
Abstract
Deleterious germline variants in the BRCA1-associated ring domain (BARD1) gene moderately elevate breast cancer risk; however, their potential association with other neoplasms remains unclear. Here, we present the case of a 43-year-old female patient diagnosed with sigmoid colon adenocarcinoma whose maternal family members met the Amsterdam Criteria II for Lynch syndrome. Comprehensive multigene panel testing revealed a heterozygous BARD1 exon 3 deletion.
Similar content being viewed by others

The full-length BRCA1-associated ring domain 1 (BARD1) protein contains an N-terminal RING domain and C-terminal tandem BRCT repeats, similar to BRCA11. BARD1 and BRCA1 form a heterodimer through the interaction of their RING-finger domains; this heterodimer plays crucial roles in several tumor-suppressive functions related to DNA repair and apoptosis. BARD1 has emerged as a moderate-risk gene for hereditary breast cancer (BC), particularly triple-negative BC (TNBC)2,3. Pathogenic/likely pathogenic (P/LP) BARD1 variant carriers have a 17–30% lifetime risk of BC, prompting the National Comprehensive Cancer Network (NCCN) guidelines to recommend annual mammograms starting at age 40, along with annual breast magnetic resonance imaging (MRI)4. The recommended timing of MRI for P/LP BARD1 variant carriers depends on additional risk factors, including age, family history, breast density, and patient preference. Risk-reducing mastectomy is not recommended for P/LP BARD1 variant carriers but may be considered depending on family history4. However, the association between P/LP BARD1 variants and an increased risk of other cancers, including colorectal cancer (CRC), remains unclear. Despite the increasing number of P/LP BARD1 variants identified in families with CRC aggregation, the data are limited, and no definitive associations have been established5,6. Recently, a germline heterozygous deletion of BARD1 exons 8–11 was reported in a family diagnosed with familial colorectal cancer type X syndrome, meeting the Revised Amsterdam Criteria (Amsterdam Criteria II) for Lynch syndrome (LS) without any P/LP variants in the DNA mismatch repair (MMR) genes7.
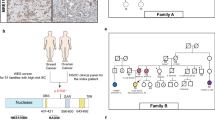
We present the case of a 42-year-old Japanese female (III-4; Fig. 1) who underwent a medical assessment following the detection of fecal occult blood during a medical check-up. Subsequent colonoscopy revealed a tumor in the sigmoid colon. The pathological diagnosis of the biopsy specimen was compatible with colon adenocarcinoma. She was referred to our hospital for a colonoscopy and contrast-enhanced whole-body computed tomography scan, which led to a diagnosis of sigmoid colon cancer (cT2N1aM0; TNM clinical staging, cStage IIIa)8. The patient underwent laparoscopic high anterior resection, and pathological examination revealed a well-differentiated adenocarcinoma (tub1 > 2; pT2N0M0; TNM pathological staging, pStage I)8. Universal MMR deficiency (dMMR) screening by immunohistochemistry (IHC) for MMR and BRAF V600E proteins in resected tumor samples revealed significant downregulation of the MLH1 and PMS2 proteins, indicating dMMR, while BRAF V600E was negative. Notably, the proband had a family history of CRC (Fig. 1A). Her grandmother (I-4, deceased in her 70s) and mother (II-12, diagnosed in her 40s and again at age 65) were both affected by CRC. As a result, this family fulfilled the Amsterdam Criteria II for LS9. Contrast-enhanced MRI revealed a small, lobulated mass in the right breast of the proband, but histopathological examination of the biopsy specimens revealed no evidence of proliferative changes.

The arrow indicates the proband (P). The filled symbols indicate individuals affected by colorectal cancer (CRC).
The patient was referred to our clinical genetics department for hereditary tumor risk assessment because her personal and family history met the Amsterdam Criteria II for LS, and the dMMR screening results were positive. A complete physical examination revealed no phenotypic features suggestive of any specific syndrome. For a definitive diagnosis of hereditary CRC, we proposed several commercially available genetic tests, including multigene panel testing (MGPT). Following pretest genetic counseling and obtaining informed consent, the patient opted for and underwent MGPT with an 84-gene hereditary cancer panel via next-generation sequencing (NGS; Invitae Multi-Cancer Panel, https://www.invitae.com/en/providers/test-catalog/test-01101) to evaluate as many CRC predisposition genes as possible. Pretest genetic counseling primarily addressed the clinically suspected genes while also discussing the potential incidental findings within the panel. The Invitae 84-gene panel was generated using Illumina NGS technology. The genes were targeted and sequenced via a custom short-read NGS assay in which genomic DNA was extracted from blood with beads designed to capture exons, 20 bases of the flanking introns, and certain noncoding regions as previously described10. A bioinformatics pipeline was used to align the sequencing reads, and community standard and custom algorithms were used to identify single-nucleotide variants, small insertions or deletions (indels), large indels, structural variants, and exon-level copy number variants (CNVs)10,11,12. The identified variants were interpreted using Sherloc13, a proprietary, point-based framework based on the joint consensus guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP)14.
No P/LP variants in established CRC susceptibility genes, including MMR genes, were detected in the proband, suggesting that the MLH1 protein expression found in our universal dMMR screening was downregulated by acquired epigenetic mechanisms such as MLH1 promoter hypermethylation. An unexpected heterozygous out-of-frame deletion of BARD1 exon 3 (NM_000465.4) was initially detected in the 84-gene MGPT (Fig. 2A) and subsequently confirmed using exon-focused array comparative genomic hybridization (aCGH). To our knowledge, this CNV of BARD1, NC_000002.12 (NM_000465.4):c.(215 + 1_216-1)_(364 + 1_347-1)del, has never been reported in disease-related databases, including the Human Gene Mutation Database (HGMD, https://my.qiagendigitalinsights.com/bbp/view/hgmd/pro/start.php); ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); or population databases, including 8.3KJPN-SV (https://jmorp.megabank.tohoku.ac.jp/) and gnomAD SVs v2.1 (https://gnomad.broadinstitute.org/). This CNV is predicted to generate an out-of-frame transcript, possibly leading to a premature termination codon and causing a loss of function event, so-called nonsense-mediated mRNA decay (NMD). The deletion of exon 3 also causes the deletion of the RING domain of the protein, which is essential for BRCA1-BARD1 heterodimer formation and E3 ubiquitin ligase activity, as well as for the stability of BRCA1 (Fig. 2B). Therefore, the functional consequences of this variant are expected to be severe, even if NMD is not caused. Segregation analysis could not be performed for all affected and unaffected relatives in this case. According to the ACMG/AMP criteria14, this CNV was classified as LP (PVS1 and PM2). Although BARD1 deletions have been reported in patients diagnosed with various cancers, including CRC (Supplementary Table S1), the germline deletion of BARD1 exon 3 represents a novel, previously unreported pathogenic variant.

A The results of the copy number analysis of BARD1 detected by the 84-gene MGPT. The red arrow indicates the deletion of BARD1 exon 3. B Structure of BARD1. Exon structure, coding exons (gray area), and domain composition of full-length BARD1 based on NM_000465.4 (green arrow) and NP_000456.2 (red arrow), with scales at the nucleotide (bp) and amino acid (aa) levels. BARD1 has an N-terminal RING-finger domain (RING), three centrally located ankyrin repeats (ANK), and two C-terminal BRCT (BRCA1 C-terminus) domains (BRCT1 and BRCT2).
While the full-length BARD1 protein participates in both BRCA1-dependent and BRCA1-independent tumor-suppressive pathways, multiple exon-skipping BARD1 isoforms, many of which have agonistic cancer susceptibility potential, have been reported15. To date, ~19 BARD1 isoforms have been identified, all of which are expressed in colon and CRC tissues15,16. Since some isoforms lack exon 3, the allele lacking exon 3 may express isoforms without specific exons, including exon 3, instead of causing NMD. Notably, several BARD1 isoforms lacking exon 3, such as BARD1β (lacking exons 2 and 3) and BARD1δ (lacking exons 2–6), antagonize full-length BARD1 and confer cancer susceptibility and oncogenicity15,17. Consequently, in the present study, the allele lacking full-length BARD1 expression due to the exon 3 deletion was likely to cause the loss or antagonism of tumor-suppressive functions, even in the presence of expressed isoforms. Because somatic pathogenic variants occurring as a second hit in the intact BARD1 allele, homologous recombination deficiency status, and the mRNA-level expression status of each isoform from each BARD1 allele were not analyzed in the tumor tissues from this patient, it is unclear whether the detected germline BARD1 variant contributed to the pathogenesis of CRC or was just an incidental alteration; if the former is true, how this variant may cause CRC is also unknown. Additional research-based analyses of tumors would be useful to clarify the involvement of BARD1 dysfunction in the pathogenesis of CRC in the present case.
Only a few CRC cases with germline BARD1 deleterious variants have been reported6,7,18,19. Of the four reported CRC cases, three had variants predicted to cause exon skipping: exon 3 deletion (this case), exons 8–11 deletion7, and NM_000465.3:c.1811-2A>G6. Although these three variants were observed in families with CRC aggregation, only two patients, including this patient, with gross BARD1 deletions met the Amsterdam Criteria II for LS7. Larger population-based studies are needed to validate the potential association between pathogenic BARD1 variants and elevated CRC risk7. Furthermore, future studies demonstrating the cosegregation of gross BARD1 deletions with the phenotype in large, affected families would be valuable in establishing an unequivocal causal link between this type of BARD1 variant and CRC aggregation. Currently, it is not feasible to assess CRC risk in at-risk family members based solely on the presence or absence of the BARD1 exonic deletion. However, if a causal relationship is established in the future, CRC risk could be estimated according to the deletion status of this gene.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3371.
References
Wu, L. C. et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 14, 430–440 (1996).
Breast Cancer Association Consortium et al. Breast cancer risk genes—association analysis in more than 113,000 women. New Engl. J. Med. 384, 428–439 (2021).
Hu, C. et al. A population-based study of genes previously implicated in breast cancer. New Engl. J. Med. 384, 440–451 (2021).
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology on Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic version:2.2024. Available at https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (2023).
Yurgelun, M. B. et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 149, 604–613.e20 (2015).
Esteban-Jurado, C. et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet Med. 17, 131–142 (2015).
Carrera, S. et al. Germline heterozygous exons 8-11 pathogenic BARD1 gene deletion reported for the first time in a family with suspicion of a hereditary colorectal cancer syndrome: more than an incidental finding? Hered. Cancer Clin. Pr. 21, 2 (2023).
Brierley, J. D., Gospodarowicz, M. K. & Wittekind, C. (eds) The TNM Classification of Malignant Tumours, 8th edn. (Wiley Blackwell, Oxford, 2017).
Vasen, H. F., Watson, P., Mecklin, J. P. & Lynch, H. T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116, 1453–1456 (1999).
Lincoln, S. E. et al. One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet. Med. 23, 1673–1680 (2021).
Truty, R. et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet. Med. 21, 114–123 (2019).
McKnight, D. et al. Multigene panel testing in a large cohort of adults with epilepsy: diagnostic yield and clinically actionable genetic findings. Neurol. Genet. 8, e650 (2021).
Nykamp, K. et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 19, 1105–1117 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Hawsawi, Y. M. et al. BARD1 mystery: tumor suppressors are cancer susceptibility genes. BMC Cancer 22, 599 (2022).
Sporn, J. C., Hothorn, T. & Jung, B. BARD1 expression predicts outcome in colon cancer. Clin. Cancer Res. 17, 5451–5462 (2011).
Irminger-Finger, I., Ratajska, M. & Pilyugin, M. New concepts on BARD1: regulator of BRCA pathways and beyond. Int J. Biochem. Cell Biol. 72, 1–17 (2016).
Yurgelun, M. B. et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J. Clin. Oncol. 35, 1086–1095 (2017).
Gong, R. et al. Mutation spectrum of germline cancer susceptibility genes among unselected Chinese colorectal cancer patients. Cancer Manag. Res. 11, 3721–3739 (2019).
Acknowledgements
The authors thank the patients who participated in this study. This work was supported in part by the Aichi Cancer Research Foundation (N. Takaiso) and the Japan Agency for Medical Research and Development (AMED; grant numbers JP22kk0305020 and JP23ck0106872 (I. Imoto)).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This study was approved by the Institutional Review Board of the Aichi Cancer Center (No. S05006). Informed consent was obtained from the patient for publication of the case details as well as genetic and genomic findings.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takaiso, N., Imoto, I., Yoshimura, A. et al. BARD1 deletion in a patient with suspected hereditary colorectal cancer. Hum Genome Var 11, 11 (2024). https://doi.org/10.1038/s41439-024-00267-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-024-00267-y
