dilution:1/1000, Santa Cruz clone F-7) in 1X PBS + 0.5% BSA overnight at 4 °C and then (ii) a secondary α-mouse antibody conjugated to Texas Red-X (working dilution: 1/1000, Invitrogen T-862) in 1X PBS + 0.5% BSA for 1 h at room temperature. A drop of ProLongTM Gold antifade mounting medium and a cover slip were successively added to the samples. S. cerevisiae strains were observed with an ApoTome microscope. Cells were visualised with Nomarski and the red channel was used to record the fluorescent signal generated by the secondary antibody.
Western blot analysis
Whole-cell extracts from both adherent S. cerevisiae and C. albicans cells were obtained with the trichloroacetic acid (TCA) extraction method. Briefly, 300 × 106 fungal cells were resuspended in 1 mL of 20% TCA and centrifuged for 4 min at 200×g. The resulting pellets were resuspended in the lysis buffer (400 μL of 6% TCA, 1 mM of PMSF and 400 μL of 0.1 mm glass beads) prior to their mechanical disruption with a Bullet Blender bead beater (six cycles of 1 min full power with 1 min incubation time on ice between each cycle). Glass beads were discarded by centrifugation (1 min at 960 × g), and then, the protein mixture was centrifuged 5 min at 2700 × g. Pellets were washed three times with 200 μL acetone and resuspended in a solution of 20 mM Tris-HCl pH8 and 1% SDS. Twenty-five micrograms of proteins from each sample were mixed with 2X Laemmli buffer (Bio-Rad) and loaded on a 10% Criterion™ XT Bis-Tris Precast Gel (Bio-Rad). Ten microlitres of PageRuler™ Plus Prestained Protein Ladder (Thermo Fisher Scientific) was used as a molecular weight marker. Proteins were transferred on a PVDF membrane with an iBlot2 dry blotting system (Thermo Fisher Scientific). The membrane was coloured with 1% (w/v) Ponceau S Red in 5% acetic acid to assess the protein load and rinsed with water. Western blotting using as primary antibodies either a mouse monoclonal α-HA antibody (working dilution: 1/1000, Santa Cruz clone F-7) or a rat monoclonal α-alpha tubulin (working dilution: 1/1000, AbD Serotec MCA78G, clone YL1/2) for S. cerevisiae or a mouse monoclonal α-GFP antibody (working dilution: 1/1000, Santa Cruz Clone B-2) for C. albicans was performed as follow: PVDF membranes were incubated with primary antibodies in 1X PBS + 1% skimmed milk overnight at 4 °C. They were then washed several times in PBS and incubated with the Peroxidase AffiniPure Goat anti-mouse secondary antibody (working dilution: 1/1000, Jackson ImmunoResearch) or HRP-conjugated Goat anti-rat secondary antibody (working dilution: 1/1000, Invitrogen, 629520) in 1X PBS + 1% skimmed milk for 1 h at room temperature. Membranes were then successively washed in 1X PBS, treated with Clarity® Western ECL Substrate (Bio-Rad) and observed using the ChemiDoc MP imaging system (Bio-Rad). Pictures are representative of three independent experiments.
RNA extraction and RT-qPCR
About 15 mL of CEC369 (WT), CEC373 (pga59Δ/pga59Δ), CEC832 (pga59Δ/pga59Δ CIpSAT1-PGA59) and CEC6217 (pga59Δ/pga59Δ CIpSAT1-PGA59V32,33N) were cultivated as described in the thioflavin T assays section. Total RNAs of adherent cells were extracted with the RNeasy mini kit (Qiagen) as per the manufacturer’s instructions. Five hundred micrograms of total RNA were used for reverse transcription using the QuantiTect Reverse Transcription kit (Qiagen) according to the manufacturer’s specifications. The qPCR reaction mixtures contained 1 μL of the resulting cDNA, 4 μL of primer mix at 10 pmol μL−1 (primers reverse and forward primers of the target genes, see Table 2), 5 μL of water and 10 μL of 2X SYBR green mix (Eurobio). Q-PCRs were performed in a CFX96 touch real-time PCR detection system (Bio-Rad) apparatus with the following cycle: 50 °C-2 min; 95 °C-10 min; (95 °C-15 s; 59 °C-1 min) x40. The relative expression level of PGA59 in each genetic background was determined using specific primers (pga59F and pga59R, Table 2) and compared to the expression level of ACT1 (primers act1F and act1R, Table 2).
Biofilm formation in a continuous flow fermentor system
Cells overexpressing either PGA59 (CEC6219) or PGA59V32,33N (CEC6220) were grown in a YPD medium with or without 50 μg mL−1 of doxycycline for 16 h. Each culture was then diluted to an OD600 of 1 in fresh GHAUM medium with or without 50 μg mL−1 of doxycycline and left at room temperature for 30 min, to allow further overexpression. For each condition, plastic slides (Thermanox; Nunc) were immersed in the diluted cultures for 90 min at room temperature to allow adherence of fungal cells to the plastic substrate. The plastic slides were then transferred to glass vessels (1 vessel per condition). Each vessel has two glass tubes inserted to drive the entry of medium and air, while the used medium is evacuated through a third tube62. The flow of the GHAUM medium is controlled by a recirculation pump (Ismatec) set at 0.6 mL min−1 and pushed by pressured air supplied at 105 Pa, conditions minimising planktonic phase growth and promoting biofilm formation. The chambers with the plastic substrate were incubated at 37 °C and biofilms were grown for 40 h. Plastic substrates were subsequently immersed in 1X PBS and vortexed to recover the fungal biofilms. The PBS solution containing the biofilms was vacuum filtered through a 1.2 μm filter (Millipore); the filter was dried out at 65 °C for 24 h and then weighed on a precision scale (Mettler AE200; Mettler Toledo) to obtain the dry mass of the biofilm. Three biological replicates were analysed through a Student’s t-test.
Biofilm formation in TPP® 6 well culture plates
C. albicans overnight cultures were washed in sterile PBS prior to their dilution at 1 × 106 cells per mL in RPMI medium; then, 6 mL were distributed in TPP® six-well plates and allowed to adhere for 90 min without agitation at 37 °C. Subsequently, non-adherent cells were washed once with sterile PBS and 6 mL of fresh RPMI medium, supplemented with either 5 μM of the WT Pga59 peptide or 5 μM of V32,33N mutant peptide, were added to each well. Fungal cells cultured without peptide were used as control. The plates were covered with BREATHseal™ and biofilms were allowed to form for 48 h at 37 °C under gentle agitation at 110 rpm. Biofilms were then gently washed with sterile PBS and vacuum filtered through a 1.2 μm filter. Filters were dried out at 65 °C for 24 h and then weighed on a precision scale (Mettler AE200; Mettler Toledo) to obtain the dry biofilm biomass.
X-ray diffraction measurement
Fibre diffraction patterns of fibrils were measured at 4 °C on a Rigaku FRX rotating anode X-ray generator equipped with a Pilatus 200 K hybrid pixel detector (DECTRIS Ltd., Baden-Dättwil, Switzerland) at the copper wavelength. The concentrated hydrated samples were mounted in a MicroLoopsTM from Mitegen (Ithaca, NY, USA) on a goniometer head under the cold nitrogen flow. Each diffraction pattern corresponds to a 360° rotation along the phi axis with an exposure time of 720 s after subtraction of a ‘blank’ image of the same exposure time with only the loop on the goniometer head.
MALDI-TOF mass spectrometry
His6-Pga59 and His6-Pga59V32,33N integrity and purity were analysed on a Bruker UltrafeXtreme MALDI-TOF/TOF instrument. A volume of 15 μL of protein was passed through a ZipTip C4 and eluted on an MTP 384 ground steel target plate (Bruker-Daltonics, Germany) with 2 μL of 20 mg mL−1 α-Cyano-4-hydroxycinnamic acid in 50% acetonitrile, 0.1% trifluoroacetic acid as matrix solution. Data were acquired using Flexcontrol software (Bruker-Daltonics, Germany) and shots were recorded in positive ion linear mode. Mass spectra were externally calibrated in the m/z range of 5–20 kDa with Protein I (Bruker-Daltonics, Germany) and analysed with the Flexanalysis software (Bruker).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are included in the article, its supplementary information files, or are available from the corresponding authors upon reasonable request.
References
Calderone, R. A. Taxonomy and Biology of Candida. Candida and Candidiasis, 1, 15–27 (2002).
Nucci, M. & Anaissie, E. Revisiting the source of candidemia: skin or gut? Clin. Infect. Dis. 33, 1959–1967 (2001).
Miranda, L. N. et al. Candida colonisation as a source for candidaemia. J. Hosp. Infect. 72, 9–16 (2009).
Pfaller, M. A. & Diekema, D. J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20, 133–163 (2007).
Horn, D. L. et al. Presentation of the PATH Alliance registry for prospective data collection and analysis of the epidemiology, therapy, and outcomes of invasive fungal infections. Diagn. Microbiol. Infect. Dis. 59, 407–414 (2007).
Azie, N. et al. The PATH (prospective antifungal therapy) Alliance® registry and invasive fungal infections: update 2012. Diagn. Microbiol. Infect. Dis. 73, 293–300 (2012).
Bongomin, F., Gago, S., Oladele, R. O. & Denning, D. W. Global and multi-national prevalence of fungal diseases-estimate precision. J. Fungi 3, 57 (2017).
Alcazar‐Fuoli, L. & Mellado, E. Current status of antifungal resistance and its impact on clinical practice. Brit. J. Haematol. 166, 471–484 (2014).
Gudlaugsson, O. et al. Attributable mortality of nosocomial candidemia, revisited. Clin. Infect. Dis. 37, 1172–1177 (2003).
Lamoth, F., Lockhart, S. R., Berkow, E. L. & Calandra, T. Changes in the epidemiological landscape of invasive candidiasis. J. Antimicrob. Chemoth 73, i4–i13 (2018).
Brown, G. D. et al. Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv13–165rv13 (2012).
Brown, G. D., Denning, D. W. & Levitz, S. M. Tackling human fungal infections. Science 336, 647–647 (2012).
Baldesi, O. et al. ICU-acquired candidaemia in France: epidemiology and temporal trends, 2004–2013 – A study from the REA-RAISIN network. J. Infect. 75, 59–67 (2017).
Mathé, L. & Dijck, P. V. Recent insights into Candida albicans biofilm resistance mechanisms. Curr. Genet. 59, 251–264 (2013).
Costa-de-Oliveira, S. & Rodrigues, A. G. Candida albicans antifungal resistance and tolerance in bloodstream infections: the triad yeast-host-antifungal. Microorganisms 8, 154 (2020).
Pereira, R., Fontenelle, R. O. S., Brito, E. H. S. & Morais, S. M. Biofilm of Candida albicans: formation, regulation and resistance. J. Appl. Microbiol. 131, 11–22 (2021).
Taff, H. T. et al. A Candida biofilm-induced pathway for matrix glucan delivery: implications for drug resistance. PLoS Pathog. 8, e1002848 (2012).
Pentland, D. R., Davis, J., Mühlschlegel, F. A. & Gourlay, C. W. CO2 enhances the formation, nutrient scavenging and drug resistance properties of C. albicans biofilms. npj Biofilms Microbiomes 7, 67 (2021).
Kumamoto, C. A. Molecular mechanisms of mechanosensing and their roles in fungal contact sensing. Nat. Rev. Microbiol. 6, 667–673 (2008).
Flanagan, P. R. et al. The Candida albicans TOR-activating GTPases Gtr1 and Rhb1 coregulate starvation responses and biofilm formation. mSphere 2, e00477–17 (2017).
Nobile, C. J. et al. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 148, 126–138 (2012).
Mourer, T., El Ghalid, M., d’Enfert, C. & Bachellier-Bassi, S. Involvement of amyloid proteins in the formation of biofilms in the pathogenic yeast Candida albicans. Res. Microbiol. 172, 103813 (2021).
de Groot, P. W. J., Bader, O., Boer, A. D., de, Weig, M. & Chauhan, N. Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot. Cell 12, 470–481 (2013).
Ramsook, C. B. et al. Yeast cell adhesion molecules have functional amyloid-forming sequences. Eukaryot. Cell 9, 393–404 (2010).
Otoo, H. N., Lee, K. G., Qiu, W. & Lipke, P. N. Candida albicans Als adhesins have conserved amyloid-forming sequences. Eukaryot. Cell 7, 776–782 (2008).
Nobbs, A. H., Vickerman, M. M. & Jenkinson, H. F. Heterologous expression of Candida albicans cell wall-associated adhesins in Saccharomyces cerevisiae reveals differential specificities in adherence and biofilm formation and in binding oral Streptococcus gordonii. Eukaryot. Cell 9, 1622–1634 (2010).
Sunde, M. et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 273, 729–739 (1997).
Otzen, D. & Riek, R. Functional amyloids. Cold Spring Harb. Perspect. Biol. 11, a033860 (2019).
Sønderby, T. V., Najarzadeh, Z. & Otzen, D. E. Functional bacterial amyloids: understanding fibrillation, regulating biofilm fibril formation and organizing surface assemblies. Molecules 27, 4080 (2022).
Saad, S. et al. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat. Cell Biol. 19, 1202–1213 (2017).
Lipke, P. N., Mathelié-Guinlet, M., Viljoen, A. & Dufrêne, Y. F. A new function for amyloid-like interactions: cross-beta aggregates of adhesins form cell-to-cell bonds. Pathogens 10, 1013 (2021).
Lipke, P. N. et al. Between amyloids and aggregation lies a connection with strength and adhesion. N. J. Sci. 2014, 815102 (2014).
de Groot, P. W. J., Hellingwerf, K. J. & Klis, F. M. Genome‐wide identification of fungal GPI proteins. Yeast 20, 781–796 (2003).
Moreno-Ruiz, E. et al. The GPI-modified proteins Pga59 and Pga62 of Candida albicans are required for cell wall integrity. Microbiology 155, 2004–2020 (2009).
Cabral, V. et al. Targeted changes of the cell wall proteome influence Candida albicans ability to form single- and multi-strain biofilms. PLoS Pathog. 10, e1004542 (2014).
Tsolis, A. C., Papandreou, N. C., Iconomidou, V. A. & Hamodrakas, S. J. A consensus method for the prediction of ‘aggregation-prone’ peptides in globular proteins. PLoS ONE 8, e54175 (2013).
Rousseau, F., Schymkowitz, J. & Serrano, L. Protein aggregation and amyloidosis: confusion of the kinds? Curr. Opin. Struct. Biol. 16, 118–126 (2006).
Garcia, M. C. et al. A role for amyloid in cell aggregation and biofilm formation. PLoS ONE 6, e17632 (2011).
Kryndushkin, D., Pripuzova, N. & Shewmaker, F. P. Isolation and analysis of prion and amyloid aggregates from yeast cells. Cold Spring Harb. Protoc. 2017, pdb.prot089045 (2017).
Monniot, C. et al. Rbt1 protein domains analysis in Candida albicans brings insights into hyphal surface modifications and Rbt1 potential role during adhesion and biofilm formation. PLoS ONE 8, e82395 (2013).
Dueholm, M. S. et al. Functional amyloid in Pseudomonas. Mol. Microbiol. 77, 1009–1020 (2010).
Wang, Y. et al. Staphylococcus epidermidis small basic protein (Sbp) forms amyloid fibrils, consistent with its function as a scaffolding protein in biofilms. J. Biol. Chem. 293, 14296–14311 (2018).
Taglialegna, A. et al. Staphylococcal Bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathog. 12, e1005711 (2016).
Ma, J. et al. Structural mechanism for modulation of functional amyloid and biofilm formation by Staphylococcal Bap protein switch. EMBO J. 40, e107500 (2021).
Ho, V. et al. An amyloid core sequence in the major Candida albicans adhesin Als1p mediates cell-cell adhesion. mBio 10, e01766-19 (2019).
Nobile, C. J. & Johnson, A. D. Candida albicans biofilms and human disease. Annu. Rev. Microbiol. 69, 71–92 (2015).
Fu, Y. et al. Candida albicans Als1p: an adhesin that is a downstream effector of the EFG1 filamentation pathway. Mol. Microbiol. 44, 61–72 (2002).
Beaussart, A. et al. Single-molecule imaging and functional analysis of Als adhesins and mannans during Candida albicans morphogenesis. ACS Nano 6, 10950–10964 (2012).
Ene, I. V. et al. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell Microbiol. 14, 1319–1335 (2012).
Eisenberg, D. S. & Sawaya, M. R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 86, 69–95 (2017).
Evans, M. L. & Chapman, M. R. Curli biogenesis: order out of disorder. Biochim. Biophys. Acta Mol. Cell Res. 1843, 1551–1558 (2014).
Shanmugam, N. et al. Microbial functional amyloids serve diverse purposes for structure, adhesion and defence. Biophys. Rev. 11, 287–302 (2019).
Besingi, R. N. et al. Functional amyloids in Streptococcus mutans, their use as targets of biofilm inhibition and initial characterization of SMU_63c. Microbiology 163, 488–501 (2017).
Murakami, K. & Ono, K. Interactions of amyloid coaggregates with biomolecules and its relevance to neurodegeneration. FASEB J. 36, e22493 (2022).
Taş, K. et al. Designed peptides as nanomolar cross-amyloid inhibitors acting via supramolecular nanofibre co-assembly. Nat. Commun. 13, 5004 (2022).
Walther, A. & Wendland, J. An improved transformation protocol for the human fungal pathogen Candida albicans. Curr. Genet. 42, 339–343 (2003).
Kawai, S., Hashimoto, W. & Murata, K. Transformation of Saccharomyces cerevisiae and other fungi. Bioeng. Bugs 1, 395–403 (2010).
Zupancic, M. L. et al. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 68, 547–559 (2008).
Gaur, N. K. & Klotz, S. A. Expression, cloning, and characterization of a Candida albicans gene, ALA1, that confers adherence properties upon Saccharomyces cerevisiae for extracellular matrix proteins. Infect. Immun. 65, 5289–5294 (1997).
Mourer, T., Sachse, M., Gazi, A. D., d’Enfert, C. & Bachellier-Bassi, S. A protocol for ultrastructural study of Candida albicans biofilm using transmission electron microscopy. Star Protoc. 3, 101514 (2022).
Mogavero, S. & Hube, B. Host-fungal interactions, methods and protocols. Methods Mol. Biol. 2260, 133–143 (2021).
García-Sánchez, S. et al. Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns. Eukaryot. Cell 3, 536–545 (2004).
Wilson, R. B., Davis, D. & Mitchell, A. P. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181, 1868–1874 (1999).
Fonzi, W. A. & Irwin, M. Y. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134, 717–728 (1993).
Firon, A. et al. The SUN41 and SUN42 genes are essential for cell separation in Candida albicans. Mol. Microbiol. 66, 1256–1275 (2007).
Brachmann, C. B. et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR‐mediated gene disruption and other applications. Yeast 14, 115–132 (1998).
Acknowledgements
T.M. is a recipient of the Pasteur-Cantarini postdoctoral fellowship from the Institut Pasteur. M.E.G. was supported by Institut Pasteur Action Initiative Concertée Mycologie and V.C. was the recipient of a PhD fellowship from the European Commission (FINSysB, PITN-GA-2008-21400). The research work was conducted in the laboratory of C.dE. is supported by the French Government’s Investissement d’Avenir programme (Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases, ANR-10-LABX-62-IBEID). This work has benefited from the Biophysical and Structural Chemistry Platform at IECB, CNRS UAR 3033, INSERM US001. We thank both UBI and PBI Core facilities from Institut Pasteur, and we are grateful for support for equipment from the French Government Programme Investissements d’Avenir France BioImaging (FBI, N° ANR-10-INSB-04-01).
Author information
Authors and Affiliations
Contributions
T.M.: Conceptualisation, methodology, experimentation and writing. M.E.G.: Conceptualisation, methodology and experimentation. G.P.-A. and B.K.: Experimentation. A.L.: Methodology, funding acquisition. S.B. and V.C.: Experimentation and methodology. C.dE. and S.B.-B.: Conceptualisation, methodology, supervision, funding acquisition and writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mourer, T., El Ghalid, M., Pehau-Arnaudet, G. et al. The Pga59 cell wall protein is an amyloid forming protein involved in adhesion and biofilm establishment in the pathogenic yeast Candida albicans. npj Biofilms Microbiomes 9, 6 (2023). https://doi.org/10.1038/s41522-023-00371-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-023-00371-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
