
Abstract
Otopalatodigital spectrum disorder (OPDSD) is characterized by variable phenotypes, including skeletal dysplasia, and is caused by pathogenic variants in filamin A-encoding FLNA. FLNA variants associated with lethal OPDSD primarily alter the CH2 subdomain of the ABD of FLNA. Herein, we report a novel FLNA mutation in a fetus with severe skeletal dysplasia in a pregnant multigravida female with a history of repeated miscarriages and terminations.
Similar content being viewed by others

Otopalatodigital spectrum disorder (OPDSD) is an X-linked inherited condition caused by pathogenic variants in filamin A-encoding FLNA. The X-linked OPDSD, characterized by skeletal malformations, includes otopalatodigital syndrome type 1 (OPD1) and type 2 (OPD2), frontometaphyseal dysplasia type 1, Melnick-Needles syndrome and terminal osseous dysplasia with pigmentary skin defects. In OPD2, most affected males die in utero or during early life owing to heart failure and coagulopathy1. Patients with OPDSD have variable skeletal dysplasia accompanied by brain malformations, cleft palate, cardiac anomalies, omphalocele, and obstructive uropathy2. Owing to the variable manifestations of OPDSD, a definitive diagnosis may often be delayed.
FLNA, previously known as the actin-binding 280 kDa protein, contains an N-terminal actin-binding domain (ABD) composed of two calponin homology domains, CH1 and CH2, and two ROD domains followed by 24 Ig repeats3,4. FLNA plays a critical role in cellular motility and signaling by cross-linking actin filaments, tethering membrane glycoproteins, and serving as a scaffold for signaling intermediates and has been associated with a wide spectrum of conditions5. Considering the overlapping phenotypes, the location of the FLNA mutation is helpful to refine the diagnosis. Pathogenic variants leading to OPD2, which is classified as a severe condition among OPDSDs, are primarily localized in CH2 and result in gain-of-function mutations that increase the actin-binding affinity6,7.
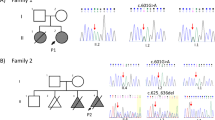
A 20-week fetus with severe skeletal dysplasia was terminated. The 38-year-old multigravida female who carried the affected fetus had a history of two abortions and two first-trimester miscarriages (Fig. 1a). Her second fetus had shortened limbs and a giant bladder (Fig. 1a, II-2). During this pregnancy, antenatal ultrasound examination revealed ventriculomegaly, shortening of long bones, abnormal digits, cleft palate, low-set ears, omphalocele, imperforate anus, and bilateral hydronephrosis at 16 weeks, leading to a strong suspicion of lethal skeletal dysplasia.

a The patient’s family pedigree. The shaded line represents the case with skeletal dysplasia. Dotted circles represent heterozygous females. b Sanger sequencing of wild-type individuals (I-1, II-3), heterozygous females (Patient I-2), and a hemizygous male (Patient II-6).
Under approval from the Ethics Committee of the National Center for Children Health and Development, Tokyo, Japan (IRB number: 926), we sequenced the genomic DNA of I-1, I-2, II-3, and II-6 (Fig. 1a). Whole-exome libraries were prepared using the Agilent SureSelect v6 Capture Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions. The HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA) was used to perform 150-bp paired-end sequencing. Variant frequencies were obtained from the 1000 Genomes Project database (http://www.internationalgenome.org), Human Genome Variation Database (HGVD; http://www.hgvd.genome.med.kyoto-u.ac.jp), and Tohoku Medical Megabank Organization (ToMMo; https://www.megabank.tohoku.ac.jp). After the selection of variants from the database, the associated phenotypes were obtained from the OMIM database (#305620, #309350, #311300, #304120) for comparison with the actual phenotypes.
Whole-exome sequencing detected a novel hemizygous FLNA variant [c.583G > A (p.G195S)] in the region encoding the CH2 subdomain of the ABD of FLNA. This variant has not been registered in variant databases in general populations or reported in individuals with FLNA-related conditions. However, it is predicted to be damaging by the protein function prediction software SIFT (score 0: damaging), PolyPhen-2 (score 1: deleterious), and CADD (score 34). The pathogenic variant-harboring domain is highly conserved, and the substitution of glycine with serine causes a significant physicochemical difference. We assessed the variant according to the ACMG guidelines and classified it as a variant of uncertain significance based on the criteria PM2 (evidence in population database), PP3 (computational evidence), and PP4 (phenotype specific for gene). This potentially deleterious variant was verified by Sanger sequencing, and it was assumed that it had been inherited by the fetus from the mother, who is a heterozygous carrier of this variant (Fig. 1b).
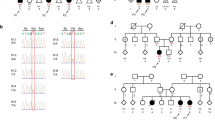
Most FLNA variants that lead to OPD2 development have been mapped to the region encoding the CH2 subdomain of the ABD of FLNA (Fig. 2a)7,8. Previous reports showed that OPDSD patients with severe phenotypes associated with lethal, prenatal, and neonatal death often have FLNA missense mutations in the region encoding this subdomain (Fig. 2b)1,8,9,10,11,12,13,14,15,16. Since only a few functional studies on lethal FLNA variants have been conducted, the pathophysiology of OPDSD should be elucidated using a large number of cases. The novel mutation in FLNA in the observed patient might be responsible for their severe phenotype. Therefore, it is assumed that the previous miscarriages were caused by the FLNA variant, which is often lethal in males, as FLNA plays a critical role in human embryonic development17. Early diagnosis will provide prospective parents with appropriate genetic counseling services regarding future pregnancies.

a Schematic representation of the missense FLNA variants in males in the region encoding the concerned protein domains. The red arrowhead indicates the case, and the black arrowhead indicates the lethal outcomes. b Clinical characterization and pathogenesis of the FLNA variants identified in nonsurviving males.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3249.
References
Feng, Y. et al. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl Acad. Sci. USA 103, 19836–19841 (2006).
Naudion, S. et al. Fetal phenotypes in otopalatodigital spectrum disorders. Clin. Genet. 89, 371–377 (2016).
Falet, H. New insights into the versatile roles of platelet FlnA. Platelets 24, 1–5 (2013).
Rosa, J. P., Raslova, H. & Bryckaert, M. Filamin A: key actor in platelet biology. Blood 134, 1279–1288 (2019).
Bandaru, S., Ala, C., Zhou, A. X. & Akyürek, L. M. Filamin A regulates cardiovascular remodeling. Int. J. Mol. Sci. 22, 6555 (2021).
Clark, A. R., Sawyer, G. M., Robertson, S. P. & Sutherland-Smith, A. J. Skeletal dysplasias due to filamin A mutations result from a gain-of-function mechanism distinct from allelic neurological disorders. Hum. Mol. Genet. 18, 4791–4800 (2009).
Cannaerts, E. et al. FLNA mutations in surviving males presenting with connective tissue findings: two new case reports and review of the literature. BMC Med. Genet. 19, 140 (2018).
Robertson, S. P. et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat. Genet. 33, 487–491 (2003).
André, M., Vigneron, J. & Didier, F. Abnormal facies, cleft palate, and generalized dysostosis: a lethal X-linked syndrome. J. Pediatr. 98, 747–752 (1981).
Sankararaman, S. et al. Otopalatodigital syndrome type 2 in a male infant: a case report with a novel sequence variation. J. Pediatr. Genet. 2, 33–36 (2013).
Joksic, I. et al. Otopalatodigital syndrome Type I: novel Characteristics and prenatal Manifestations in two Siblings. Balk. J. Med. Genet. 22, 83–88 (2019).
Mariño-Enríquez, A., Lapunzina, P., Robertson, S. P. & Rodríguez, J. I. et al. Otopalatodigital syndrome type 2 in two siblings with a novel filamin A 629G>T mutation: Clinical, pathological, and molecular findings. Am. J. Med. Genet. 143A, 1120–1125 (2007).
Savarirayan, R. et al. Oto-palato-digital syndrome, type II: report of three cases with further delineation of the chondro-osseous morphology. Am. J. Med. Genet. 95, 193–200 (2000).
Robertson, S., Gunn, T., Allen, B., Chapman, C. & Becroft, D. Are Melnick-Needles syndrome and oto-palato-digital syndrome type II allelic? Observations in a four-generation kindred. Am. J. Med. Genet. 71, 341–347 (1997).
Young, K., Barth, C. K., Moore, C. & Weaver, D. D. Otopalatodigital syndrome type II associated with omphalocele: report of three cases. Am. J. Med. Genet. 45, 481–347 (1993).
Robertson, S. P. et al. Frontometaphyseal dysplasia: mutations in FLNA and phenotypic diversity. Am. J. Med. Genet. A 140, 1726–1736 (2006).
Hart, A. W. et al. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 15, 2457–2467 (2006).
Acknowledgements
This study was supported by grant 21ek0109489h0002 from the Japan Agency for Medical Research and Development (AMED). This study was supported by grants 21ek0109489h0002 and JP22ek0109489 from the Japan Agency for Medical Research and Development (AMED) and National Center for Child Health and Development (NCCHD 2022A-3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oshina, K., Kamei, Y., Hori, A. et al. A novel FLNA variant in a fetus with skeletal dysplasia. Hum Genome Var 9, 45 (2022). https://doi.org/10.1038/s41439-022-00224-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00224-7
