
Abstract
Microbiome profiling through 16S rRNA gene sequence analysis has proven to be a useful research tool in the study of C. difficile infection (CDI); however, CDI microbiome studies typically report results at the genus level or higher, thus precluding identification of this pathogen relative to other members of the gut microbiota. Accurate identification of C. difficile relative to the overall gut microbiome may be useful in assessments of colonization in research studies or as a prognostic indicator for patients with CDI. To investigate the burden of C. difficile at the species level relative to the overall gut microbiome, we applied a high-resolution method for 16S rRNA sequence assignment to previously published gut microbiome studies of CDI and other patient populations. We identified C. difficile in 131 of 156 index cases of CDI (average abundance 1.78%), and 18 of 211 healthy controls (average abundance 0.008%). We further detected substantial levels of C. difficile in a subset of infants that persisted over the first two to 12 months of life. Correlation analysis of C. difficile burden compared to other detected species demonstrated consistent negative associations with C. scindens and multiple Blautia species. These analyses contribute insight into the relative burden of C. difficile in the gut microbiome for multiple patient populations, and indicate that high-resolution 16S rRNA gene sequence analysis may prove useful in the development and evaluation of new therapies for CDI.
Similar content being viewed by others

Introduction
Clostridium difficile infection (CDI) poses a major healthcare burden to the global population, with an estimated 450,000 cases and 29,000 deaths in the United States annually.1,2 CDI is often associated with antibiotic treatment and is frequently acquired by patients during hospitalization. Multiple diagnostic tests for CDI are available and hospitals commonly use a combination of enzyme immunoassay (EIA) and glutamate dehydrogenase (GDH) testing in tandem with real-time polymerase chain reaction (PCR) for increased sensitivity and shorter turnaround time.3
After diagnosis, patients with CDI are typically treated with metronidazole and/or vancomycin depending on symptom severity.3 Treatment failure is estimated to occur in 20% of patients, resulting in a recurrent CDI population that may require other treatment strategies.4,5 The development of microbial-based therapeutics, such as fecal microbiota transplantation (FMT) and combinations of selected microbes for the treatment of recurrent CDI suggests that mixtures of commensal microbes may be routinely utilized in the future as an alternative to powerful antibiotics.6,7
Microbiome profiling through 16S rRNA gene sequencing has proven to be a valuable tool to characterize the diversity and composition of gut microbial communities, including in studies of CDI development and recurrence.8 Given the intricate relationship between the gut microbiota and CDI, accurate identification of C. difficile directly from 16S rRNA profiles in patient populations could be a valuable measure in future studies. However, a fundamental challenge to studying C. difficile through these approaches has been the level of taxonomic resolution provided through short 16S rRNA sequences. As a result, most microbiome sequencing studies of CDI utilize higher aggregate taxonomic categories (e.g., the Clostridium XI cluster, which encompasses many other organisms related to C. difficile) as a proxy for the organism itself or simply avoid quantification altogether.9,10,11,12,13,14,15,16,17
Here we utilize a high-resolution method (Resphera Insight) for assigning species-level context to 16S rRNA gene sequence data to estimate C. difficile burden in different patient populations. This method was recently validated for detection of Listeria monocytogenes 18 and Salmonella enterica,19,20 and was applied in this study to determine the relative abundance of C. difficile in several clinically relevant patient groups. Re-examining published 16S rRNA gene sequence datasets has confirmed previous associations of C. difficile with C. scindens, and identified new positive and negative correlations of C. difficile with other species, both of which may help provide insight into community aspects of C. difficile colonization and resistance against CDI.
Results
Evaluation of sensitivity and specificity for C. difficile identification
One of the challenges of 16S rRNA gene sequencing is the limited information available in these short DNA fragments to distinguish related microbial members below the genus-level. To accurately assess C. difficile at the species level from 16S rRNA gene sequence data, we used a method developed specifically for species level characterization (Resphera Insight, see Methods). We first validated this approach by obtaining full-length 16S rRNA gene sequences from 804 novel C. difficile isolates derived from multiple sources, and subsequently simulated noisy 16S rRNA gene sequence reads for taxonomic assignment (see Methods). Performance was measured using the Diagnostic True Positive Rate (DTP), defined as the percentage of sequences with an unambiguous assignment to C. difficile. The method achieved an average DTP of 99.9% (ranging from 98.92 to 100% per isolate, Table S1), indicating sufficient sensitivity to detect C. difficile from short 16S rRNA gene sequence reads.
In addition to establishing sufficient sensitivity to detect C. difficile, we also sought to evaluate false positive rates in which the method incorrectly assigns a sequence to C. difficile. As this species is a member of the Clostridium XI cluster, a false positive assessment was performed based on in silico simulations of 22 other members of this group, including the very similar Clostridium irregulare. Simulating 10,000 16S rRNA gene sequence reads per species with a 0.5% error rate, 20 of 22 species resulted in zero false positive assignments to C. difficile, with the highest false positive rate (0.07%) attributed to Clostridium irregulare (Table 1).
Representation of C. difficile relative to the microbiota in adult cases of CDI and healthy individuals
To examine the presence of C. difficile in different human populations, we re-examined existing published 16S rRNA gene sequencing datasets with our validated method. We first compared the relative abundance of C. difficile across a cohort of healthy individuals to two cohorts of patients diagnosed with CDI (symptomatic index cases) from Seekatz et al.10 and Khanna et al.21 (Table S2). The Seekatz protocol for CDI diagnosis followed a two-stage algorithm employing enzyme immunoassay for GDH antigen and toxins A and B, with confirmation of tcdB gene presence via PCR if toxin and GDH results were discordant; the Khanna et al. protocol for CDI diagnosis was not reported in the original publication. The healthy patient cohort and Seekatz CDI datasets were generated using equivalent processing and sequencing methods.10 Average analyzed sequencing depths per sample for CDI and healthy groups were 16,114 and 14,937, respectively.
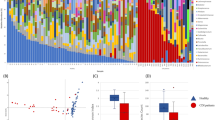
Overall, C. difficile was detected in 58 of 70 CDI index patients (82.9%) in the Seekatz study with an average abundance of 3.04% (Fig. 1a). In the Khanna dataset, C. difficile was detected in 73 of 86 CDI index patients (84.9%) with an average abundance of 0.76% (Fig. 1b). Among healthy controls, only 18 of 211 (8.5%) harbored detectable levels of C. difficile, with an average abundance of 0.008%, significantly less than both Seekatz and Khanna index cases (P < 2e-16; Mann–Whitney test).

Relative burden of C. difficile in the gut microbiome of two cohorts of CDI index patients and healthy controls. a Index cases of recurrent CDI (Seekatz et al.) and b CDI index patients (Khanna et al.) frequently harbored moderate to high levels of C. difficile. c Healthy controls. Overall 91.5% of controls had no detectable C. difficile and 0.9% maintained C. difficile levels higher than 0.1%
We were further interested in determining whether the ability to detect C. difficile or varying levels of C. difficile relative abundance from 16S rRNA gene sequences was related to disease outcome. The Seekatz dataset included samples collected from patients that went on to develop recurrent CDI, a serious outcome following primary diagnosis, or from patients who were later reinfected with CDI beyond the standard time recurrence window.10 Additionally, a severity score22 was available for some of the patients. Across the full Seekatz CDI positive sample set, our method detected C. difficile above 0.1% abundance in 59.2% of samples (Table 2). On average, patients with CDI for index (at primary diagnosis), recurrence or reinfection events had C. difficile abundances greater than 1% regardless of the calculated severity status using Infectious Diseases Society of America (IDSA) standards. We found no significant associations of C. difficile abundance with IDSA severity status among index samples or at the time of recurrence or reinfection (P > 0.05, Mann–Whitney test).
Representation of C. difficile relative to the microbiota in infants
To assess the levels of C. difficile carriage among infants relative to the total gut microbiome, we re-examined 16S rRNA gene sequence datasets describing longitudinal studies of pre-term infants in the neonatal intensive care unit (NICU) by Zhou et al.23 and a single infant profiled during the first 18 months of life by Davis et al.16 In the Zhao dataset, 12 necrotizing enterocolitis (NEC) cases and 26 age-matched controls (all treated at Brigham and Women’s Hospital NICU, Boston, MA) were sequenced with an average of seven samples per subject. The Davis asymptomatic case study consisted of profiling 50 fecal samples over time, during which researchers noted colonization switching between toxigenic and non-toxigenic strains and observed 100,000-fold fluctuations of C. difficile spore counts.16
In these two 16S rRNA gene sequence datasets, moderate levels of C. difficile (>1.0% abundance) appeared consistently within infants over time. In the Zhao dataset, C. difficile was detected in 25 of 38 (66%) infants, including 6 of 12 (50%) infants with NEC, and 19 of 26 (73%) normal infants. There was no significant difference in overall C. difficile presence between NEC and normal infants (P = 0.27, Fisher’s exact test), and both groups maintained statistically similar C. difficile abundance distributions relative to their total gut microbial communities under multivariate regression after adjustment for patient source (Fig. 2a). As the original Davis case study determined C. difficile carriage using spore counts and GDH concentration, we detected substantial representation of C. difficile (up to 7.1% abundance) until the time of weaning and transition to cow’s milk (Fig. 2b). We further found a statistically significant correlation between our C. difficile relative abundance estimates and GDH concentration measurements from the Davis study (Spearman correlation = 0.817; P = 5e-13).

Distribution of C. difficile during longitudinal gut microbiome sampling of infants. a Pre-term infants in a NICU, including those developing necrotizing enterocolitis (purple) and normal (grey). Each boxplot reflects a single patient with multiple time points (total samples per patient shown along the x-axis). b A longitudinal case study of an infant before (red) and after (blue) weaning during the first 18 months of life. During the transition to cow’s milk, C. difficile relative abundance fell to undetectable levels
Correlations of C. difficile with other bacterial species
Recent studies in animal models have indicated that certain species may generate metabolites that inhibit C. difficile, such as the production of secondary bile acids by C. scindens.15 However, previous studies correlating the abundance of C. difficile with other taxa did not utilize the microbiome-based abundances directly, but rather quantified C. difficile abundance through other means such as real-time PCR, colony forming units through culture, measuring GDH concentration or spore counts.15,16,17
We sought to determine whether high-resolution analysis of the 16S rRNA gene sequence data itself could reveal the same associations, and perhaps other relevant species. Computing correlations using Compositionality Corrected by REnormalization and PErmutation (CCREPE)24 across our re-analyzed cohorts, we found a significant negative association between C. difficile and C. scindens for the Khanna CDI patient cohort and the Davis infant longitudinal study (P < 0.02 for both datasets), with a supporting trend in the other studies (Fig. 3, Table S3). Additionally, multiple members of Blautia spp. displayed a consistent negative correlation like that of C. scindens (Fig. 3, Table S3). In contrast, other Clostridia such as C. neonatale and C. paraputrificum and members of Veillonella showed strong positive associations with C. difficile abundance. In silico simulations of noisy 16S rRNA gene sequence reads from these species confirmed a low mis-assignment rate (average 0.08%; see Table S4).

Correlation analysis identifies species positively or negatively associated with C. difficile. The CCREPE N-dimensional checkerboard score (y-axis) incorporates the ratio of co-variation to co-exclusion patterns normalized to a range of (−1, +1). In addition to C. scindens, we identify significant negative correlations with C. difficile for members of Blautia and positive correlations with other Clostridia and Veillonella spp. (*P ≤ 0.05). Ambiguous species level assignments are denoted by slashes. Key for re-analyzed datasets from the following studies: Recurrent CDI=10, Index CDI=21, FMT=9, Infant longitudinal=16, NICU=23 (Table S2)
Discussion
In this study, we sought to identify species-level abundances of C. difficile in 16S rRNA gene sequence datasets from different patient populations using a validated algorithm (Resphera Insight). Similar to previous studies of Listeria monocytogenes 18 and Salmonella enterica,19,20 validation using a high-resolution taxonomic assignment method from 804 novel C. difficile isolates established an overall sensitivity of 99.9% with a marginal false positive rate less than 0.1%, suggesting that C. difficile could be distinguished from other related microbiota members.
Compared to the microbiota of healthy individuals, we observed a higher presence and relative abundance of C. difficile in microbiota data collected from two CDI patient cohorts. 8.5% of healthy individuals were positive for C. difficile using our approach, supporting previous epidemiological assessments of asymptomatic carriage rates.25,26,27,28 Although analysis of CDI datasets revealed a wide distribution of C. difficile relative abundances (ranging from virtually undetectable to above 50% of total sequences), the relative abundance of detected C. difficile in relation to other members of the microbiota was significantly lower in healthy individuals than that of CDI patients. The ability to assess C. difficile levels as part of the microbiota community is potentially more important within population surveys compared to diagnosis using traditional PCR or GDH/EIA tests that merely account for the presence of C. difficile using toxin B or GDH as a proxy.
While detection of C. difficile from 16S rRNA gene sequence data is limited by sequencing depth, our results suggest that C. difficile does not generally reside in healthy adults. In contrast, we did not detect C. difficile in all patients with CDI. The relative presence of C. difficile in these patients is likely below the detection limit given the available sequencing depth, however some of the samples collected from patients in the Seekatz dataset were collected during antibiotic treatment, thus potentially limiting growth of C. difficile during those time points. Indeed, Seekatz et al. report that they were unable to retrieve C. difficile strains from all patient time points via anaerobic cultivation, generally the gold standard for C. difficile detection and diagnosis.
In a third cohort of 14 recurrent CDI patients receiving fecal microbiota transplantation from nine healthy donors (FMT; Table S2, Fig. 3), C. difficile was less frequently detected than the Seekatz and Khanna index CDI patient groups. Only 4 of 14 FMT patients had any detectable levels of C. difficile before treatment, and 3 of 14 had observations of C. difficile post-FMT. Notably, Resphera Insight detected C. difficile presence in both patients who went on to develop symptomatic CDI post-FMT (recipient IDs 005 and 006).9 Prior to FMT, all patients were treated with vancomycin (125 mg 4× per day) for at least 4 days before and the day of transplantation. Thus, we attribute the reduced detection of C. difficile in this cohort to differences in patient treatment before sampling.
Applying our approach to a longitudinal dataset of 38 premature infants in a single NICU, we identified C. difficile in two-thirds of this patient cohort. Asymptomatic carriage of C. difficile among infants has been observed to be higher than for adults, and it remains unknown whether infant cases of CDI represent true disease.29,30 While CDI testing of infants is not recommended,30 recent epidemiological studies indicate 26% of children hospitalized with CDI are infants under 12 months of age, and 5% are neonates.31 In one study of 753 pediatric patients 0 to 12 years of age, 2.9% of CDI outpatients, 4.6% of CDI inpatients, and 6.6% of healthy controls were positive for C. difficile toxin B.32 Another recent study of C. difficile in 338 healthy infants (<2 yrs) in the United Kingdom found 10% were colonized at enrollment with a toxigenic strain, and 49% became colonized with a toxigenic strain post-enrollment.33 Symptomatic Clostridium difficile infections are believed not to occur in infants due to the expected lack of specific toxin receptors and under-developed signaling pathways in the gut; however, these proposed mechanisms have not been rigorously evaluated in studies of humans.34,35,36 Multiple case studies have argued that CDI can occur in this patient population,36 and there is ongoing debate about the appropriate policy for treatment of symptomatic children who test positive for C. difficile.37,38
Our analysis of an infant case study of asymptomatic colonization during the first 18 months of life identified a reduction in C. difficile relative abundance after abrupt transition from human milk to cow’s milk. Yet in a large longitudinal study by Stoesser and colleagues, multivariate analysis demonstrated that breastfeeding (mixed with formula or exclusively) was protective against asymptomatic C. difficile colonization.33 As noted by Davis and colleagues,16 C. difficile does not carry the functional capacity for cleaving monosaccharides from oligosaccharide side chains and thus depends on the generation of monomeric glucose by other commensal members of the gut microbiome.39 Additionally, C. difficile relies on sialic acid as a carbon source for expansion made available by other commensals such as Bifidobacterium species.40 Therefore, the reduction of C. difficile after transition to cow’s milk is potentially the result not of milk source alone, but shifting microbial community composition and the presence of substrates by which C. difficile may thrive.
We were also able to identify a significant negative correlation between the abundance of C. difficile and C. scindens in one of the CDI cohorts, confirming similar trends reported by Buffie et al.15 C. scindens, a secondary bile acid producer of deoxycholic acid which has been shown to protect against CDI, may have important translational implications.13,41 New and consistent negative correlations were also identified between C. difficile and multiple species within the Blautia genus including B. faecis, B. luti, B. schinkii, and B. wexlerae. Notably, some members of the Blautia genus are known for 7α-dehydroxylating activity of primary bile acids,42,43,44 however this remains to be evaluated for the species we identified in this study. These data suggest that species other than C. scindens may provide relevant functional capabilities in the context of CDI and prove to be informative in the development of future microbial-based therapeutics. One exception to these findings was the lack of negative correlations identified within the NICU infant cohort, which can be attributed to the very limited observations of these Blautia species and C. scindens in the overall dataset (Table S3). Indeed, among the 322 NICU infant samples analyzed, only B. luti and B. wexlerae were observed at all, and only in 5 (1.6%) and 2 (0.6%) samples, respectively, which precluded their evaluation with the CCREPE method.
While microbiome profiling through 16S rRNA gene sequencing is unlikely to replace existing methods for routine diagnosis of CDI, sequence-based assessment of C. difficile levels in the context of microbiota profiling rather than presence alone may prove valuable in surveillance of C. difficile in patient populations, prediction of disease outcome, or the development of new therapies for CDI. Although our study is limited to 16S rRNA gene-based identification of C. difficile and cannot predict whether a strain produces toxin or carries a functional pathogenicity locus,45 consideration for accurate identification of C. difficile and related members may be useful in assessing clinical outcomes of new microbial therapies that rely on 16S rRNA gene sequencing to validate recovery of the microbiota.
Methods
Validation of Resphera Insight for identification of C. difficile
Whole-genome shotgun sequence datasets available from (i) The Wellcome Trust Sanger Institute and (ii) The University of Maryland Institute for Genome Sciences designated as novel C. difficile isolates were downloaded from the NCBI Sequence Read Archive (see Table S1 for accessions), trimmed for quality using Trimmomatic46 and assembled into contigs using Minia.47 Contigs containing portions of 16S rRNA genes were identified using BLASTN48 and extracted for amplicon simulations. For each isolate, we subsequently simulated 16S rRNA amplicon sequence reads (10,000 per isolate) from the V4 region (the primary amplicon region selected in the real datasets) with a random nucleotide error rate of 0.5%. The Diagnostic True Positive Rate was computed as the percentage of sequences unambiguously assigned by Resphera Insight to C. difficile.
For false positive assessment, simulated V4 sequences were generated from reference 16S rRNA genes for 22 unique species within the Clostridium XI cluster (10,000 per species, 0.5% nucleotide error rate). False positives were defined as unambiguous assignments to C. difficile.
Processing of 16S rRNA gene sequence datasets
Raw 16S rRNA gene sequence datasets were processed as follows: Raw overlapping paired-end reads were merged into consensus fragments by FLASH49 requiring a minimum 20 bp overlap with 5% maximum mismatch density, and subsequently filtered for quality (targeting error rates < 1%) and length (minimum 200 bp) using Trimmomatic46 and QIIME.50 Spurious hits to the PhiX control genome were identified using BLASTN and removed. Sequences were then trimmed of their associated primers, evaluated for chimeras with UCLUST (de novo mode),51 and screened for human-associated contaminants using Bowtie252 searches of NCBI Homo sapiens Annotation Release 106. Mitochondrial contaminants were detected and filtered using the RDP classifier53 with a confidence threshold of 50%, and passing high-quality 16S rRNA gene sequences were subsequently assigned to a high-resolution taxonomic lineage using Resphera Insight (Baltimore, MD).18,19,20,54,55 Briefly, the method relies on (i) a manually curated 16S rRNA gene database including 11,000 unique species and (ii) a hybrid global-local alignment strategy to assign sequences a species-level taxonomic lineage. While the method attempts to achieve species-level resolution, if the internal statistical model indicates uncertainty in final species membership, the tool minimizes false positives by providing “ambiguous assignments” i.e., a list of species reflecting all relevant candidates. For example, if a 16S rRNA gene fragment is ambiguous between Veillonella atypica and Veillonella dispar, the algorithm will provide the ambiguous assignment: “Veillonella_atypica:Veillonella_dispar.”
Statistical analyses
Correlations between C. difficile and other species were computed using CCREPE (v.1.10.0)24 (http://huttenhower.sph.harvard.edu/ccrepe). CCREPE (Compositionality Corrected by REnormalization and PErmutation) utilizes an N-dimensional extension of the checkerboard score particularly suited to similarity score calculations between compositions derived from ecological relative abundance measurements of co-occurrence or co-exclusion. Two sample statistical comparisons utilized the Mann-Whitney U test unless otherwise noted.
In silico evaluation for species identified in CCREPE analysis
For single species reported in CCREPE correlation analysis, we simulated noisy 16S rRNA gene sequences (V4 region; 0.5% error rate; 1000 seqs per species), and calculated the frequency of (1) assignments that included the correct species (allowing for ambiguous assignments), (2) unambiguous assignments to the correct species, and (iii) mis-assignments that did not include the correct species (Table S4).
Ethics approvals and consent to participate
IRB approval and patient consent statements from each study: Recurrent CDI (Seekatz et al.10)—All subjects signed written consent to participate in this study. This study was approved by the University of Michigan Institutional Review Board (Study HUM33286; originally approved 8/26/2009).
Index CDI (Khanna et al.21)—We prospectively recruited 88 patients (median age 52.7 years, interquartile range 36.9–65.1; 60.2% female) with their first CDI episode (from 3/2012–9/2013) as identified from the Clinical Microbiology Laboratory at Mayo Clinic, Rochester, Minnesota and collected an aliquot from the stool samples that led to the diagnosis. Clinical data including demographics, hospitalization status, concomitant medications, CDI severity, laboratory parameters, prior and concomitant antibiotic use, initial CDI treatment, treatment response and recurrent CDI were obtained by a review of the electronic medical record.
NICU Infants (Zhou et al.23)—Samples were collected following a protocol that was approved by the Partner’ s Human Research Committee (IRB) for Brigham and Women’ s Hospital. All study procedures were approved by the IRBs at both Brigham and Women’ s Hospital in Boston, MA and at The Genome Institute in St. Louis, MO. The IRB deemed this study to be of minimal risk with no interaction and no intervention with human subjects and thus, was exempt from consent.
Infant Longitudinal (Davis et al.16)—The study was approved by the TechLab Institutional Review Board and included informed consent obtained from the mother.
FMT (Seekatz et al.9)—Informed consent was received from all participants under an approved Institutional Review Board (IRB) protocol at Essentia Health Duluth Clinic (IRB no. SMDC-09068; principal investigator, Timothy Rubin, FDA Investigational New Drug [IND] no. 15460).
Healthy Controls (Seekatz et al. submitted)—All subjects signed written consent to participate in this study. This study was approved by the University of Michigan Institutional Review Board (Study HUM33286; originally approved 8/26/2009).
Data availability
NCBI BioProject accessions of publicly available 16S rRNA gene sequence datasets used in this study: PRJNA307992, PRJNA342347, PRJNA264177, PRJNA331150, PRJNA238042, and PRJNA386260 (Table S2).
References
Leffler, D. A. & Lamont, J. T. Clostridium difficile Infection. N. Engl. J. Med. 373, 287–288 (2015).
Lessa, F. C. et al. Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 372, 825–834 (2015).
Surawicz, C. M. et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 108, 478–498 (2013).
Pepin, J. et al. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin. Infect. Dis. 40, 1591–1597 (2005).
Vincent, Y., Manji, A., Gregory-Miller, K. & Lee, C. A review of management of Clostridium difficile Infection: primary and recurrence. Antibiotics 4, 411–423 (2015).
Seekatz, A. M. & Young, V. B. Clostridium difficile and the microbiota. J. Clin. Invest. 124, 4182–4189 (2014).
Kelly, C. R. et al. Update on fecal microbiota transplantation 2015: indications, methodologies, mechanisms, and outlook. Gastroenterology 149, 223–237 (2015).
Lynch, S. V. & Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369–2379 (2016).
Seekatz, A. M. et al. Recovery of the gut microbiome following fecal microbiota transplantation. MBio 5, e00893–00814 (2014).
Seekatz, A. M., Rao, K., Santhosh, K. & Young, V. B. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 8, 47 (2016).
Seekatz, A. M. et al. Fecal microbiota transplantation eliminates Clostridium difficile in a murine model of relapsing disease. Infect. Immun. 83, 3838–3846 (2015).
Zackular, J. P. et al. Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection. Nat. Med. 22, 1330–1334 (2016).
Theriot, C. M. et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 5, 3114 (2014).
Weingarden, A. R. et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G310–G319 (2014).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208 (2015).
Davis, M. Y., Zhang, H., Brannan, L. E., Carman, R. J. & Boone, J. H. Rapid change of fecal microbiome and disappearance of Clostridium difficile in a colonized infant after transition from breast milk to cow milk. Microbiome 4, 53 (2016).
Schubert, A. M., Sinani, H. & Schloss, P. D. Antibiotic-induced alterations of the murine gut microbiota and subsequent effects on colonization resistance against Clostridium difficile. MBio 6, e00974 (2015).
Ottesen, A. et al. Enrichment dynamics of Listeria monocytogenes and the associated microbiome from naturally contaminated ice cream linked to a listeriosis outbreak. BMC Microbiol. 16, 275 (2016).
Daquigan, N., Grim, C. J., White, J. R., Hanes, D. E. & Jarvis, K. G. Early recovery of Salmonella from food using a 6-hour non-selective pre-enrichment and reformulation of tetrathionate broth. Front. Microbiol. 7, 2103 (2016).
Grim, C. J. et al. High-resolution microbiome profiling for detection and tracking of Salmonella enterica. Front. Microbiol. 8, 1587 (2017).
Khanna, S. et al. Gut microbiome predictors of treatment response and recurrence in primary Clostridium difficile infection. Aliment. Pharmacol. Ther. 44, 715–727 (2016).
Cohen, S. H. et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 31, 431–455 (2010).
Zhou, Y. et al. Longitudinal analysis of the premature infant intestinal microbiome prior to necrotizing enterocolitis: a case-control study. PLoS One 10, e0118632 (2015).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell. Host. Microbe 15, 382–392 (2014).
Furuya-Kanamori, L. et al. Asymptomatic Clostridium difficile colonization: epidemiology and clinical implications. BMC Infect. Dis. 15, 516 (2015).
McNamara, S. E. et al. Carriage of Clostridium difficile and other enteric pathogens among a 4-H avocational cohort. Zoonoses Public Health 58, 192–199 (2011).
Miyajima, F. et al. Characterisation and carriage ratio of Clostridium difficile strains isolated from a community-dwelling elderly population in the United Kingdom. PLoS One 6, e22804 (2011).
Ozaki, E. et al. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. J. Med. Microbiol. 53, 167–172 (2004).
Rousseau, C. et al. Clostridium difficile carriage in healthy infants in the community: a potential reservoir for pathogenic strains. Clin. Infect. Dis. 55, 1209–1215 (2012).
Schutze, G. E. & Willoughby, R. E. Committee on infectious diseases and American academy of pediatrics. Clostridium difficile infection in infants and children. Pediatrics 131, 196–200 (2013).
Kim, J. et al. Epidemiological features of Clostridium difficile-associated disease among inpatients at children’s hospitals in the United States, 2001–2006. Pediatrics 122, 1266–1270 (2008).
Cerquetti, M., Luzzi, I., Caprioli, A., Sebastianelli, A. & Mastrantonio, P. Role of Clostridium difficile in childhood diarrhea. Pediatr. Infect. Dis. J. 14, 598–603 (1995).
Stoesser, N. et al. Epidemiology of Clostridium difficile in infants in Oxfordshire, UK: Risk factors for colonization and carriage, and genetic overlap with regional C. difficile infection strains. PLoS One 12, e0182307 (2017).
Chang, T. W., Sullivan, N. M. & Wilkins, T. D. Insusceptibility of fetal intestinal mucosa and fetal cells to Clostridium difficile toxins. Zhongguo Yao Li Xue Bao 7, 448–453 (1986).
Eglow, R. et al. Diminished Clostridium difficile toxin A sensitivity in newborn rabbit ileum is associated with decreased toxin A receptor. J. Clin. Invest. 90, 822–829 (1992).
Kuiper, G. A. et al. Clostridium difficile infections in young infants: case presentations and literature review. IDCases 10, 7–11 (2017).
Nicholson, M. R., Thomsen, I. P. & Edwards, K. M. Controversies surrounding Clostridium difficile infection ininfants and young children. Children. 1, 40–47 (2014).
El Feghaly, R. E., Stauber, J. L., Tarr, P. I. & Haslam, D. B. Intestinal inflammatory biomarkers and outcome in pediatric Clostridium difficile infections. J. Pediatr. 163, 1697–1704 (2013).
Wilson, K. H. & Perini, F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect. Immun. 56, 2610–2614 (1988).
Baumler, A. J. & Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85–93 (2016).
Greathouse, K. L., Harris, C. C. & Bultman, S. J. Dysfunctional families: Clostridium scindens and secondary bile acids inhibit the growth of Clostridium difficile. Cell. Metab. 21, 9–10 (2015).
Ridlon, J. M., Alves, J. M., Hylemon, P. B. & Bajaj, J. S. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes 4, 382–387 (2013).
Kakiyama, G. et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 58, 949–955 (2013).
Theriot, C. M., Bowman, A. A. & Young, V. B. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere 1, e00045-15 (2016).
Natarajan, M., Walk, S. T., Young, V. B. & Aronoff, D. M. A clinical and epidemiological review of non-toxigenic Clostridium difficile. Anaerobe 22, 1–5 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Chikhi, R. & Rizk, G. Space-efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms Mol. Biol. 8, 22 (2013).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336 (2010).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Abernethy, M. G. et al. Urinary microbiome and cytokine levels in women with interstitial cystitis. Obstet. Gynecol. 129, 500–506 (2017).
Guerrero-Preston, R. et al. High-resolution microbiome profiling uncovers Fusobacterium nucleatum, Lactobacillus gasseri/johnsonii, and Lactobacillus vaginalis associated to oral and oropharyngeal cancer in saliva from HPV positive and HPV negative patients treated with surgery and chemo-radiation. Oncotarget. https://doi.org/10.18632/oncotarget.20677 (2017).
Acknowledgements
We thank Cynthia Sears, Karen Carroll, and David Cook for helpful suggestions on this work. This work was supported in part by the ERIN CRC (Enteric Research Investigative Network Cooperative Research Center), (U19AI09087, NIAID), awarded to V.B.Y. A.M.S. supported by the National Center for Advancing Translational Sciences (UL1TR000433).
Author information
Authors and Affiliations
Contributions
A.M.S. and J.R.W. conceived the study. A.M.S. and J.R.W. designed and performed the experiments. N.D., A.M.S., V.B.Y., and J.R.W. interpreted the data. All authors wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.R.W. has an ownership interest in Resphera Biosciences. N.D. is a consultant for Resphera Biosciences. The remaining authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Daquigan, N., Seekatz, A.M., Greathouse, K.L. et al. High-resolution profiling of the gut microbiome reveals the extent of Clostridium difficile burden. npj Biofilms Microbiomes 3, 35 (2017). https://doi.org/10.1038/s41522-017-0043-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-017-0043-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
