
Abstract
Congenital contractual arachnodactyly (CCA) is a genetic connective tissue disorder that is characterized by arachnodactyly, kyphoscoliosis, marfanoid habitus, and crumpled ears. We report a case of a boy with suspected Marfan syndrome. Genetic analysis revealed c.3207_3217+9del in a heterozygote form of the fibrillin-2 (FBN2) gene. This patient was diagnosed with CCA based on his phenotype, and the pathogenicity of this variant was classified according to cDNA analysis and protein modeling.
Similar content being viewed by others
Congenital contractual arachnodactyly (CCA) is a genetic connective tissue disorder. Patients with CCA tend to have a distinctive Marfan-like appearance and present with joint contractures, arachnodactyly, crumpled ears, and kyphoscoliosis1. Early diagnosis and follow-up of this disorder ensure that patients can refrain from physical contact sports or activities and LASIK eye surgery2. In particular, it is important to differentiate CCA from Marfan syndrome (MFS), which has cardiovascular involvement1. CCA is caused by pathogenic variants in the FBN2 (OMIM#121050) gene, which is located on chromosome 5q23.3. It encodes fibrillin-2, which is a large extracellular matrix protein that multimerizes into linear structures called microfibrils. These calcium-binding structures have intrinsic elasticity and can associate with elastin to form elastic fibers. Microfibrils provide support for both elastic and nonelastic connective tissues, are involved in cell-matrix communication, and contribute to extracellular growth factor sequestration and regulation. Fibrillin-2 is homologous to fibrillin-1, which is encoded by FBN1 (of which pathogenic variants cause MFS), but it is mainly expressed during early embryonic development. Notably, the disease-associated variants in FBN2 are located in the central region of the gene (exons 24–35), which encode for a central stretch of calcium-binding epidermal growth factor-like (cbEGF-like) domains3.
We examined a case of a 14-year-old boy with a height of 177.4 cm, an arm span of 179.5 cm, and a weight of 51.9 kg, with long slim limbs, long spider-like fingers, and scoliosis. He had a positive wrist sign and a positive thumb sign on his left hand. No ectopia lentis or myopia had been observed thus far, although the patient was suspected to have MFS. His heartbeat was normal, and he did not have the typical abnormally crumpled ears. Informed consent for molecular analysis was obtained from the patient, and this study was approved by the Ethics Committee of Gifu University Hospital (approval reference number 2018-181). A gene panel analysis that targeted connective tissue diseases using next-generation sequencing was performed at the Kazusa DNA Research Institute. The target genes were FBN1, FBN2, TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, ACTA2, MYH11, MYLK, COL3A1, EFEMP2, FLNA, and SLC2A10. Genomic DNA was extracted from the leukocytes of the patient’s peripheral blood. Total RNA was isolated from the patient’s fresh blood sample and a fibroblast sample (Kurabo, Osaka, Japan) as a healthy control using an Isogen kit (Nippon Gene, Tokyo, Japan). Sanger sequencing was performed using the 5′-GCATCTTGCCCACCAGTACA-3′ (forward) and 5′-CATCTCCGGTTGCAGAGGAA-3′ (reverse) primers. Reverse transcription was performed using the FBN2-specific antisense primers Ex24RTr (5′-AAGAACATCCCCTCGGTTAGC-3′), Ex26RTr (5′-GAGGTCAGGAGAAATCCTGC-3′), and an oligo (dT) primer (Thermo Fisher Scientific, Waltham, MA, USA). For the amplification of cDNA, a forward primer for exon 21 (5′-CTGAAATCTGAATGCTGTGCC-3′) and a reverse primer for exon 26 (5′-GAGGTCAGGAGAAATCCTGC-3′) were used. Conservation analysis of the variant was performed in Mus musculus (house mouse) NM_010181.2, Rattus norvegicus (Norway rat) NM_031826.2, Bos taurus (cattle) NM_001278588.1, Canis lupus familiaris (dog) XM_038681416.1, and Pan troglodytes (chimpanzee) XM_016953727.2. SpliceAI was used for the mutation prediction. For the fibrillin-2 protein, the structures of the wild-type and variant were predicted using the SWISS-MODEL automated protein homology modeling server (http://swissmodel.expasy.org).
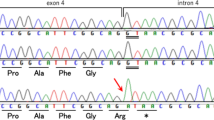
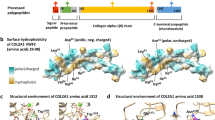
Genetic analysis of the patient’s sample revealed a novel heterozygous deletion of FBN2 genomic description NC_000005.10:g.128345351_128345370del. This variant is not present in population databases (gnomAD and jMorp). We referred to ClinVar_20230820 to determine whether the variant identified in the patient was novel. To the best of our knowledge, this variant has not yet been reported as a responsible mutation for CCA in the Japanese population or elsewhere. In addition, this variant was confirmed by Sanger sequencing (data not shown). We performed cDNA analysis because the patient’s genomic variant included a splicing site and in silico data indicated the predominant splice donor loss within his transcripts in SpliceAI. The patient’s RNA revealed two bands: the wild-type and a fragment that was shorter than the control (Fig. 1a). Although we did not check the relative expression levels of these two PCR products, the shorter band appears to be more abundant. Direct sequencing of the shorter PCR fragment from the patient showed a deletion (108 bp) at the end of exon 24, which resulted in an r.3110_3217 deletion (Fig. 1b). This is predicted to cause the deletion of 36 amino acids and substitutions in the FBN2 protein (p.(Cys1037_Lys1073delinsTyr)) but preserves the integrity of the reading frame (Supplementary Fig. 1). This large deletion of amino acids in fibrillin-2, including an antiparallel β-sheet, may cause structural protein folding instability in the model (Fig. 2) and the loss of the RGD (arginine-glycine-aspartic acid) sequence. Because RGD is a common sequence for integrin binding, which is important for cell adhesion4, the variant is predicted to decrease cell adhesion ability. Moreover, this variant is present in the TB3-cbEGF11 domain, where another pathogenic missense mutation, p.Gly1057Asp, was previously reported5. This variant is also located in a hotspot for most CCA-causing mutations3. The amino acid sequence of this variant position is highly conserved among species, which indicates that this region may play an important role in fibrillin-2 (Supplementary Fig. 2). There is possible for other forms of FBN2 transcripts in the patient because we performed RT-PCR in only exons 21–26, which can be considered as a limitation of our analysis. His unaffected parents did not approve genetic testing for themselves. Therefore, this variant was not confirmed to be de novo. The variant was classified as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines6. The pathogenicity criteria fulfilled the PM1 (pathogenic, moderate), PM2 (pathogenic, moderate), and PM4 (pathogenic, moderate) categories.

a Agarose gel electrophoresis of the RT-PCR product, the arrow indicates the shorter band of the patient sample. Transcripts from the control fibroblasts showed only the predicted wild-type amplicon size of 630 bp. b Schematic representation of the abnormal FBN2 transcripts. The line indicates the partial sequencing of the exon 24 exon 25 boundary of the mutant allele cDNA. The cDNA sequencing of the aberrant amplification band revealed a truncated exon 24.

Modeled structures of the TB3-cbRGF11 domains of the FBN2 protein are illustrated in the wild-type and variant forms. TB3 is colored yellow, cbEGF11 is colored blue, and the RGD sequence is colored red.
To conclude, the present study reports a novel heterozygous non-canonical FBN2 variant, c.3207_3217+9del, which was identified in a Japanese patient with CCA. Considering all the factors together, including the patient phenotype, transcript alterations, and protein modeling, the variant was suggested as a pathogenic variant that resulted in CCA.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3362.
Data availability
The data described in this report are available upon request. Supplementary Information is available online through the Human Genome Variation.
Materials availability
The materials described in this report are available upon request. Supplementary Information is available online through the Human Genome Variation.
References
Viljoen, D. Congenital contractural arachnodactyly (Beals syndrome). J. Med. Genet. 31, 640–643 (1994).
Callewaert, B. Congenital Contractural Arachnodactyly (University of Washington, Seattle, 2023).
Zhou, S. et al. A novel FBN2 mutation cosegregates with congenital contractural arachnodactyly in a five-generation Chinese family. Clin. Case Rep. 6, 1612–1617 (2018).
Jovanovic, J., Iqbal, S., Jensen, S., Mardon, H. & Handford, P. Fibrillin-integrin interactions in health and disease. Biochem Soc. Trans. 36, 257–262 (2008).
Park, E. S., Putnam, E. A., Chitayat, D., Child, A. & Milewicz, D. M. Clustering of FBN2 mutations in patients with congenital contractural arachnodactyly indicates an important role of the domains encoded by exons 24 through 34 during human development. Am. J. Med. Genet. 78, 350–355 (1998).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet. Med. 17, 405–424 (2015).
Acknowledgements
We sincerely thank the patient for his participation. We thank Sienna F. from Enago (Crimson Interactive Pvt. Ltd.) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nakama, M., Miwa, Y., Manabe, S. et al. Novel variant of FBN2 in a patient with congenital contractual arachnodactyly. Hum Genome Var 11, 7 (2024). https://doi.org/10.1038/s41439-024-00264-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-024-00264-1

{kind=link}
{kind=link}